
Abstract
Background
Pulmonary arterial hypertension (PAH), Group 1 pulmonary hypertension (PH), is a type of pulmonary vascular disease characterized by abnormal contraction and remodeling of the pulmonary arterioles, manifested by pulmonary vascular resistance (PVR) and increased pulmonary arterial pressure, eventually leading to right heart failure or even death. The mechanisms involved in this process include inflammation, vascular matrix remodeling, endothelial cell apoptosis and proliferation, vasoconstriction, vascular smooth muscle cell proliferation and hypertrophy. In this study, we review the mechanisms of action of prostaglandins and their receptors in PAH.
Main body
PAH-targeted therapies, such as endothelin receptor antagonists, phosphodiesterase type 5 inhibitors, activators of soluble guanylate cyclase, prostacyclin, and prostacyclin analogs, improve PVR, mean pulmonary arterial pressure, and the six-minute walk distance, cardiac output and exercise capacity and are licensed for patients with PAH; however, they have not been shown to reduce mortality. Current treatments for PAH primarily focus on inhibiting excessive pulmonary vasoconstriction, however, vascular remodeling is recalcitrant to currently available therapies. Lung transplantation remains the definitive treatment for patients with PAH. Therefore, it is imperative to identify novel targets for improving pulmonary vascular remodeling in PAH. Studies have confirmed that prostaglandins and their receptors play important roles in the occurrence and development of PAH through vasoconstriction, vascular smooth muscle cell proliferation and migration, inflammation, and extracellular matrix remodeling.
Conclusion
Prostacyclin and related drugs have been used in the clinical treatment of PAH. Other prostaglandins also have the potential to treat PAH. This review provides ideas for the treatment of PAH and the discovery of new drug targets.
Similar content being viewed by others


Background
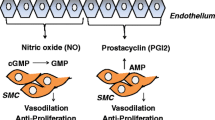
Pulmonary hypertension (PH) is a pathophysiological condition characterized by an abnormal increase in pulmonary arterial pressure caused by a combination of causes that can lead to dyspnea, right heart failure, and even death [1]. The global prevalence of PH is approximately 1%, and it can reach as high as 10% in individuals aged 65 and above as a result of cardiovascular and respiratory factors [2]. Furthermore, right heart failure is present in at least 50% of PH cases [2]. Left heart disease and lung disease are the two leading causes of PH [2]. PH hemodynamics are defined as a mean pulmonary arterial pressure (mPAP) > 20 mmHg measured by the right cardiac catheter at sea level and at resting state [3]. Figure 1 shows in detail the hemodynamic indices of PH, including pre-capillary PH, isolated post-capillary PH, isolated post-capillary PH, and exercise PH [4]. Based on the pathological findings, hemodynamic features, and clinical management strategies, the World Health Organization classified PH into five groups: Group 1, pulmonary arterial hypertension (PAH); Group 2, PH associated with left heart disease; Group 3, PH associated with lung diseases and/or hypoxia; Group 4, PH associated with pulmonary artery obstructions; and Group 5, PH with unclear and/or multifactorial mechanisms [3, 4]. Table 1 provides a detailed description of the classification of PH. This review primarily focused on PAH. The pathophysiological characteristics of PAH include vasoconstriction, extracellular matrix remodeling, and inflammation (Fig. 1). Pulmonary vascular remodeling in PAH is associated with some cellular dysfunction [5]. Abnormal endothelial cells (ECs) apoptosis and proliferation are common pathological features of pulmonary vessels in patients with PAH. Additionally, the proliferation, hypertrophy, and migration of pulmonary artery smooth muscle cells (PASMCs) contribute to severe remodeling of the pulmonary artery, resulting in increased pulmonary arterial pressure (PAP) [6]. The normal interaction between ECs and PASMCs is crucial for maintaining the homeostasis of the lung circulation. Endothelial cells release bioactive agents, including nitric oxide (NO) and endothelin-1 (ET-1), to regulate the function of the underlying smooth muscle cells (SMCs) [7, 8]. Under pathological conditions, the interaction between ECs and SMCs may be changed and certain molecules secreted by endothelial cells exert an influence on the contraction and proliferation of smooth muscle cells. For example, apoptotic ECs release TGFβ1 to induce SMCs proliferation [9] and the endothelial-derived factor CXCL12 induced SMCs proliferation [10].

Hemodynamic classification and pathology of PH. The hemodynamics indices of PH include the pre-capillary PH, isolated post-capillary PH, isolated post-capillary PH, and exercise PH. Pathological changes of PH include inflammation, vascular matrix remodeling, EC apoptosis and proliferation, and VSMC proliferation and migration. Abbreviation: CO, cardiac output; Cpc PH, combined post- and pre-capillary pulmonary hypertension; ECs, endothelial cells; Ipc PH, isolated post-capillary pulmonary hypertension; mPAP, mean pulmonary arterial pressure; PAWP, pulmonary arterial wedge pressure; PH, pulmonary hypertension; PVR, pulmonary vascular resistance; VSMC, vascular smooth muscle cells; WU, Wood units
Some signaling pathways (e.g., NO, endothelin, and prostacyclin pathways) and their modulators have been shown to play key roles in pulmonary vascular tone regulation and remodeling [11, 12]. The overexpression of ET-1 leads to vasoconstriction and vascular cell proliferation [13, 14]. However, NO and prostacyclin (PGI2) can lead to vascular dilation and changes in anti-proliferative mechanisms [15]. Currently, drugs targeting the prostacyclin, ET-1, and NO pathways are used in patients with PAH and have been shown to relieve the associated symptoms [16]. Prostacyclin analogs and prostacyclin receptor agonists have shown the potential to enhance exercise capacity, improve quality of life and Borg dyspnea score, and positively impact hemodynamic variables, including mPAP, cardiac index, and pulmonary vascular resistance (PVR) [17]. Endothelin receptor antagonists significantly improve the six-minute walk distance (6MWD), time to clinical worsening, cardiac index, and PVR of patients with PAH [14, 18]. Drugs targeting NO signaling pathway (phosphodiesterase type 5 inhibitors, activators of soluble guanylate cyclase) have demonstrated the potential to improve several clinical parameters, including 6MWD, mPAP, PVR, Borg dyspnea score, and time to clinical worsening [4, 19, 20]. There are five main types of PAH therapeutics: endothelin receptor antagonists, phosphodiesterase type 5 inhibitors, activators of soluble guanylate cyclase, prostacyclin and prostacyclin analogs, and prostacyclin receptor agonists [4]. Current treatments for PAH primarily focus on inhibiting excessive pulmonary vasoconstriction, however, vascular remodeling is recalcitrant to currently available therapies, and lung transplantation remains the definitive treatment for patients with PAH [21, 22]. Therefore, it is imperative to identify novel targets for improving pulmonary vascular remodeling in PAH.
Prostaglandins
In 1935, von Euler preliminarily isolated and extracted PGs from semen and named them PGs [23], which were subsequently successfully isolated [24]. At that time, PGs were thought to be a part of prostate secretion and were eventually found to be produced by seminal vesicles. Subsequently, PGs were found to exist widely in humans and other animals. PGs are a class of lipid-active proteins derived from arachidonic acid (AA), an eicosanoic unsaturated fatty acid. PGs biosynthesis is achieved through three successive enzymatic reactions. First, AA is released from membrane phospholipids by phospholipase A2 (PLA2) under various physiological and pathological stimuli. Subsequently, under the action of prostaglandin H synthase (PGHS), also known as cyclooxygenase (COX), PGs intermediate metabolites PGG2 and PGH2 are successively transformed. Finally, prostaglandin terminal synthetases/isomerases including prostaglandin D synthase (PGDS), prostaglandin E synthase (PGES), prostaglandin F synthase (PGFS), prostaglandin I synthase (PGIS), and thromboxane A synthase (TXAS) convert PGH2 into various bioactive PGs [25]. In mammals, PGs mainly include prostaglandin D2 (PGD2), prostaglandin E2 (PGE2), prostaglandin F2α (PGF2α), prostaglandin I2 (PGI2), and thromboxane A2 (TXA2) (Fig. 2). After synthesis, prostaglandins are transported into the extracellular microenvironment through simple diffusion. Subsequently, they bind to the prostaglandin receptors to perform various physiological functions [26]. Structural differences among PGs result in different biological activities. They are generally autocrine or paracrine factors and their target cells are located near their secretory sites. In some cases, PGs have different or even opposite effects on different tissues, depending on the type of receptors to which they bind. PGs receptors are a subfamily of G-protein-coupled receptors (GPCRs) known as PGD2 receptor 1 (DP1), PGD2 receptor 2 (DP2), PGE2 receptor 1 (EP1), PGE2 receptor 2 (EP2), PGE2 receptor 3 (EP3), PGE2 receptor 4 (EP4), prostaglandin F receptor (FP), prostacyclin receptor (IP), and TXA2 receptor (TP). EP3 and DP2 receptors inhibit cyclic adenosine monophosphate (cAMP) signaling, whereas EP2, EP4, DP1, and IP receptors activate cAMP signaling [27]. EP1, FP, and TP receptors mainly activate protein kinase C (PKC) and Ca2+ pathways [27]. The TP and EP3 receptors also activate Rho. EP2 and EP4 receptors also activate the phosphoinositide 3-kinase (PI3K) and β-arretin pathways [28]. PGs, prostaglandin-synthesis-related enzymes, and PGs receptors are associated with inflammation, cancer, and systemic disease [29]. PGD2 has been found to induce sleep, elicit allergic responses, inhibit platelet aggregation, and induce relaxation of both vascular and non-vascular smooth muscle [30]. PGE2 promotes tumor development [31], regulates blood pressure homeostasis (with activation of EP2 and EP4 receptors decreasing blood pressure, and activation of EP1 and EP3 receptors increasing blood pressure) [32], facilitates tissue repair and regeneration [33], and contributes to inflammation [34]. PGF2α promotes uterine contraction and vasoconstriction [35, 36]. PGI2 plays a role in promoting vasodilation and bronchial relaxation, as well as inhibiting platelet aggregation, inflammation, and proliferation [37]. TXA2 promotes platelet aggregation, airway constriction, and arterial contraction [38].

The prostaglandin synthesis pathway and corresponding receptors. AA is released from membrane phospholipids by PLA2, and is metabolized to PGH2 by COX-1 and COX-2. PGH2 is metabolized to TXA2 by TXAS, PGI2 by PGIS, PGE2 by PGES, PGF2α by PGFS, and PGD2 by PGDS. TXA2 binds to TP, PGI2 binds to IP and PPARs, PGE2 binds to EPs, PGF2α binds to FP, and PGD2 binds to DPs. Abbreviation: AA, arachidonic acid; COX, cyclooxygenase; CYP450, cytochrome P450; LOX, lipoxygenase; PLA2, phospholipase A2;
A single-cell study demonstrated that activation of EP3 receptor enhanced adhesion and cytotoxicity of NK cells toward hepatic stellate cells [39], and various molecules involved in cell adhesion and toxicity can significantly impact the progression of PAH [40, 41]. Another single-cell study showed that PGE2 treatment inhibited senescence of decidual cells [42], and senescence-related molecules play a crucial role in the pathogenesis of PAH [43]. However, further research is needed to thoroughly investigate the role of PGs in PAH using single-cell analysis.
Prostaglandins and pulmonary Hypertension
Currently, research on prostaglandins and their receptors in pulmonary hypertension predominantly focuses on animal models. Common animal models of PH are induced by monocrotaline (MCT), chronic hypoxia, and hypoxia/SU5416. MCT can be used to simulate Group 1 PH, while hypoxia can simulate Group 3 PH. Additionally, hypoxia combined with SU5416 can simulate Group 1/3 PH [44]. MCT induces endothelial damage in pulmonary blood vessels, resulting in the narrowing or occlusion of the vascular lumen [45]. In the MCT-induced model of PH, there is an observed increase in apoptosis of endothelial cells and proliferation of PASMCs [46]. Hypoxia is associated with the development of PH in patients with chronic lung diseases, including interstitial lung disease and chronic obstructive pulmonary disease [47]. Hypoxia-induced PH leads to the thickening of the pulmonary artery wall and increased vasoconstriction. However, the hypoxia model often leads to less severe manifestations of PH with weak VSMCs proliferation and obstructive intimal lesions [48]. In our previous study, we observed that deficiency of DP1 promoted the proliferation of PASMCs in the pulmonary blood vessels of rats treated with MCT; however, we did not observe a significant increase in PASMC proliferation in DP1 knockout mice treated with hypoxia [49]. This finding implies that compared with the hypoxia model, the MCT model is more helpful to observe the anti-proliferation effect of DP1 in vascular remodeling. The hypoxia model does not accurately reflect the same degree of pathological changes observed in patients with PAH, thus further development is needed. Sugen 5416, an antagonist of the VEGF receptor-2 (VEGFR-2), can induce apoptosis in endothelial cells and proliferation in SMCs [50]. The combination of hypoxia and Sugen 5416 leads to severe and progressive remodeling of the pulmonary vasculature, providing a more accurate simulation of Group 1 PH [51]. The more severe PH phenotype animal model makes it more conducive to studying the therapeutic effects of molecules and drugs, such as PGs, in PAH.
AA causes vascular contraction, phenotypic transformation of SMCs, and an imbalance in endothelial cell proliferation and apoptosis, mainly through various derivatives including PGH2, PGE2, TXA2, 12-HETE (12-hydroxy-5,8,10,14-eicosatetraenoic acid), 15-HETE, LTB4 (leukotriene B4), epoxyeicosatrienoic acids (EETs), ultimately leading to vascular remodeling [52,53,54]. AA has three metabolic pathways: COX, lipoxygenase (LOX), and cytochrome P450 (CYP450). There are two isoforms of cyclooxygenase: COX-1 and COX-2. COX-1 is constitutively expressed in the majority of tissues, whereas COX-2 is constitutively expressed at lower levels but is induced in inflammation and hypoxia [55]. COX-2 plays a role in cardiovascular diseases, including changes in PH [53]. The COX-2 protein is associated with PH in multiple species. COX-2 is overexpressed in the lung tissues of children with PH, increased in hypoxia-induced human pulmonary artery smooth muscle cells (HPASMCs) in vitro, and has an anti-proliferative function [56, 57]. In rats, a significant increase in COX-2 expression in pulmonary vessels and SMCs was observed after hypoxia induction; however, COX-1 expression did not significantly change. Moreover, SC236, a selective COX-2 inhibitor, aggravated PH [58]. COX-2-dependent contractile factors caused abnormal pulmonary artery responses in piglets exposed to hypoxia for three days [59]. In mice, both genetic deletion of COX-2 and the pharmacological inhibition of COX-2 by nimesulide exacerbated hypoxia-induced PH by acting on vascular remodeling, specifically characterized by PASMCs hypertrophy, without inducing cell proliferation [60]. In addition, in vitro experiments have demonstrated that COX-2 deficiency enhances the contractility of hypoxia-induced vascular SMCs and their interactions with the extracellular matrix [60]. In addition to its role in hypoxia-induced PH, COX-2 plays a role in MCT-induced PAH. In MCT-induced PAH mouse models, COX-2 knockdown exacerbates oxidative stress-derived endothelial dysfunction, vasoconstriction, and mild inflammation, thereby aggravating PAH [61]. Bone marrow-derived endothelial progenitor cells (BMEPCs) effectively attenuated MCT-induced PAH in rat models, and the protective effects of BMEPCs on pulmonary vessels may be mediated by the COX-2/PGI2/cAMP pathway [62]. Although COX-1 expression in PH lung tissue did not change significantly, endotracheal administration of COX-1 alleviated MCT-induced PAH and right ventricular hypertrophy in rats [63]. Drugs that target COX-2 have side effects because there are numerous downstream molecules of COX-2, such as prostaglandin and prostaglandin receptors. Drugs that target downstream molecules have improved safety and efficacy [53]. Prostacyclin, prostacyclin analogs and receptor agonists, including selexipag, epoprostenol, beraprost, iloprost, and treprostinil, have been clinically used to treat PAH. Prostacyclin analogs exhibited heterogeneous binding affinities to other PG receptors, which can result in varying clinical efficacy [64, 65]. Prostacyclin and prostacyclin analogs do not act solely on IP receptor. Epoprostenol mainly acts on IP and EP3; beraprost mainly acts on IP; iloprost mainly act on IP, EP1and EP3; and treprostinil mainly acts on IP, EP2, and DP1. (Tables 2 and 3). DP1, EP2, EP4, and IP signaling pathways mainly improve pulmonary vascular remodeling, thus improving PAH (Fig. 3), while DP2, EP1, EP3, and TP signaling pathways aggravate PAH (Fig. 4). Compared to iloprost, treprostinil demonstrated a more sustained effect on PVR and exhibited better tolerance, due to its differential affinity for specific prostaglandin receptors [66]. Various prostaglandin receptors play distinct roles in the development and progression of PH. Therefore, in this review, we summarized the progress of prostaglandins and prostaglandin receptors in the study of PAH.

Prostaglandin receptor-related pathways in alleviating pulmonary hypertension. Activation of DP1, EP2, EP4, and IP promotes vasodilation and inhibits the proliferation of pulmonary vascular smooth muscle cells (PVSMCs) through the AC/cAMP/PKA pathway. DP1 activation also attenuates hypertrophy of PVSMCs through PKA-mediated dissociation of raptor from the mTORC1 complex. EP4 also inhibits PVSMC proliferation and migration through PKA/PPARγ and Kv channels. Niacin stimulates the expression of H-PGDS in macrophages and increases the release of PGD2. PGI2 plays an anti-apoptotic role through PPARβ in endothelial cells and PPARα in VSMCs. Abbreviation: AC, adenylate cyclase; ATP, adenosine triphosphate; cAMP, cyclic adenosine monophosphate; CREB, cAMP-response element binding protein; PKA, protein kinase A; PPAR, peroxisome proliferator-activated receptor

How prostaglandin receptor-related pathways are involved in aggravating pulmonary hypertension. Activation of TP and EP1 promotes vasoconstriction through the PLC/PKC pathway. Via the Rho pathway, EP3 leads to extracellular matrix remodeling and TP leads to vasoconstriction. EP3 can inhibit the cAMP/PKA pathway and TP can inhibit Kv channels. CRTH2 activation in Th2 cells promotes PASMC proliferation by activating STAT6. Abbreviation: CREB, cAMP-response element binding protein; DAG, diacylglycerol; ERK: extracellular signal-regulated kinase; GEF, guanine nucleotide exchange factor; IP3, inositol triphosphate; Jak, Janus kinase; LAP: latency-associated protein; MLC: myosin light chain; MMP, Matrix metalloproteinase; MRTF-A, myocardin-related transcription factor A; MT1-MMP, membrane type 1-matrix metalloproteinase; PIP2, phosphatidylinositol (4,5) bisphosphate; PKC, protein kinase C; PLC, phospholipase C; Rock, Rho-associated protein kinase; SMAD, small mother against decapentaplegic; STAT6, signal transducer and activator of transcription 6; TGF-β1: transforming growth factor beta
Prostaglandin D2 and prostaglandin D2 receptors
Prostaglandin D2
There are two distinct types of PGD2 synthases: lipocalin-type PGD2 (L-PGDS) and hematopoietic PGD2 (H-PGDS) [67]. L-PGDS is mainly expressed in endothelial cells and cardiomyocytes of the cardiovascular system [68]. H-PGDS is highly expressed in macrophages and mast cells [69]. Systemic biosynthesis of PGD2 occurs mainly via H-PGDS in mice [70]. The physiological function of PGD2 varies depending on the cell and tissue type, mainly according to the receptor type to which it binds. PGD2 was significantly elevated in patients with primary PH [71]. A large infusion of PGD2 specifically reversed induced PH and prevented hypoxic pulmonary vasoconstriction in newborn lambs without changing their systemic blood pressure [72]. However, although PGD2 is a specific pulmonary vasodilator in fetuses and newborn animals, it does not reduce pulmonary blood pressure in newborn infants with persistent PH syndrome or improve oxygenation [73]. We have found that niacin prevents the progression of MCT-induced and hypoxia/SU5416-induced PAH in rats and suppresses the development of hypoxia/SU5416-induced PAH in mice by enhancing the expression of H-PGDS in lung tissue macrophages and increasing the release of PGD2, which inhibits the hypertrophy of pulmonary vein smooth muscle cells (PVSMCs) and improves the remodeling of pulmonary vessels. Deletion of H-PGDS exacerbated hypoxia/SU5416-induced PAH in mice and eliminated the protective effects of niacin against PAH, but not L-PGDS [74]. However, in this study there was no direct evidence of the mechanisms underlying receptor PGD2 function.
Prostaglandin D2 receptor 1
DP1 is a protein encoded by the PTGDR1 gene (also known as PTGDR) located at q22.1 on chromosome 14, a chromosomal site associated with asthma and other allergic diseases [75, 76]. PGD2, PGE2, PGF2α, PGI2, and TXA2 prostaglandins are endogenous ligands of DP1, of which PGD2 has the highest affinity and is also a DP1 ligand in other rodents [64, 65, 77,78,79,80]. DP1 is mainly expressed in cells involved in mediating allergic and inflammatory responses, namely mast cells, basophil and eosinophils, Th2 cells, and dendritic cells in humans and rodents, and cells that contribute to these responses, namely human and rodent airway epithelial cells, vascular endothelial cells, and goblet cells [81, 82]. DP1 has been linked to allergic diseases such as rhinitis and asthma [83]. Also, DP1 plays an important role in neurological diseases [84], reproductive development [85], digestive tract diseases [86], cardiovascular diseases [69], and maintaining hemodynamics in rodents and humans, including ischemia-reperfusion injury and niacin induced vasodilation [87, 88]. DP1 is expressed in both the pulmonary artery and veins, and its activation induces vascular relaxation [89]. Treprostinil induces pulmonary venous relaxation in part by acting through DP1, and its effect on DP1 in human pulmonary veins may contribute to the treatment of PAH [90]. Recently, we have been reported that DP1 activation prevents hypoxia-induced PH through PKA/raptor-dependent mTORC1 (mammalian target of rapamycin complex 1) dissociation. The DP1 expression level is downregulated in the pulmonary arteries (PAs) of various PAH animal models and patients with idiopathic PAH. Furthermore, DP1 receptor knockout in mice aggravated hypoxia/SU5416-induced PAH by increasing mTORC1 activity; therefore, DP1 activation provides protection against hypoxia-induced PH through PKA/raptor-dependent mTORC1 dissociation. DP1 activation also attenuates hypoxia-induced PA remodeling and hypertrophy of pulmonary VSMCs through the PKA-mediated dissociation of raptor from the mTORC1 complex [49]. This provides direct evidence for the pathogenesis of DP1 in PAH.
Prostaglandin D2 receptor 2
The PTGDR2 gene and GPR44 together code for the protein known as prostaglandin D2 receptor 2 (DP2 or CRTH2) [91]. It is selectively expressed in Th2 cells, and is therefore a chemoattractant receptor homologous molecule expressed in Th2 cells (CRTH2) [92, 93]. PTGDR2, located at q12.2 on human chromosome 11, consists of two introns and three exons, and encodes a GPCR composed of 472 amino acids [92]. CRTH2 is also expressed in eosinophils [94], mast cells [95] and group 2 innate lymphoid cells (ILC2s) [96]. PGD2 induces chemotaxis of these immune cells through CRTH2 signaling, which is the main pathway driving type 2 inflammation [96,97,98]. The PGD2/CRTH2 signaling axis has been extensively studied in type 2 inflammation-related diseases such as asthma and atopic dermatitis [83, 99]. Therefore, CRTH2 antagonists may develop into a novel type of anti-inflammatory medication [100,101,102,103]. The infiltration of inflammatory cells around the pulmonary vessels is usually observed in patients with PH [104], and similar pulmonary inflammation has been observed in experimental PH models [105, 106]. In addition, some patients with immune diseases (such as systemic sclerosis and systemic lupus erythematosus) also develop PH [107,108,109]. Inflammation and immune disorders may exacerbate PH development, and anti-inflammatory therapy may improve PH development in patients [110]. For instance, acetazolamide prevented PVSMCs dedifferentiation and proliferation in the hypoxia/SU5416 induced rat PAH model through the inhibition of macrophage carbonic anhydrase [111]. Interferon regulatory factor 7 (IRF7) inhibited inflammation by suppressing NF-κBp65 signaling and improved vascular remodeling in MCT-induced rat models of PAH [112]. It has been found that patients with idiopathic PAH and rodents with PAH models exhibit higher levels of CRTH2 expression in their circulating CD4 T cells. CRTH2 knockout significantly improved pulmonary remodeling and PAH in different PAH mouse models through inhibiting Th2 activity via reducing the secretion of IL-4 and IL-13 by Th2 cells [113]. Furthermore, CRTH2 activation in Th2 cells promoted PASMC proliferation by activating signal transducer and the activator of transcription 6 (STAT6) [113].
PGD2 primarily exerts its effects through DP1 and DP2 receptors, which exhibit antagonistic roles in PAH. DP1, expressed in PAs and veins, contributes to the improvement of PAH upon activation [89]. Conversely, activation of DP2, predominantly present in Th2 cells, aggravates PAH. Selective activation of DP1 with inhibition of DP2 becomes crucial in the treatment of PAH associated with PGD2.
Prostaglandin E2 and prostaglandin E2 receptors
Prostaglandin E2
PGE2 is produced by various cell types in the body, such as epithelial cells, fibroblasts, and infiltrating inflammatory cells [33] and mediates many physiological and pathological processes, including vascular homeostasis, inflammation, pain, and kidney function [114, 115]. PGE2 performs various complex biological functions by binding to different downstream prostaglandin E receptors, including EP1, EP2, EP3, and EP4 [116]. PGE2 expression was elevated in a hypoxia/SU5416-induced rat PAH model [117]. PGE2 mediates anoxic constriction of the rat intrapulmonary artery (IPA) [118] and may inhibit intense constriction of PASMCs in response to hypoxia in mice [60]. In addition, intravenous infusion of PGE2 reduced PAP by reducing cardiac output (CO) in broiler chickens but did not dilate the pulmonary vasculature [119]. Moreover, in samples isolated from human patients, impairment of PGE2-induced bronchodilation may be involved in Group 3 PH pathogenesis [120]. The pathway of action of PGE2 depends on the cell type and the receptor to which it binds.
Prostaglandin E2 receptor 1
The PTGER1 gene encodes the protein known as EP1, located at the p13.12 locus of human chromosome 19, and contains two introns and three exons and encodes a GPCR [121]. PGE2 activates phospholipase C (PLC), which then triggers PKC, increasing phosphatidylinositol hydrolysis and intracellular calcium concentrations through binding to the EP1 receptors [122]. EP1 is widely expressed in rodent tissues and cells including the kidneys, lung, stomach, thalamus, and central nervous system, but is only distributed in a few human organs and cells, such as the pulmonary vasculature, myometrium, and colonic longitudinal muscles [123,124,125,126,127]. To date, animal studies on EP1 have mainly focused on pain, cancer, and renal function with few studies on cardiovascular [34, 128,129,130]. In terms of cardiovascular function, EP1 plays a major role in blood pressure regulation. PGE2 acts on EP1 receptor to cause vascular contraction and increased blood pressure [131]. Gene knockout of the EP1 receptor significantly reduces basal blood pressure and inhibits Ang II-induced hypertension and associated organ damage in mice. Sulprostone, a nonselective agonist of EP1/EP3 receptors, promotes vasoconstriction and increases blood pressure; this vasoconstriction effect is significantly inhibited in EP1/EP3 receptor-knockout mice [132]. However, the role of EP1 in PH has not yet been elucidated. The Th1-mediated immune reaction in vivo is influenced by the PGE2- EP1 pathway, which alters the Th1/Th2 balance toward Th1 dominance [133]. The Th1 cytokine IFN-γ is required for the development of the pneumocystis-associated PH mouse model [134]. Therefore, EP1 may affect the development of PH by promoting the differentiation of CD4 T cells toward Th1 cells; however, direct evidence of this mechanism is lacking.
Prostaglandin E2 receptor 2
The human gene PTGER2 encodes EP2, which is the prostaglandin receptor of PGE2. It is located at the p22.1 position of human chromosome 14 and contains two introns and three exons, encoding GPCRs [135]. EP2 receptors mediate the increase in cAMP levels and cAMP levels can increase EP2 receptor expression [136]. After binding to PGE2, the EP2 receptor initiates the PI3K/protein kinase B (PKB) pathway through the dissociated Gβγ subunit, which phosphorylates and inactivates glycogen synthetase kinase 3 (GSK-3), stabilizing β-catenin and causing nuclear translocation as well as the expression of genes that promote inflammation and growth [137, 138]. The transcription factor nuclear factor κB (NF-κB), which is then transported to the nucleus and mediates the transcription of a range of genes implicated in inflammation, may also be phosphorylated by activated PI3K/PKB [139, 140]. The EP2 receptor prevents neutrophil phospholipase D pathway activation [141] and causes Th1 cell differentiation [142], which is dependent on PI3K. The EP2 receptor is extensively expressed in humans and has been found to be concentrated in the cerebral cortex, articular cartilage, lungs, and smooth muscle, where it influences a number of physiological processes including neural plasticity, immunoregulation, and vasodilation [27, 143]. In addition, PGE2/EP2 signaling promotes cell proliferation [140, 144]. A growing number of studies have revealed that EP2 is crucial for PAH. Treprostinil has a high affinity for DP1, EP2, and IP receptors, and part of its effect on PAH therapy is mediated by EP2 receptors [65]. In humans, the majority of inherited PAH cases are linked to mutations in members of the transforming growth factor (TGF) receptor superfamily [89]. The expression of transforming growth factor β1 (TGF-β1) in ECs can promote the differentiation of SMCs into a synthetic phenotype [145]. TGF-β1 plays a crucial role in the development of PAH in animal models. SD-208, TGF-beta receptor I inhibitor, can improve pulmonary vascular remodeling, leading to an amelioration of PH [146, 147]. Therefore, the inhibition of TGF beta 1 is important to prevent pulmonary vascular remodeling and PAH development. The EP2 receptor antagonist PF-04418948 inhibits the expression of TGF-β1 of lung fibroblasts in humans, and enhances the expression of the genes for collagen production (COL1A1 and COL1A2) and fibroblast contractility (ACTG2) when treprostinil is present [147]. By reducing the production and accumulation of type I collagen and fibronectin, treprostinil has a positive preventative effect on pulmonary vascular wall remodeling [148]. PF-04418948 partially or completely reversed treprostinil-induced decline in COL1A1, COL1A2, and ACTG2 expression [147]. In addition, activation of the prostaglandin EP2 receptor inhibits fibroblast functions, including proliferation, migration, and the transition from fibroblasts to myofibroblasts [149]. EP2 is a major subtype of the prostaglandin receptor in PH that inhibits fibrosis and fibrosis-induced remodeling. Moreover, EP2 receptor was upregulated in HPASMC and PAs from PAH patients, suggesting that EP2 may play a role in the development of PH [150]. Butaprost, a highly selective EP2 receptor agonist, was utilized to investigate the potential role of EP2 in PH development. Butaprost administration led to a concentration-dependent decrease in HPASMC proliferation. PF-04418948 abolishes the antiproliferative effects of butaprost. The anti-proliferative effect of the therapeutic dose of treprostinil appears to depend primarily on the activation of the EP2 receptor in HPASMCs in patients with PAH [150]. Therefore, targeting EP2 receptors is a promising strategy for the treatment of PAH.
Prostaglandin E2 receptor 3
The human gene PTGER3 encodes the prostaglandin EP3 receptor, commonly known as EP3. The gene PTGER3 generates GPCRs from the rhodopsin-like receptor family and is found at the p31.1 region of human chromosome 1 [151, 152]. In humans, PTGER3 encodes at least eight different isomers, namely PTGER3-1 to PTGER3-8 (EP3-1, EP3-2, EP3-3, EP3-4, EP3-5, EP3-6, EP3-7, and EP3-8). In mice, PTGER3 encodes at least three isomers, Ptger1-Ptger3 (i.e. Ep3-α, Ep3-β, and Ep3-γ) [81, 153]. These isomers are variations created by selective 5’-end splicing of DNA to create proteins with altered C-termini or regions therein [154, 155] and may perform different functions due to differences in tissue expression and the signaling pathways they activate [156]. EP3 receptor is widely distributed in human tissues and play crucial functions in the kidneys, urinary bladder, reproductive system, brain, and cardiovascular system [157]. Similar to most other prostaglandin receptors, the EP3 receptor has been involved in cancer, inflammation, and immune regulation. Investigation of the pharmacological characteristics of the EP3 receptor demonstrated smooth muscle contractility [157]. EP3 receptors mediate vasoconstriction in human arteries, including PAs [158], and are also implicated in mediating the pulmonary vascular contractions brought on by isoprostanes, which are oxidative polyunsaturated fatty acid metabolites that are significantly elevated in patients with PH [159, 160]. Five of the ten different splicing variants of EP3 (EP3-1a, EP3-1b, EP3-1c, EP3-4, and EP3-5) were markedly elevated in human PA (hPA) exposed to 1% O2 for 24 h [161]. EP3 expression increased in the PAs of hypoxia-induced mice and MCT-treated rats compared to normoxia mice and control rats, respectively [161, 162]. By the inhibition of Rho-dependent extracellular MMP-2/TGF-β1 signaling, disruption of EP3 improved pulmonary vascular remodeling and alleviated both hypoxia-induced and hypoxia/SU5416-induced PAH in mice. More significantly, therapy with an EP3 antagonist L-798,106 suppressed the progression of PAH in the MCT rat model [161]. In vitro or in vivo, PASMC proliferation or hypertrophy in animals with PAH is not significantly affected by EP3. In summary, overexpression of the EP3 receptor in PAs contributes to pulmonary vascular remodeling in PAH.
Prostaglandin E2 receptor 4
The PTGER4 gene encodes prostaglandin E2 receptor 4 (EP4), the prostaglandin receptor of human PGE2, located at the p13.1 position of human chromosome 5 and containing seven exons encoding GPCRs of the rhodopsin-like receptor family [163]. EP4 is broadly expressed in human tissues [27] and has a high expression level in the gastrointestinal tract, uterus, hematopoietic tissues, and skin [164]. The relaxation of the pulmonary veins is mediated by EP4 receptors, although PAs are unaffected [165]. Bradykinin stimulates the expression of COX-2 in human PASMCs by activating the cAMP response element with EP4 and EP2 agonists [166]. Compared to the control group, IP receptor expression was decreased in the lung samples of patients with idiopathic PAH and in the lungs of MCT-induced 28d rats, but the expression of the EP4 receptor was stable [167]. An EP4 receptor antagonist, AH23848, dose-dependently decreased the rise of cytoplasmic cAMP caused by iloprost in PASMCs, but AH6809 (an EP2 receptor antagonist) had no such effect [167]. This finding indicated that under the condition of lower IP receptor expression related to PAH, iloprost exerts vasodilatory activities via the EP4 receptor. Another study reported that in the presence of lower IP expression related to PAH, the EP4-PKA-PPARγ signaling pathway exerts a significant regulatory role in suppressing PASMC proliferation and migration. L-902,688, an EP4 agonist, was effective in severe experimental PAH rodent models by raising PPARγ expression [168]. Furthermore, EP4-specific agonist L-902,688 reduced right ventricular [93] fibrosis and prevented TGF-β1-induced endothelial-mesenchymal transition (EndMT) [169]. Therefore, EP4 is a potential therapeutic target for improving pulmonary vascular remodeling and enhancing RV function of lower IP expression related to PAH. The beneficial effects of another PGI2 analog, beraprost (BPS), on vascular contraction in PH may be mediated, partly through interacting with the EP4 receptor and reactivating Kv channels [170]. The expression and functional reduction of Kv channels are included in the pathogenesis of hypoxia-induced PH, ultimately causing vascular remodeling and pulmonary vasoconstriction, and Kv channel upregulation has therapeutic value for PH [171]. In addition to its benefits in patients with Group 1 PAH, EP4 is also beneficial in patients with Group 3 PH with respiratory diseases. EP4 agonists can improve outcomes in patients with Group 3 PH by decreasing dyspnea and improving the capacity to perform physical effort (6 MWD).
Together, EP2 and EP4 have vasodilatory effects, while EP1 and EP3 contribute to vasoconstriction. The function of PGE2 on PH is complex, depending on the dominant receptor subtype at that time. Thus, selective targeting PGE2 receptors or combination of PGE2 with EP1 and EP3 inhibitors may be a promising treatment for patients with PH.
Prostaglandin F2α and prostaglandin F receptors
Prostaglandin F2α
PGF2α plays a significant role in the female reproductive system, and participates in physiological processes such as pregnancy physiology, the onset of labor, and postpartum uterine contraction [172]. PGF2α is known to be a potent vasoconstrictor derived from the prostanoids family [173]. It is associated with hypertrophic growth of cardiomyocytes, vascular smooth muscle cells, and skeletal muscle cells [174]. ROS is required for PGF2α-associated vascular smooth muscle hypertrophy [175]. An analog of prostaglandin F2, 15-F2t-isoprostane (15-F2t-IsoP) mediates the vasoconstriction of PAs and resistance microvessels, as well as mitogenesis in VSMCs [176]. 15-F2t-IsoP stimulates endothelial cell proliferation and ET-1 synthesis in PAs [177]. Patients with PH have been observed to have higher urine concentrations of 15-F2t-IsoP and it is associated with survival [160]. One study reported elevated plasma concentrations of 15-F2t-isoP in 80 patients with IPAH. Moreover, individuals with baseline plasma concentrations of 15-F2t-isoP > 97 pg/ml had a considerably reduced chance of surviving, and that concentrations were elevated in patients who died during follow-up (30 ± 12 months) and reduced in those who survived [178]. It has been reported that 15-F2t-IsoP acts through the TP receptor [179].
Prostaglandin F receptor
PGF2α biological effects are mediated by FP, which comprises seven exons and is encoded by the PTGFR gene, which is found on human chromosome 1 at location p31.1 and belongs to the family of GPCRs called rhodopsin-like receptors [180, 181]. FPA and FPB are different C-terminal length isoforms of PTGFR encoded by alternatively spliced transcripts [158, 182]. FP receptors are widely distributed in human tissues and have important functions in reproduction, the central nervous system, kidneys, eyes, cardiovascular system, cancer, and bone. The systolic blood pressure of FP knockout mice was significantly lower compared to that of wild-type mice, and this was accompanied by significant reductions in plasma concentrations of renin and angiotensin-1 [183]. However, there is a lack of research on the role and function of FP in PH, necessitating further in vivo and in vitro studies in this area.
Prostaglandin I2 and prostaglandin I2 receptor
Prostaglandin I2
PGI2 is mostly generated by endothelial cells and VSMCs [184, 185], and has various pharmacological effects including vasodilation, inhibition of smooth muscle cell proliferation, and platelet aggregation [186]. PGI2 and some of its analogs are PPARα and PPARβ/δ ligands, as are some prostaglandin receptors. It is no longer possible to conceptualize PGI2 as a hormone that only acts biologically by activating the IP receptor [187]. PPARs are members of the nuclear receptor superfamily, and three of them (PPARα, PPARβ/δ, and PPARγ) have been identified in mammalian cells [188]. Pretreatment of human umbilical vein endothelial cells (HUVEC) with PGI2 prevented H2O2-induced apoptosis, while suppression of PPARβ/δ eliminated the anti-apoptotic effect of PGI2 [189].PGI2 was found to protect vascular smooth muscle cells from oxidative-induced apoptosis, although its anti-apoptotic effect was eliminated by PPARα inhibitors [190]. Therefore, PGI2 may exert its effects on PAH through the PPAR pathway. Prostacyclin analogs and prostacyclin receptor agonists are well established in the treatment of PAH [191]. Currently, prostacyclin analogs and prostacyclin receptor agonists are treatment options for PAH and are recommended for patients with functional Grade II–IV PAH based on the latest PAH treatment recommendations [16]. When monotherapy and other treatments fail to control symptoms in patients with PAH, these analogs are taken into consideration for combination therapy. Moreover, different prostacyclin analogs and prostacyclin receptor agonists and their preparations can be administered intravenously, subcutaneously, or via inhalation to patients with PAH. Prostacyclin analogs include iloprost, treprostinil, and beraprost. Selexipag is a prostacyclin receptor agonist. Epoprostenol, a synthetic prostacyclin, mainly acts on IP, EP3, primarily inducing vasodilation and inhibiting platelet aggregation [192]. It has been shown to significantly improve dyspnea, fatigue symptoms, and exercise capacity in patients with PAH, as well as improve hemodynamic parameters including PVR, mPAP, and cardiac index [193, 194]. Due to its short half-life (2–3 min), epoprostenol can only be administered through continuous intravenous infusion [195], which poses a risk of infection. Iloprost, acts on mainly IP, EP1, EP3, is a vasodilator and also has antiplatelet properties [196]. Inhaled iloprost improves PVR, mPAP, and 6MWD in patients with PAH [197, 198]. Treprostinil, a prostacyclin analog, acts on mainly IP, EP2, DP1, and also exerting vasodilatory effects and inhibiting platelet aggregation [199, 200]. Treprostinil has a prolonged half-life (4 h) and can be administered via intravenous, subcutaneous, inhalation, or oral routes [16]. Treprostinil administered via the intravenous, subcutaneous, or inhalation route can also improve PVR, mPAP, and 6MWD in patients with PAH [201,202,203]. Beraprost, a prostacyclin analog, mainly acts on IP, has vascular dilatation and antiplatelet effect [204]. Oral prostaglandin can improve PVR, mPAP, and 6MWD in patients with PAH [205, 206]. The therapeutic indications, administration, side effects, and corresponding receptors of prostacyclin analogs and prostacyclin receptor agonists are shown in Table 2. Each drug has a different affinity for the prostaglandin receptors (Table 3), which may be related to their effects.
Prostaglandin I2 receptor
The prostacyclin receptor, also known as the prostaglandin I2 receptor or IP, modulates the biological activity of prostacyclins or PGI2 by binding to them. The PTGIR gene in humans encodes the IP receptor, being located at locus q13.32 on human chromosome 19 and contains six exons encoding GPCRs of the rhodopsin-like receptor family [207, 208]. Northern blot analysis revealed that the expression level of IP receptor mRNA was highest in the thymus, and high levels of IP mRNA expression were found in the lung, heart, and spleen [208, 209]. Activation of IP receptors triggers the formation of intracellular cyclic adenosine phosphate and activates protein kinase A, which mediates pulmonary artery vasodilation, inhibits platelet aggregation, and relaxes smooth muscles [210]. A knockout mouse model of the IP receptor confirmed the blood pressure-lowering and anti-aggregatory capabilities of cicaprost, as well as its anti-proliferative activities in cultured mouse PASMCs [211]. In addition to its function in vascular dilatation, proliferation inhibition, and pulmonary remodeling protection, it has been reported that PGI2 regulates the immune response through IP signaling [212], and also by promoting Th17 differentiation in vivo, which may have clinical significance for the application of PGI2 and its analogs in the treatment of PAH [213]. Selexipag, a prostacyclin receptor agonist, selectively binds to IP, can improve the prognosis of patients with PAH [214, 215]. Ralinepag is another prostacyclin receptor agonist, and a Phase 3 study for Ralinepag is ongoing. In the Phase 2 trial, Ralinepag dramatically decreased PVR in patients with PAH [216].
Thromboxane A2 and thromboxane A2 receptor
Thromboxane A2
TXA2 is mainly produced by platelets, lung, kidney, and intestinal parenchymal cells, and has various pharmacological effects, including platelet aggregation, airway constriction, and contraction of different types of arteries, including PAs [38]. In vitro, TXA2 pretreatment significantly promotes hypoxic pulmonary vasoconstriction. In vivo, administration of TXA2-mimicking drug U46619 caused pulmonary vasoconstriction, leading to an increase in PVR and mPAP, decrease in CO, and U46619 can be utilized to construct PH in dogs [217, 218]. In addition, TXA2 production and disruption of TP signaling attenuated the detrimental impact on pulmonary and cardiac metrics in a porcine hypoxia-induced PH model [219]. TXA2 exerts its pharmacological effects via the TP receptor.
Thromboxane A2 receptor
The TBXA2R gene, which is found on chromosome 19 at location p13.3, encodes TP, generally referred to as the TXA2 receptor [220]. TBXA2R encodes a member of the G protein-coupled superfamily of seven-transmembrane receptors [221]. There are two subtypes of human TP receptor: TPα and TPβ [222]. Platelets exhibit significant levels of the α isoform, but the β isoform is not identified [221]. The β isoform is generated in human endothelial cells. Rodents exhibit the TPα isoform only. These rodents are utilized as animal models to clarify how genes and their byproducts work; however, they do not have two TP isoforms, which limits our recognizing of the various roles played by each TP receptor isoform [158]. Research has mainly focused on the role of TP receptors in platelet function. Nonetheless, it is now evident that TP receptors are widely distributed in various systems and cells types [221]. TP receptors have been identified in heart, immunological, reproductive, lung, and nervous system tissues [27]. Several studies have demonstrated the significance of TP signaling in the onset and development of PH [223,224,225,226,227]. In a porcine model, the TP antagonist daltroban attenuated hypoxic pulmonary vasoconstriction by decreasing mPAP [219]. In rats, TP receptor activation induces a contractile response through Rho kinase signal [228]. NTP42, an antagonist of TP, attenuated the MCT-induced PAH, comparable effectiveness to that of Selexipag or Sildenafil, which are the standard treatments. In addition, in MCT-treated rats, NTP42 significantly improved pulmonary vascular remodeling, inflammatory mast cell infiltration, and fibrosis with greater effects than those observed with Sildenafil and Selexipag [229]. Furthermore, NTP42:KVA4, an oral formulation of NTP42, was found to alleviate pulmonary pathologies, reduce right ventricular remodeling, and improve hypertrophy, by antagonizing TP signaling, thereby reducing PAH pathophysiology and improving cardiac function [230]. Moreover, the elevations in mPAP and right systolic ventricular pressure (RSVP) induced by hypoxia/SU5416 was significantly decreased by the combination of NTP42 and Sildenafil; however, Sildenafil or NTP42 mono-therapy did not reduce the increase in mPAP and RSVP. Comprehensive histological analyses suggested that combined treatment with NTP42 and Sildenafil was considerably more beneficial for pulmonary vessel remodeling, right ventricular hypertrophy, and fibrosis than monotherapy with either drug alone. Sildenafil exhibits vasodilatory and anti-proliferative effects by NO signaling pathway, while NTP42 effectively inhibits excessive vasoconstriction and microvascular thrombosis by antagonizing TP signaling. Both NTP42 and Sildenafil contribute to the reduction of PH through distinct yet complementary mechanisms [231]. It is considerably more beneficial to treat or counteract the main etiologies underlying PAH when NTP42 and Sildenafil are used together in dual treatment [231]. Increased availability of TXA2 as well as increased contractile sensitivity to TXA2 may increase pulmonary circulation burden in PAH. The thickening of pulmonary arterial walls increases right ventricular afterload and peripheral resistance with functional changes in PAH [38]. Therefore, TP plays an important role in PAH and its progression and can influence right heart function. This is a promising intervention target for PAH.
Conclusions
In this review, we investigated the role of the PGs pathway in the occurrence and development of PAH (Table 4). PGs exert their effects on pulmonary vascular remodeling by mediating the proliferation and hypertrophy of PVSMCs, endothelial cells, and other cell types. PGs are essential PAH mediators and, to date, research has focused on identifying the signaling pathways that PGs receptors activate in PAH. Different receptors have different physiological effects: DP1, EP2, EP4, and IP signaling pathways mainly improve pulmonary vascular remodeling and thus improve PAH (Fig. 3), while DP2, EP3, and TP signaling pathways aggravate PAH (Fig. 4). In the extracellular microenvironment, PGs bind to GPCRs and influence PAH development. We further summarized the potential drugs targeting PGs and their receptors, which might be involved in PAH (Fig. 5). Prostacyclin analogs and prostacyclin receptor agonists have effectively improved the quality of life of patients with PAH, although they have not been able to inhibit or reverse pulmonary vascular remodeling [232]. The mortality rate of patients with PH is up to 10% in one year and has not declined [233]. Prostacyclin and prostacyclin analogs, including epoprostenol, beraprost, iloprost, and treprostinil, do not act solely on the IP receptors. Epoprostenol mainly acts on IP and EP3; beraprost mainly acts on IP; iloprost mainly act on IP, EP1and EP3; and treprostinil mainly acts on IP, EP2, and DP1. Since the activation of EP3 receptors can aggravate PAH, whether their effect on EP3 receptors has any clinical effect remains unknown. However, combination therapy with EP3 receptor antagonists has not yet been reported. The treatment for patients with PAH is heterogeneous, and more targeted treatment is needed. Selexipag acts specifically on IP receptors. However, studies have shown no significant differences in 6 MWD, PVR, mortality, or adverse events compared with the control groups [234]. Thus, the specific binding of IP receptors does not reduce adverse events or mortality in patients with PAH. PGI2 has been shown to function via the PPAR signaling pathway. The presence of other receptors for prostacyclin analogs and prostacyclin receptor agonists remains unknown. Furthermore, the downstream signaling pathways of prostaglandin receptors are promising targets for PAH therapy. Everolimus, an mTOR inhibitor, improves the 6MWD and PVR in patients with PAH [235]. Fasudil, a Rho kinase inhibitor, is effective and safe for improving mPAP, PVR, and CI in patients with PAH in the short- and medium-term [236]. Combination therapy with prostaglandin receptor agonists, antagonists, or drugs targeting downstream sites may improve PAH. As, evidenced by many of the studies in this review, most of the evidence for the role of PGs receptors comes from in vitro or animal studies; clinical transformation is slow. In particular, the roles of EP1 and FP in PH remain unclear. Existing medical therapies primarily target Group 1 PH (PAH). Moreover, treprostinil has been shown to improve 6MWD in patients with Group 3 PH (PH associated with interstitial lung disease) and Group 4 PH (chronic thromboembolic PH) [237, 238]. Further research is needed to investigate the therapeutic effects of prostaglandins and related drugs on different groups of PH. In summary, we examined the function of PGs signaling pathways in PAH and suggest that targeting PGs pathways may provide opportunities for PAH prevention and treatment.

Prostacyclin drugs and prostaglandin receptor agonists/antagonists are involved in the pathogenesis of pulmonary arterial hypertension (PAH) through acting on distinct prostaglandin receptors. DP1 (activated by BW245C, treprostinil), EP2 (activated by butaprost, treprostinil), EP4, IP (activated by epoprostenol, beraprost, treprostinil, iloprost, selexipag) promoted vasodilation, anti-proliferation, and anti-thrombotic effects through the cAMP signaling pathway. EP1 (activated by iloprost), EP3 (activated by epoprostenol, iloprost, inhibited by L-798,106), TP (inhibited by NTP42) promoted vasoconstriction and proliferation through Rho and PKC signaling pathways. BW245C, a DP1-specific agonist; Butaprost, a highly selective EP2 receptor agonist; L-798,106, an EP3 antagonist; NTP42, a TP antagonist. Abbreviation: AC, adenylate cyclase; ATP, adenosine triphosphate; cAMP, cyclic adenosine monophosphate; DAG, diacylglycerol; IP3, inositol triphosphate; PIP2, phosphatidylinositol (4,5) bisphosphate;PKA, protein kinase A; PKC, protein kinase C; PLC, phospholipase C
Data Availability
Not applicable.
Abbreviations
- 12-HETE:
-
12-hydroxy-5,8,10,14-eicosatetraenoic acid
- 15-F2t-IsoP:
-
15-F2t-isoprostane
- 6MWD:
-
Six-minute walk distance
- AA:
-
Arachidonic acid
- AC:
-
Adenylate cyclase
- ATP:
-
Adenosine triphosphate
- BMEPCs:
-
Bone marine-derived endothelial progenitor cells
- cAMP:
-
Cyclic adenosine monophosphate
- CO:
-
Cardiac output
- COX:
-
Cyclooxygenase
- Cpc PH:
-
Combined post- and pre-capillary pulmonary hypertension
- CREB:
-
cAMP-response element binding protein
- CRTH2:
-
Chemoattractant receptor homologous molecule expressed on Th2
- CYP:
-
Cytochrome P450
- DAG:
-
Diacylglycerol
- DEGs:
-
Differentially expressed genes
- DP1:
-
PGD2 receptor 1
- DP2:
-
PGD2 receptor 2
- EETs:
-
Epoxyeicosatrienoic acids
- EP1:
-
PGE2 receptor 1
- EP2:
-
PGE2 receptor 2
- EP3:
-
PGE2 receptor 3
- EP4:
-
PGE2 receptor 4
- ET-1:
-
Endothelin-1
- FP:
-
Prostaglandin F receptor
- GEF:
-
Guanine nucleotide exchange factor
- GPCR:
-
G-protein-coupled receptor
- GSK-3β:
-
Glycogen synthetase kinase 3β
- HPASMCs:
-
Human pulmonary artery smooth muscle cells
- H-PGDS:
-
Hematopoietic PGDS
- HUVEC:
-
Human umbilical vein endothelial cells
- i.v.:
-
Intravenous
- ILC2:
-
The group 2 innate lymphoid cell
- IP:
-
Prostacyclin receptor
- IP3:
-
Inositol triphosphate
- IPA:
-
Intrapulmonary artery
- IRF7:
-
Interferon regulatory factor 7
- Jak:
-
Janus kinase
- LOX:
-
Lipoxygenase
- L-PGDS:
-
Lipocalin-type PGDS
- LTB4:
-
Leukotriene B4
- MCT:
-
Monocrotaline
- MMP:
-
Matrix metalloproteinase
- mPAP:
-
Mean pulmonary arterial pressure
- MRTF-A:
-
Myocardin-related transcription factor A
- MT1-MMP:
-
Membrane type 1-matrix metalloproteinase
- mTORC1:
-
Mammalian target of rapamycin complex 1
- NF-κB:
-
Nuclear factor κB
- NO:
-
Nitric oxide
- PAECs:
-
Pulmonary artery endothelial cells
- PAH:
-
Pulmonary arterial hypertension
- PAP:
-
Pulmonary arterial pressure
- PAs:
-
Pulmonary arteries
- PASMCs:
-
Pulmonary artery smooth muscle cells
- PGD2:
-
Prostaglandin D2
- PGDS:
-
Prostaglandin D synthase
- PGE2:
-
Prostaglandin E2
- PGES:
-
Prostaglandin E synthase
- PGF2α:
-
Prostaglandin F2α
- PGFS:
-
Prostaglandin F synthase
- PGHS:
-
Prostaglandin H synthase
- PGI2:
-
Prostaglandin I2
- PGIS:
-
Prostaglandin I synthase
- PGs:
-
Prostaglandins
- PH:
-
Pulmonary hypertension
- PI3K:
-
Phosphoinositide 3-kinase
- PIP2:
-
Phosphatidylinositol (4,5) bisphosphate
- PKB:
-
Protein kinase B
- PKC:
-
Protein kinase C
- PLA2:
-
Phospholipase A2
- PLC:
-
Phospholipase C
- PPAR:
-
Peroxisome proliferator-activated receptor
- PVR:
-
Pulmonary vascular resistance
- PVSMCs:
-
Pulmonary vein smooth muscle cells
- Rock:
-
Rho-associated coiled-coil-containing protein kinase
- RSVP:
-
Right systolic ventricular pressure
- SERT:
-
Serotonin
- SMCs:
-
Smooth muscle cells
- SOCC:
-
Store-operated calcium channels
- STAT6:
-
Signal transducer and activator of transcription 6
- TGF-β1:
-
Transforming growth factor β1
- TP:
-
TXA2 receptor
- TXA2:
-
Thromboxane A2
- TXAS:
-
Thromboxane A synthase
- VEGFR-2:
-
The VEGF receptor-2
- VOCC:
-
Voltage-operated calcium channels
- vWF:
-
Von Willebrand factor
References
Sockrider M. What is pulmonary Hypertension? Am J Respir Crit Care Med. 2021;203(5):P12–P3.
Hoeper MM, Humbert M, Souza R, Idrees M, Kawut SM, Sliwa-Hahnle K, et al. A global view of pulmonary Hypertension. Lancet Respir Med. 2016;4(4):306–22.
Simonneau G, Montani D, Celermajer DS, Denton CP, Gatzoulis MA, Krowka M et al. Haemodynamic definitions and updated clinical classification of pulmonary Hypertension. Eur Respir J. 2019;53(1).
Humbert M, Kovacs G, Hoeper MM, Badagliacca R, Berger RMF, Brida M, et al. 2022 ESC/ERS guidelines for the diagnosis and treatment of pulmonary Hypertension. Eur Heart J. 2022;43(38):3618–731.
Bisserier M, Janostiak R, Lezoualc’h F, Hadri L. Targeting epigenetic mechanisms as an emerging therapeutic strategy in pulmonary Hypertension Disease. Vasc Biol. 2020;2(1):R17–R34.
Shimoda LA, Laurie SS. Vascular remodeling in pulmonary Hypertension. J Mol Med (Berl). 2013;91(3):297–309.
Lai YC, Potoka KC, Champion HC, Mora AL, Gladwin MT. Pulmonary arterial Hypertension: the clinical syndrome. Circ Res. 2014;115(1):115–30.
Sylvester JT, Shimoda LA, Aaronson PI, Ward JP. Hypoxic pulmonary vasoconstriction. Physiol Rev. 2012;92(1):367–520.
Sakao S, Taraseviciene-Stewart L, Wood K, Cool CD, Voelkel NF. Apoptosis of pulmonary microvascular endothelial cells stimulates vascular smooth muscle cell growth. Am J Physiol Lung Cell Mol Physiol. 2006;291(3):L362–8.
Dai Z, Zhu MM, Peng Y, Jin H, Machireddy N, Qian Z, et al. Endothelial and smooth muscle cell Interaction via FoxM1 Signaling mediates vascular remodeling and pulmonary Hypertension. Am J Respir Crit Care Med. 2018;198(6):788–802.
Ryerson CJ, Nayar S, Swiston JR, Sin DD. Pharmacotherapy in pulmonary arterial Hypertension: a systematic review and meta-analysis. Respir Res. 2010;11(1):12.
Hoeper MM, Simon RGJ. The changing landscape of pulmonary arterial Hypertension and implications for patient care. Eur Respir Rev. 2014;23(134):450–7.
Liu R, Yuan T, Wang R, Gong D, Wang S, Du G et al. Insights into Endothelin receptors in Pulmonary Hypertension. Int J Mol Sci. 2023;24(12).
Correale M, Ferraretti A, Monaco I, Grazioli D, Di Biase M, Brunetti ND. Endothelin-receptor antagonists in the management of pulmonary arterial Hypertension: where do we stand? Vasc Health Risk Manag. 2018;14:253–64.
Montani D, Chaumais MC, Guignabert C, Gunther S, Girerd B, Jais X, et al. Targeted therapies in pulmonary arterial Hypertension. Pharmacol Ther. 2014;141(2):172–91.
Dhoble S, Patravale V, Weaver E, Lamprou DA, Patravale T. Comprehensive review on novel targets and emerging therapeutic modalities for pulmonary arterial Hypertension. Int J Pharm. 2022;621:121792.
Bisserier M, Pradhan N, Hadri L. Current and emerging therapeutic approaches to pulmonary Hypertension. Rev Cardiovasc Med. 2020;21(2):163–79.
Channick R, Badesch DB, Tapson VF, Simonneau G, Robbins I, Frost A, et al. Effects of the dual endothelin receptor antagonist bosentan in patients with pulmonary Hypertension: a placebo-controlled study. J Heart Lung Transplant. 2001;20(2):262–3.
Ghofrani HA, Galie N, Grimminger F, Grunig E, Humbert M, Jing ZC, et al. Riociguat for the treatment of pulmonary arterial Hypertension. N Engl J Med. 2013;369(4):330–40.
Ghofrani HA, Osterloh IH, Grimminger F. Sildenafil: from angina to erectile dysfunction to pulmonary Hypertension and beyond. Nat Rev Drug Discov. 2006;5(8):689–702.
Humbert M, Guignabert C, Bonnet S, Dorfmuller P, Klinger JR, Nicolls MR et al. Pathology and pathobiology of pulmonary Hypertension: state of the art and research perspectives. Eur Respir J. 2019;53(1).
Spiekerkoetter E, Kawut SM, de Jesus Perez VA. New and Emerging therapies for Pulmonary arterial Hypertension. Annu Rev Med. 2019;70:45–59.
von Euler US. On the specific vaso-dilating and plain muscle stimulating substances from accessory genital glands in man and certain animals (prostaglandin and vesiglandin). J Physiol. 1936;88(2):213–34.
Eglinton G, Raphael RA, Smith GN, Hall WJ, Pickles VR. Isolation and identification of two smooth muscle stimulants from Menstrual Fluid. Nature. 1963;200:960–PASSIM.
Yang G, Chen L. An update of microsomal prostaglandin E Synthase-1 and PGE2 receptors in Cardiovascular Health and Diseases. Oxid Med Cell Longev. 2016;2016:5249086.
Schuster VL. Prostaglandin transport. Prostaglandins Other Lipid Mediat. 2002;68–69:633–47.
Woodward DF, Jones RL, Narumiya S. International Union of Basic and Clinical Pharmacology. LXXXIII: classification of prostanoid receptors, updating 15 years of progress. Pharmacol Rev. 2011;63(3):471–538.
Yao C, Narumiya S. Prostaglandin-cytokine crosstalk in chronic inflammation. Br J Pharmacol. 2019;176(3):337–54.
Wang Q, Morris RJ, Bode AM, Zhang T. Prostaglandin pathways: opportunities for Cancer Prevention and Therapy. Cancer Res. 2022;82(6):949–65.
Saito S, Tsuda H, Michimata T. Prostaglandin D2 and reproduction. Am J Reprod Immunol. 2002;47(5):295–302.
O’Callaghan G, Houston A. Prostaglandin E2 and the EP receptors in malignancy: possible therapeutic targets? Br J Pharmacol. 2015;172(22):5239–50.
Swan CE, Breyer RM. Prostaglandin E2 modulation of blood pressure homeostasis: studies in rodent models. Prostaglandins Other Lipid Mediat. 2011;96(1–4):10–3.
Cheng H, Huang H, Guo Z, Chang Y, Li Z. Role of prostaglandin E2 in tissue repair and regeneration. Theranostics. 2021;11(18):8836–54.
Kawahara K, Hohjoh H, Inazumi T, Tsuchiya S, Sugimoto Y. Prostaglandin E2-induced inflammation: relevance of prostaglandin E receptors. Biochim Biophys Acta. 2015;1851(4):414–21.
Fujimoto N, Shio H, Miyoshi T, Suzuki H. [Prostaglandin F2 alpha and its metabolites]. Nihon Rinsho. 1995;53(Su Pt):713–5.
Liu B, Li J, Yan H, Tian D, Li H, Zhang Y, et al. TP and/or EP3 receptors mediate the vasoconstrictor and pressor responses of prostaglandin F(2alpha) in mice and/or humans. FASEB J. 2019;33(2):2451–9.
Olschewski H, Prostacyclins. Handb Exp Pharmacol. 2013;218:177–98.
Cho S, Namgoong H, Kim HJ, Vorn R, Yoo HY, Kim SJ. Downregulation of Soluble Guanylate Cyclase and protein kinase G with upregulated ROCK2 in the pulmonary artery leads to thromboxane A2 sensitization in Monocrotaline-Induced Pulmonary Hypertensive rats. Front Physiol. 2021;12:624967.
Tao X, Zhang R, Du R, Yu T, Yang H, Li J et al. EP3 enhances adhesion and cytotoxicity of NK cells toward hepatic stellate cells in a murine liver fibrosis model. J Exp Med. 2022;219(5).
Moiseeva EP. Adhesion receptors of vascular smooth muscle cells and their functions. Cardiovasc Res. 2001;52(3):372–86.
Guignabert C, Phan C, Seferian A, Huertas A, Tu L, Thuillet R, et al. Dasatinib induces lung vascular toxicity and predisposes to pulmonary Hypertension. J Clin Invest. 2016;126(9):3207–18.
Stadtmauer DJ, Wagner GP. Single-cell analysis of prostaglandin E2-induced human decidual cell in vitro differentiation: a minimal ancestral deciduogenic signaldagger. Biol Reprod. 2022;106(1):155–72.
van der Feen DE, Bossers GPL, Hagdorn QAJ, Moonen JR, Kurakula K, Szulcek R et al. Cellular senescence impairs the reversibility of pulmonary arterial Hypertension. Sci Transl Med. 2020;12(554).
Maarman G, Lecour S, Butrous G, Thienemann F, Sliwa K. A comprehensive review: the evolution of animal models in pulmonary Hypertension research; are we there yet? Pulm Circ. 2013;3(4):739–56.
Shah M, Patel K, Sehgal PB. Monocrotaline pyrrole-induced endothelial cell megalocytosis involves a golgi blockade mechanism. Am J Physiol Cell Physiol. 2005;288(4):C850–62.
Gomez-Arroyo JG, Farkas L, Alhussaini AA, Farkas D, Kraskauskas D, Voelkel NF, et al. The monocrotaline model of pulmonary Hypertension in perspective. Am J Physiol Lung Cell Mol Physiol. 2012;302(4):L363–9.
Yan S, Resta TC, Jernigan NL. Vasoconstrictor mechanisms in Chronic Hypoxia-Induced Pulmonary Hypertension: role of oxidant signaling. Antioxid (Basel). 2020;9(10).
Voelkel NF, Tuder RM, Wade K, Hoper M, Lepley RA, Goulet JL, et al. Inhibition of 5-lipoxygenase-activating protein (FLAP) reduces pulmonary vascular reactivity and pulmonary Hypertension in hypoxic rats. J Clin Invest. 1996;97(11):2491–8.
He Y, Zuo C, Jia D, Bai P, Kong D, Chen D, et al. Loss of DP1 aggravates vascular remodeling in pulmonary arterial Hypertension via mTORC1 Signaling. Am J Respir Crit Care Med. 2020;201(10):1263–76.
Bikou O, Hajjar RJ, Hadri L, Sassi Y. Induction and characterization of Pulmonary Hypertension in mice using the Hypoxia/SU5416 model. J Vis Exp. 2020(160).
Taraseviciene-Stewart L, Kasahara Y, Alger L, Hirth P, Mc Mahon G, Waltenberger J, et al. Inhibition of the VEGF receptor 2 combined with chronic hypoxia causes cell death-dependent pulmonary endothelial cell proliferation and severe pulmonary Hypertension. FASEB J. 2001;15(2):427–38.
Imig JD. Prospective for cytochrome P450 epoxygenase cardiovascular and renal therapeutics. Pharmacol Ther. 2018;192:1–19.
Mitchell JA, Kirkby NS. Eicosanoids, prostacyclin and cyclooxygenase in the cardiovascular system. Br J Pharmacol. 2019;176(8):1038–50.
Zhang C, Ma C, Yao H, Zhang L, Yu X, Liu Y, et al. 12-Lipoxygenase and 12-hydroxyeicosatetraenoic acid regulate hypoxic angiogenesis and survival of pulmonary artery endothelial cells via PI3K/Akt pathway. Am J Physiol Lung Cell Mol Physiol. 2018;314(4):L606–L16.
Rouzer CA, Marnett LJ. Cyclooxygenases: structural and functional insights. J Lipid Res. 2009;50(SupplSuppl):29–34.
Loukanov T, Jaschinski C, Kirilov M, Klimpel H, Karck M, Gorenflo M. Cyclooxygenase-2 expression in lung in patients with congenital heart malformations and pulmonary arterial Hypertension. Thorac Cardiovasc Surg. 2013;61(4):307–11.
Yang X, Sheares KK, Davie N, Upton PD, Taylor GW, Horsley J, et al. Hypoxic induction of cox-2 regulates proliferation of human pulmonary artery smooth muscle cells. Am J Respir Cell Mol Biol. 2002;27(6):688–96.
Pidgeon GP, Tamosiuniene R, Chen G, Leonard I, Belton O, Bradford A, et al. Intravascular Thrombosis after hypoxia-induced pulmonary Hypertension: regulation by cyclooxygenase-2. Circulation. 2004;110(17):2701–7.
Fike CD, Kaplowitz MR, Zhang Y, Pfister SL. Cyclooxygenase-2 and an early stage of chronic hypoxia-induced pulmonary Hypertension in newborn pigs. J Appl Physiol (1985). 2005;98(3):1111–8. discussion 091.
Fredenburgh LE, Liang OD, Macias AA, Polte TR, Liu X, Riascos DF, et al. Absence of cyclooxygenase-2 exacerbates hypoxia-induced pulmonary Hypertension and enhances contractility of vascular smooth muscle cells. Circulation. 2008;117(16):2114–22.
Seta F, Rahmani M, Turner PV, Funk CD. Pulmonary oxidative stress is increased in cyclooxygenase-2 knockdown mice with mild pulmonary Hypertension induced by monocrotaline. PLoS ONE. 2011;6(8):e23439.
Jiang DM, Han J, Zhu JH, Fu GS, Zhou BQ. Paracrine effects of bone marrow-derived endothelial progenitor cells: cyclooxygenase-2/prostacyclin pathway in pulmonary arterial Hypertension. PLoS ONE. 2013;8(11):e79215.
Somanna NK, Worner PM, Murthy SN, Pankey EA, Schachtele DJ, St Hilaire RC, et al. Intratracheal administration of cyclooxygenase-1-transduced adipose tissue-derived stem cells ameliorates monocrotaline-induced pulmonary Hypertension in rats. Am J Physiol Heart Circ Physiol. 2014;307(8):H1187–95.
Abramovitz M, Adam M, Boie Y, Carriere M, Denis D, Godbout C, et al. The utilization of recombinant prostanoid receptors to determine the affinities and selectivities of prostaglandins and related analogs. Biochim Biophys Acta. 2000;1483(2):285–93.
Whittle BJ, Silverstein AM, Mottola DM, Clapp LH. Binding and activity of the prostacyclin receptor (IP) agonists, treprostinil and iloprost, at human prostanoid receptors: treprostinil is a potent DP1 and EP2 agonist. Biochem Pharmacol. 2012;84(1):68–75.
Voswinckel R, Enke B, Reichenberger F, Kohstall M, Kreckel A, Krick S, et al. Favorable effects of inhaled treprostinil in severe pulmonary Hypertension: results from randomized controlled pilot studies. J Am Coll Cardiol. 2006;48(8):1672–81.
Seo MJ, Oh DK. Prostaglandin synthases: molecular characterization and involvement in prostaglandin biosynthesis. Prog Lipid Res. 2017;66:50–68.
Eguchi Y, Eguchi N, Oda H, Seiki K, Kijima Y, Matsu-ura Y, et al. Expression of lipocalin-type prostaglandin D synthase (beta-trace) in human heart and its accumulation in the coronary circulation of angina patients. Proc Natl Acad Sci U S A. 1997;94(26):14689–94.
Kong D, Yu Y, Prostaglandin. D(2) signaling and cardiovascular homeostasis. J Mol Cell Cardiol. 2022;167:97–105.
Song WL, Ricciotti E, Liang X, Grosser T, Grant GR, FitzGerald GA. Lipocalin-Like Prostaglandin D synthase but not hemopoietic prostaglandin D synthase deletion causes Hypertension and accelerates Thrombogenesis in mice. J Pharmacol Exp Ther. 2018;367(3):425–32.
Robbins IM, Barst RJ, Rubin LJ, Gaine SP, Price PV, Morrow JD, et al. Increased levels of prostaglandin D(2) suggest macrophage activation in patients with primary pulmonary Hypertension. Chest. 2001;120(5):1639–44.
Soifer SJ, Morin FC 3rd, Heymann MA. Prostaglandin D2 reverses induced pulmonary Hypertension in the newborn lamb. J Pediatr. 1982;100(3):458–63.
Soifer SJ, Clyman RI, Heymann MA. Effects of prostaglandin D2 on pulmonary arterial pressure and oxygenation in newborn infants with persistent pulmonary Hypertension. J Pediatr. 1988;112(5):774–7.
Jia D, Bai P, Wan N, Liu J, Zhu Q, He Y, et al. Niacin attenuates pulmonary Hypertension through H-PGDS in macrophages. Circ Res. 2020;127(10):1323–36.
Boie Y, Sawyer N, Slipetz DM, Metters KM, Abramovitz M. Molecular cloning and characterization of the human prostanoid DP receptor. J Biol Chem. 1995;270(32):18910–6.
Lee YH, Choi SJ, Ji JD, Song GG. PTGDR polymorphisms and susceptibility to Asthma: a meta-analysis. Mol Biol Rep. 2013;40(3):2195–203.
Wright DH, Metters KM, Abramovitz M, Ford-Hutchinson AW. Characterization of the recombinant human prostanoid DP receptor and identification of L-644,698, a novel selective DP agonist. Br J Pharmacol. 1998;123(7):1317–24.
Wright DH, Nantel F, Metters KM, Ford-Hutchinson AW. A novel biological role for prostaglandin D2 is suggested by distribution studies of the rat DP prostanoid receptor. Eur J Pharmacol. 1999;377(1):101–15.
Syed NI, Jones RL. Assessing the agonist profiles of the prostacyclin analogues treprostinil and naxaprostene, particularly their DP(1) activity. Prostaglandins Leukot Essent Fatty Acids. 2015;95:19–29.
Kiriyama M, Ushikubi F, Kobayashi T, Hirata M, Sugimoto Y, Narumiya S. Ligand binding specificities of the eight types and subtypes of the mouse prostanoid receptors expressed in Chinese hamster ovary cells. Br J Pharmacol. 1997;122(2):217–24.
Matsuoka T, Narumiya S. Prostaglandin receptor signaling in Disease. ScientificWorldJournal. 2007;7:1329–47.
Claar D, Hartert TV, Peebles RS. Jr. The role of prostaglandins in allergic lung inflammation and Asthma. Expert Rev Respir Med. 2015;9(1):55–72.
Kupczyk M, Kuna P. Targeting the PGD(2)/CRTH2/DP1 signaling pathway in Asthma and allergic Disease: current status and future perspectives. Drugs. 2017;77(12):1281–94.
Ahmad AS, Ottallah H, Maciel CB, Strickland M, Dore S. Role of the L-PGDS-PGD2-DP1 receptor axis in sleep regulation and neurologic outcomes. Sleep. 2019;42(6).
Rossitto M, Ujjan S, Poulat F, Boizet-Bonhoure B. Multiple roles of the prostaglandin D2 signaling pathway in reproduction. Reproduction. 2015;149(1):R49–58.
Li J, Kong D, Wang Q, Wu W, Tang Y, Bai T, et al. Niacin ameliorates ulcerative Colitis via prostaglandin D(2)-mediated D prostanoid receptor 1 activation. EMBO Mol Med. 2017;9(5):571–88.
Cheng K, Wu TJ, Wu KK, Sturino C, Metters K, Gottesdiener K, et al. Antagonism of the prostaglandin D2 receptor 1 suppresses nicotinic acid-induced vasodilation in mice and humans. Proc Natl Acad Sci U S A. 2006;103(17):6682–7.
Ahmad AS. PGD2 DP1 receptor stimulation following Stroke ameliorates cerebral blood flow and outcomes. Neuroscience. 2014;279:260–8.
Walch L, Labat C, Gascard JP, de Montpreville V, Brink C, Norel X. Prostanoid receptors involved in the relaxation of human pulmonary vessels. Br J Pharmacol. 1999;126(4):859–66.
Benyahia C, Boukais K, Gomez I, Silverstein A, Clapp L, Fabre A, et al. A comparative study of PGI2 mimetics used clinically on the vasorelaxation of human pulmonary arteries and veins, role of the DP-receptor. Prostaglandins Other Lipid Mediat. 2013;107:48–55.
Marchese A, Sawzdargo M, Nguyen T, Cheng R, Heng HH, Nowak T, et al. Discovery of three novel orphan G-protein-coupled receptors. Genomics. 1999;56(1):12–21.
Nagata K, Tanaka K, Ogawa K, Kemmotsu K, Imai T, Yoshie O, et al. Selective expression of a novel surface molecule by human Th2 cells in vivo. J Immunol. 1999;162(3):1278–86.
Sawyer N, Cauchon E, Chateauneuf A, Cruz RP, Nicholson DW, Metters KM, et al. Molecular pharmacology of the human prostaglandin D2 receptor, CRTH2. Br J Pharmacol. 2002;137(8):1163–72.
Nagata K, Hirai H, Tanaka K, Ogawa K, Aso T, Sugamura K, et al. CRTH2, an orphan receptor of T-helper-2-cells, is expressed on basophils and eosinophils and responds to mast cell-derived factor(s). FEBS Lett. 1999;459(2):195–9.
Moon TC, Campos-Alberto E, Yoshimura T, Bredo G, Rieger AM, Puttagunta L, et al. Expression of DP2 (CRTh2), a prostaglandin D(2) receptor, in human mast cells. PLoS ONE. 2014;9(9):e108595.
Wojno ED, Monticelli LA, Tran SV, Alenghat T, Osborne LC, Thome JJ, et al. The prostaglandin D(2) receptor CRTH2 regulates accumulation of group 2 innate lymphoid cells in the inflamed lung. Mucosal Immunol. 2015;8(6):1313–23.
Xue L, Gyles SL, Wettey FR, Gazi L, Townsend E, Hunter MG, et al. Prostaglandin D2 causes preferential induction of proinflammatory Th2 cytokine production through an action on chemoattractant receptor-like molecule expressed on Th2 cells. J Immunol. 2005;175(10):6531–6.
Kostenis E, Ulven T. Emerging roles of DP and CRTH2 in allergic inflammation. Trends Mol Med. 2006;12(4):148–58.
Pelaia C, Crimi C, Vatrella A, Busceti MT, Gaudio A, Garofalo E, et al. New treatments for Asthma: from the pathogenic role of prostaglandin D(2) to the therapeutic effects of fevipiprant. Pharmacol Res. 2020;155:104490.
Kuna P, Bjermer L, Tornling G. Two phase II randomized trials on the CRTh2 antagonist AZD1981 in adults with Asthma. Drug Des Devel Ther. 2016;10:2759–70.
Bateman ED, Guerreros AG, Brockhaus F, Holzhauer B, Pethe A, Kay RA et al. Fevipiprant, an oral prostaglandin DP(2) receptor (CRTh2) antagonist, in allergic Asthma uncontrolled on low-dose inhaled corticosteroids. Eur Respir J. 2017;50(2).
Ortega H, Fitzgerald M, Raghupathi K, Tompkins CA, Shen J, Dittrich K, et al. A phase 2 study to evaluate the safety, efficacy and pharmacokinetics of DP2 antagonist GB001 and to explore biomarkers of airway inflammation in mild-to-moderate Asthma. Clin Exp Allergy. 2020;50(2):189–97.
Asano K, Sagara H, Ichinose M, Hirata M, Nakajima A, Ortega H, et al. A phase 2a study of DP(2) antagonist GB001 for Asthma. J Allergy Clin Immunol Pract. 2020;8(4):1275–83e1.
Price LC, Wort SJ, Perros F, Dorfmuller P, Huertas A, Montani D, et al. Inflammation in pulmonary arterial Hypertension. Chest. 2012;141(1):210–21.
Ito T, Okada T, Miyashita H, Nomoto T, Nonaka-Sarukawa M, Uchibori R, et al. Interleukin-10 expression mediated by an adeno-associated virus vector prevents monocrotaline-induced pulmonary arterial Hypertension in rats. Circ Res. 2007;101(7):734–41.
Frid MG, Brunetti JA, Burke DL, Carpenter TC, Davie NJ, Reeves JT, et al. Hypoxia-induced pulmonary vascular remodeling requires recruitment of circulating mesenchymal precursors of a monocyte/macrophage lineage. Am J Pathol. 2006;168(2):659–69.
Radstake TR, van Bon L, Broen J, Wenink M, Santegoets K, Deng Y, et al. Increased frequency and compromised function of T regulatory cells in systemic sclerosis (SSc) is related to a diminished CD69 and TGFbeta expression. PLoS ONE. 2009;4(6):e5981.
Flament T, Bigot A, Chaigne B, Henique H, Diot E, Marchand-Adam S. Pulmonary manifestations of Sjogren’s syndrome. Eur Respir Rev. 2016;25(140):110–23.
Bonelli M, Savitskaya A, Steiner CW, Rath E, Smolen JS, Scheinecker C. Phenotypic and functional analysis of CD4 + CD25- Foxp3 + T cells in patients with systemic Lupus Erythematosus. J Immunol. 2009;182(3):1689–95.
Voelkel NF, Tamosiuniene R, Nicolls MR. Challenges and opportunities in treating inflammation associated with pulmonary Hypertension. Expert Rev Cardiovasc Ther. 2016;14(8):939–51.
Hudalla H, Michael Z, Christodoulou N, Willis GR, Fernandez-Gonzalez A, Filatava EJ, et al. Carbonic anhydrase inhibition ameliorates inflammation and experimental pulmonary Hypertension. Am J Respir Cell Mol Biol. 2019;61(4):512–24.
Deng Y, Guo SL, Li JQ, Xie SS, Zhou YC, Wei B, et al. Interferon regulatory factor 7 inhibits rat vascular smooth muscle cell proliferation and inflammation in monocrotaline-induced pulmonary Hypertension. Life Sci. 2021;264:118709.
Chen G, Zuo S, Tang J, Zuo C, Jia D, Liu Q, et al. Inhibition of CRTH2-mediated Th2 activation attenuates pulmonary Hypertension in mice. J Exp Med. 2018;215(8):2175–95.
Kondeti V, Al-Azzam N, Duah E, Thodeti CK, Boyce JA, Paruchuri S. Leukotriene D4 and prostaglandin E2 signals synergize and potentiate vascular inflammation in a mast cell-dependent manner through cysteinyl leukotriene receptor 1 and E-prostanoid receptor 3. J Allergy Clin Immunol. 2016;137(1):289–98.
Finetti F, Travelli C, Ercoli J, Colombo G, Buoso E, Trabalzini L. Prostaglandin E2 and Cancer: insight into Tumor Progression and Immunity. Biology (Basel). 2020;9(12).
Rastogi S, Willmes DM, Nassiri M, Babina M, Worm M. PGE(2) deficiency predisposes to anaphylaxis by causing mast cell hyperresponsiveness. J Allergy Clin Immunol. 2020;146(6):1387–96. e13.
Al-Husseini A, Wijesinghe DS, Farkas L, Kraskauskas D, Drake JI, Van Tassel B, et al. Increased eicosanoid levels in the Sugen/chronic hypoxia model of severe pulmonary Hypertension. PLoS ONE. 2015;10(3):e0120157.
Benza RL, Miller DP, Gomberg-Maitland M, Frantz RP, Foreman AJ, Coffey CS, et al. Predicting survival in pulmonary arterial Hypertension: insights from the Registry to Evaluate Early and Long-Term Pulmonary arterial Hypertension Disease Management (REVEAL). Circulation. 2010;122(2):164–72.
Stebel S, Wideman RF. Pulmonary hemodynamic responses to intravenous prostaglandin E2 in broiler chickens. Poult Sci. 2008;87(1):138–45.
Ozen G, Benyahia C, Mani S, Boukais K, Silverstein AM, Bayles R, et al. Bronchodilation induced by PGE(2) is impaired in Group III pulmonary Hypertension. Br J Pharmacol. 2020;177(1):161–74.
Funk CD, Furci L, FitzGerald GA, Grygorczyk R, Rochette C, Bayne MA, et al. Cloning and expression of a cDNA for the human prostaglandin E receptor EP1 subtype. J Biol Chem. 1993;268(35):26767–72.
Tang CH, Yang RS, Fu WM. Prostaglandin E2 stimulates fibronectin expression through EP1 receptor, phospholipase C, protein kinase calpha, and c-Src pathway in primary cultured rat osteoblasts. J Biol Chem. 2005;280(24):22907–16.
Senior J, Marshall K, Sangha R, Baxter GS, Clayton JK. In vitro characterization of prostanoid EP-receptors in the non-pregnant human myometrium. Br J Pharmacol. 1991;102(3):747–53.
Konger RL, Billings SD, Prall NC, Katona TM, Dasilva SC, Kennedy CR, et al. The EP1 subtype of prostaglandin E2 receptor: role in keratinocyte differentiation and expression in non-melanoma Skin cancer. Prostaglandins Leukot Essent Fatty Acids. 2009;81(4):279–90.
Norel X, de Montpreville V, Brink C. Vasoconstriction induced by activation of EP1 and EP3 receptors in human lung: effects of ONO-AE-248, ONO-DI-004, ONO-8711 or ONO-8713. Prostaglandins Other Lipid Mediat. 2004;74(1–4):101–12.
Wang XS, Lau HY. Prostaglandin E potentiates the immunologically stimulated histamine release from human peripheral blood-derived mast cells through EP1/EP3 receptors. Allergy. 2006;61(4):503–6.
Smid SD, Svensson KM. Inhibition of cyclooxygenase-2 and EP1 receptor antagonism reduces human colonic longitudinal muscle contractility in vitro. Prostaglandins Other Lipid Mediat. 2009;88(3–4):117–21.
Kawabata A. Prostaglandin E2 and pain–an update. Biol Pharm Bull. 2011;34(8):1170–3.
Chen C, Guan J, Gu X, Chu Q, Zhu H. Prostaglandin E2 and receptors: insight into Tumorigenesis, Tumor Progression, and Treatment of Hepatocellular Carcinoma. Front Cell Dev Biol. 2022;10:834859.
Nasrallah R, Hebert RL. Prostacyclin signaling in the kidney: implications for health and Disease. Am J Physiol Renal Physiol. 2005;289(2):F235–46.
Yang T, Du Y. Distinct roles of central and peripheral prostaglandin E2 and EP subtypes in blood pressure regulation. Am J Hypertens. 2012;25(10):1042–9.
Guan Y, Zhang Y, Wu J, Qi Z, Yang G, Dou D, et al. Antihypertensive effects of selective prostaglandin E2 receptor subtype 1 targeting. J Clin Invest. 2007;117(9):2496–505.
Nagamachi M, Sakata D, Kabashima K, Furuyashiki T, Murata T, Segi-Nishida E, et al. Facilitation of Th1-mediated immune response by prostaglandin E receptor EP1. J Exp Med. 2007;204(12):2865–74.
Swain SD, Siemsen DW, Pullen RR, Han S. CD4 + T cells and IFN-gamma are required for the development of Pneumocystis-associated pulmonary Hypertension. Am J Pathol. 2014;184(2):483–93.
Regan JW, Bailey TJ, Pepperl DJ, Pierce KL, Bogardus AM, Donello JE, et al. Cloning of a novel human prostaglandin receptor with characteristics of the pharmacologically defined EP2 subtype. Mol Pharmacol. 1994;46(2):213–20.
Sagana RL, Yan M, Cornett AM, Tsui JL, Stephenson DA, Huang SK, et al. Phosphatase and tensin homologue on chromosome 10 (PTEN) directs prostaglandin E2-mediated fibroblast responses via regulation of E prostanoid 2 receptor expression. J Biol Chem. 2009;284(47):32264–71.
Hsu HH, Lin YM, Shen CY, Shibu MA, Li SY, Chang SH et al. Prostaglandin E2-Induced COX-2 expressions via EP2 and EP4 signaling pathways in human LoVo Colon Cancer cells. Int J Mol Sci. 2017;18(6).
Singh N, Bansal M, Pal S, Alam S, Jagdale P, Ayanur A, et al. COX-2/EP2-EP4/beta-catenin signaling regulates patulin-induced intestinal cell proliferation and inflammation. Toxicol Appl Pharmacol. 2018;356:224–34.
Aoki T, Frosen J, Fukuda M, Bando K, Shioi G, Tsuji K et al. Prostaglandin E2-EP2-NF-kappaB signaling in macrophages as a potential therapeutic target for intracranial aneurysms. Sci Signal. 2017;10(465).
Sun X, Li Q. Prostaglandin EP2 receptor: novel therapeutic target for human cancers (review). Int J Mol Med. 2018;42(3):1203–14.
Burelout C, Thibault N, Levasseur S, Simard S, Naccache PH, Bourgoin SG. Prostaglandin E2 inhibits the phospholipase D pathway stimulated by formyl-methionyl-leucyl-phenylalanine in human neutrophils. Involvement of EP2 receptors and phosphatidylinositol 3-kinase gamma. Mol Pharmacol. 2004;66(2):293–301.
Yao C, Sakata D, Esaki Y, Li Y, Matsuoka T, Kuroiwa K, et al. Prostaglandin E2-EP4 signaling promotes immune inflammation through Th1 cell differentiation and Th17 cell expansion. Nat Med. 2009;15(6):633–40.
Markovic T, Jakopin Z, Dolenc MS, Mlinaric-Rascan I. Structural features of subtype-selective EP receptor modulators. Drug Discov Today. 2017;22(1):57–71.
Donnini S, Finetti F, Solito R, Terzuoli E, Sacchetti A, Morbidelli L, et al. EP2 prostanoid receptor promotes squamous cell carcinoma growth through epidermal growth factor receptor transactivation and iNOS and ERK1/2 pathways. FASEB J. 2007;21(10):2418–30.
Xia SB, Tian ZB, Zhang W, Zhang H. NORAD promotes the viability, Migration, and phenotypic switch of human vascular smooth muscle cells during Aortic Dissection via LIN28B-Mediated TGF-beta Promotion and subsequent enhanced glycolysis. Biomed Res Int. 2022;2022:5333928.
Zaiman AL, Podowski M, Medicherla S, Gordy K, Xu F, Zhen L, et al. Role of the TGF-beta/Alk5 signaling pathway in monocrotaline-induced pulmonary Hypertension. Am J Respir Crit Care Med. 2008;177(8):896–905.
Corboz MR, Salvail W, Gagnon S, LaSala D, Laurent CE, Salvail D, et al. Prostanoid receptor subtypes involved in treprostinil-mediated vasodilation of rat pulmonary arteries and in treprostinil-mediated inhibition of collagen gene expression of human lung fibroblasts. Prostaglandins Other Lipid Mediat. 2021;152:106486.
Lambers C, Roth M, Jaksch P, Murakozy G, Tamm M, Klepetko W, et al. Treprostinil inhibits proliferation and extracellular matrix deposition by fibroblasts through cAMP activation. Sci Rep. 2018;8(1):1087.
Kolodsick JE, Peters-Golden M, Larios J, Toews GB, Thannickal VJ, Moore BB. Prostaglandin E2 inhibits fibroblast to myofibroblast transition via E. prostanoid receptor 2 signaling and cyclic adenosine monophosphate elevation. Am J Respir Cell Mol Biol. 2003;29(5):537–44.
Patel JA, Shen L, Hall SM, Benyahia C, Norel X, McAnulty RJ et al. Prostanoid EP(2) receptors are Up-Regulated in human pulmonary arterial Hypertension: a key anti-proliferative target for Treprostinil in smooth muscle cells. Int J Mol Sci. 2018;19(8).
Kotani M, Tanaka I, Ogawa Y, Usui T, Tamura N, Mori K, et al. Structural organization of the human prostaglandin EP3 receptor subtype gene (PTGER3). Genomics. 1997;40(3):425–34.
Adam M, Boie Y, Rushmore TH, Muller G, Bastien L, McKee KT, et al. Cloning and expression of three isoforms of the human EP3 prostanoid receptor. FEBS Lett. 1994;338(2):170–4.
Narumiya S. Prostanoids and inflammation: a new concept arising from receptor knockout mice. J Mol Med (Berl). 2009;87(10):1015–22.
Schmid A, Thierauch KH, Schleuning WD, Dinter H. Splice variants of the human EP3 receptor for prostaglandin E2. Eur J Biochem. 1995;228(1):23–30.
Kotelevets L, Foudi N, Louedec L, Couvelard A, Chastre E, Norel X. A new mRNA splice variant coding for the human EP3-I receptor isoform. Prostaglandins Leukot Essent Fatty Acids. 2007;77(3–4):195–201.
Kim SO, Dozier BL, Kerry JA, Duffy DM. EP3 receptor isoforms are differentially expressed in subpopulations of primate granulosa cells and couple to unique G-proteins. Reproduction. 2013;146(6):625–35.
Coleman RA, Smith WL, Narumiya S. International Union of Pharmacology classification of prostanoid receptors: properties, distribution, and structure of the receptors and their subtypes. Pharmacol Rev. 1994;46(2):205–29.
Ricciotti E, FitzGerald GA. Prostaglandins and inflammation. Arterioscler Thromb Vasc Biol. 2011;31(5):986–1000.
Cracowski JL, Cracowski C, Bessard G, Pepin JL, Bessard J, Schwebel C, et al. Increased lipid peroxidation in patients with pulmonary Hypertension. Am J Respir Crit Care Med. 2001;164(6):1038–42.
Cracowski JL, Degano B, Chabot F, Labarere J, Schwedhelm E, Monneret D, et al. Independent association of urinary F2-isoprostanes with survival in pulmonary arterial Hypertension. Chest. 2012;142(4):869–76.
Lu A, Zuo C, He Y, Chen G, Piao L, Zhang J, et al. EP3 receptor deficiency attenuates pulmonary Hypertension through suppression of Rho/TGF-beta1 signaling. J Clin Invest. 2015;125(3):1228–42.
Morrison K, Studer R, Ernst R, Haag F, Kauser K, Clozel M. Differential effects of Selexipag and prostacyclin analogs in rat pulmonary artery. J Pharmacol Exp Ther. 2012;343(3):547–55.
Bastien L, Sawyer N, Grygorczyk R, Metters KM, Adam M. Cloning, functional expression, and characterization of the human prostaglandin E2 receptor EP2 subtype. J Biol Chem. 1994;269(16):11873–7.
Konya V, Marsche G, Schuligoi R, Heinemann A. E-type prostanoid receptor 4 (EP4) in Disease and therapy. Pharmacol Ther. 2013;138(3):485–502.
Foudi N, Kotelevets L, Louedec L, Leseche G, Henin D, Chastre E, et al. Vasorelaxation induced by prostaglandin E2 in human pulmonary vein: role of the EP4 receptor subtype. Br J Pharmacol. 2008;154(8):1631–9.
Bradbury DA, Newton R, Zhu YM, El-Haroun H, Corbett L, Knox AJ. Cyclooxygenase-2 induction by bradykinin in human pulmonary artery smooth muscle cells is mediated by the cyclic AMP response element through a novel autocrine loop involving endogenous prostaglandin E2, E-prostanoid 2 (EP2), and EP4 receptors. J Biol Chem. 2003;278(50):49954–64.
Lai YJ, Pullamsetti SS, Dony E, Weissmann N, Butrous G, Banat GA, et al. Role of the prostanoid EP4 receptor in iloprost-mediated vasodilatation in pulmonary Hypertension. Am J Respir Crit Care Med. 2008;178(2):188–96.
Li HH, Hsu HH, Chang GJ, Chen IC, Ho WJ, Hsu PC, et al. Prostanoid EP(4) agonist L-902,688 activates PPARgamma and attenuates pulmonary arterial Hypertension. Am J Physiol Lung Cell Mol Physiol. 2018;314(3):L349–L59.
Lai YJ, Chen IC, Li HH, Huang CC. EP4 agonist L-902,688 suppresses EndMT and attenuates right ventricular Cardiac Fibrosis in Experimental Pulmonary arterial Hypertension. Int J Mol Sci. 2018;19(3).
Fan F, Tian H, Geng J, Deng J, Liu Y, Chen C, et al. Mechanism of Beraprost effects on Pulmonary Hypertension: contribution of cross-binding to PGE2 receptor 4 and modulation of O(2) sensitive voltage-gated K(+) channels. Front Pharmacol. 2018;9:1518.
Dogan MF, Yildiz O, Arslan SO, Ulusoy KG. Potassium channels in vascular smooth muscle: a pathophysiological and pharmacological perspective. Fundam Clin Pharmacol. 2019;33(5):504–23.
Chen Y, Jiang W, Zhao Y, Sun D, Zhang X, Wu F, et al. Prostaglandins for Postpartum Hemorrhage: Pharmacology, Application, and current opinion. Pharmacology. 2021;106(9–10):477–87.
Feletou M, Huang Y, Vanhoutte PM. Vasoconstrictor prostanoids. Pflugers Arch. 2010;459(6):941–50.
Markworth JF, Cameron-Smith D. Prostaglandin F2α stimulates PI3K/ERK/mTOR signaling and skeletal myotube hypertrophy. Am J Physiol Cell Physiol. 2011;300(3):C671-82.
Rice KM, Uddemarri S, Desai DH, Morrison RG, Harris R, Wright GL, et al. PGF2alpha-associated vascular smooth muscle hypertrophy is ROS dependent and involves the activation of mTOR, p70S6k, and PTEN. Prostaglandins Other Lipid Mediat. 2008;85(1–2):49–57.
Montuschi P, Barnes PJ, Roberts LJ 2. Isoprostanes: markers and mediators of oxidative stress. FASEB J. 2004;18(15):1791–800.
Lahaie I, Hardy P, Hou X, Hassessian H, Asselin P, Lachapelle P, et al. A novel mechanism for vasoconstrictor action of 8-isoprostaglandin F2 alpha on retinal vessels. Am J Physiol. 1998;274(5):R1406–16.
Zhang R, Sun ML, Fan YF, Jiang X, Zhao QH, He J, et al. Plasma 15-F2t-isoprostane in idiopathic pulmonary arterial Hypertension. Int J Cardiol. 2014;175(2):268–73.
Braun H, Hauke M, Eckenstaler R, Petermann M, Ripperger A, Kuhn N, et al. The F2-isoprostane 8-iso-PGF(2alpha) attenuates atherosclerotic lesion formation in Ldlr-deficient mice - potential role of vascular thromboxane A(2) receptors. Free Radic Biol Med. 2022;185:36–45.
Abramovitz M, Boie Y, Nguyen T, Rushmore TH, Bayne MA, Metters KM, et al. Cloning and expression of a cDNA for the human prostanoid FP receptor. J Biol Chem. 1994;269(4):2632–6.
Betz R, Lagercrantz J, Kedra D, Dumanski JP, Nordenskjold A. Genomic structure, 5’ flanking sequences, and precise localization in 1P31.1 of the human prostaglandin F receptor gene. Biochem Biophys Res Commun. 1999;254(2):413–6.
Zhang J, Gong Y, Yu Y. PG F(2alpha) receptor: a Promising Therapeutic Target for Cardiovascular Disease. Front Pharmacol. 2010;1:116.
Yu Y, Lucitt MB, Stubbe J, Cheng Y, Friis UG, Hansen PB, et al. Prostaglandin F2alpha elevates blood pressure and promotes Atherosclerosis. Proc Natl Acad Sci U S A. 2009;106(19):7985–90.
Alfranca A, Iniguez MA, Fresno M, Redondo JM. Prostanoid signal transduction and gene expression in the endothelium: role in Cardiovascular Diseases. Cardiovasc Res. 2006;70(3):446–56.
Vane J, Corin RE. Prostacyclin: a vascular mediator. Eur J Vasc Endovasc Surg. 2003;26(6):571–8.
Pluchart H, Khouri C, Blaise S, Roustit M, Cracowski JL. Targeting the Prostacyclin Pathway: Beyond Pulmonary arterial Hypertension. Trends Pharmacol Sci. 2017;38(6):512–23.
Clapp LH, Gurung R. The mechanistic basis of prostacyclin and its stable analogues in pulmonary arterial Hypertension: role of membrane versus nuclear receptors. Prostaglandins Other Lipid Mediat. 2015;120:56–71.
Kersten S, Desvergne B, Wahli W. Roles of PPARs in health and Disease. Nature. 2000;405(6785):421–4.
Liou JY, Lee S, Ghelani D, Matijevic-Aleksic N, Wu KK. Protection of endothelial survival by peroxisome proliferator-activated receptor-delta mediated 14-3-3 upregulation. Arterioscler Thromb Vasc Biol. 2006;26(7):1481–7.
Yu H, Clarke MC, Figg N, Littlewood TD, Bennett MR. Smooth muscle cell apoptosis promotes vessel remodeling and repair via activation of cell migration, proliferation, and collagen synthesis. Arterioscler Thromb Vasc Biol. 2011;31(11):2402–9.
Ruopp NF, Cockrill BA. Diagnosis and treatment of pulmonary arterial Hypertension: a review. JAMA. 2022;327(14):1379–91.
Doran A, Harris S, Goetz B. Advances in prostanoid infusion therapy for pulmonary arterial Hypertension. J Infus Nurs. 2008;31(6):336–45.
Barst RJ, Rubin LJ, Long WA, McGoon MD, Rich S, Badesch DB, et al. A comparison of continuous intravenous epoprostenol (prostacyclin) with conventional therapy for primary pulmonary Hypertension. N Engl J Med. 1996;334(5):296–301.
Klings ES, Hill NS, Ieong MH, Simms RW, Korn JH, Farber HW. Systemic sclerosis-associated pulmonary Hypertension: short- and long-term effects of epoprostenol (prostacyclin). Arthritis Rheum. 1999;42(12):2638–45.
Kallen AJ, Lederman E, Balaji A, Trevino I, Petersen EE, Shoulson R, et al. Bloodstream Infections in patients given treatment with intravenous prostanoids. Infect Control Hosp Epidemiol. 2008;29(4):342–9.
Krug S, Sablotzki A, Hammerschmidt S, Wirtz H, Seyfarth HJ. Inhaled iloprost for the control of pulmonary Hypertension. Vasc Health Risk Manag. 2009;5(1):465–74.
Olschewski H, Hoeper MM, Behr J, Ewert R, Meyer A, Borst MM, et al. Long-term therapy with inhaled iloprost in patients with pulmonary Hypertension. Respir Med. 2010;104(5):731–40.
Olschewski H, Simonneau G, Galie N, Higenbottam T, Naeije R, Rubin LJ, et al. Inhaled iloprost for severe pulmonary Hypertension. N Engl J Med. 2002;347(5):322–9.
Budev MM, Minai OA, Arroliga AC. Overview of treprostinil sodium for the treatment of pulmonary arterial Hypertension. Drugs Today (Barc). 2004;40(3):225–34.
Skoro-Sajer N, Lang I. Treprostinil for the treatment of pulmonary Hypertension. Expert Opin Pharmacother. 2008;9(8):1415–20.
Channick RN, Olschewski H, Seeger W, Staub T, Voswinckel R, Rubin LJ. Safety and efficacy of inhaled treprostinil as add-on therapy to bosentan in pulmonary arterial Hypertension. J Am Coll Cardiol. 2006;48(7):1433–7.
Lang I, Gomez-Sanchez M, Kneussl M, Naeije R, Escribano P, Skoro-Sajer N, et al. Efficacy of long-term subcutaneous treprostinil sodium therapy in pulmonary Hypertension. Chest. 2006;129(6):1636–43.
Tapson VF, Gomberg-Maitland M, McLaughlin VV, Benza RL, Widlitz AC, Krichman A, et al. Safety and efficacy of IV treprostinil for pulmonary arterial Hypertension: a prospective, multicenter, open-label, 12-week trial. Chest. 2006;129(3):683–8.
Melian EB, Goa KL. Beraprost: a review of its pharmacology and therapeutic efficacy in the treatment of peripheral arterial Disease and pulmonary arterial Hypertension. Drugs. 2002;62(1):107–33.
Galie N, Humbert M, Vachiery JL, Vizza CD, Kneussl M, Manes A, et al. Effects of beraprost sodium, an oral prostacyclin analogue, in patients with pulmonary arterial Hypertension: a randomized, double-blind, placebo-controlled trial. J Am Coll Cardiol. 2002;39(9):1496–502.
Vizza CD, Sciomer S, Morelli S, Lavalle C, Di Marzio P, Padovani D, et al. Long term treatment of pulmonary arterial Hypertension with beraprost, an oral prostacyclin analogue. Heart. 2001;86(6):661–5.
Boie Y, Rushmore TH, Darmon-Goodwin A, Grygorczyk R, Slipetz DM, Metters KM, et al. Cloning and expression of a cDNA for the human prostanoid IP receptor. J Biol Chem. 1994;269(16):12173–8.
Nakagawa O, Tanaka I, Usui T, Harada M, Sasaki Y, Itoh H, et al. Molecular cloning of human prostacyclin receptor cDNA and its gene expression in the cardiovascular system. Circulation. 1994;90(4):1643–7.
Zhou W, Hashimoto K, Goleniewska K, O’Neal JF, Ji S, Blackwell TS, et al. Prostaglandin I2 analogs inhibit proinflammatory cytokine production and T cell stimulatory function of dendritic cells. J Immunol. 2007;178(2):702–10.
Humbert M, Ghofrani HA. The molecular targets of approved treatments for pulmonary arterial Hypertension. Thorax. 2016;71(1):73–83.
Falcetti E, Hall SM, Phillips PG, Patel J, Morrell NW, Haworth SG, et al. Smooth muscle proliferation and role of the prostacyclin (IP) receptor in idiopathic pulmonary arterial Hypertension. Am J Respir Crit Care Med. 2010;182(9):1161–70.
Zhou W, Goleniewska K, Zhang J, Dulek DE, Toki S, Lotz MT, et al. Cyclooxygenase inhibition abrogates aeroallergen-induced immune tolerance by suppressing prostaglandin I2 receptor signaling. J Allergy Clin Immunol. 2014;134(3):698–705e5.
Zhou W, Dowell DR, Huckabee MM, Newcomb DC, Boswell MG, Goleniewska K, et al. Prostaglandin I2 signaling drives Th17 differentiation and exacerbates experimental autoimmune encephalomyelitis. PLoS ONE. 2012;7(5):e33518.
White RJ, Jerjes-Sanchez C, Bohns Meyer GM, Pulido T, Sepulveda P, Wang KY, et al. Combination therapy with oral Treprostinil for Pulmonary arterial Hypertension. A double-blind placebo-controlled clinical trial. Am J Respir Crit Care Med. 2020;201(6):707–17.
Asaki T, Kuwano K, Morrison K, Gatfield J, Hamamoto T, Clozel M. Selexipag: an oral and selective IP prostacyclin receptor agonist for the treatment of pulmonary arterial Hypertension. J Med Chem. 2015;58(18):7128–37.
Torres F, Farber H, Ristic A, McLaughlin V, Adams J, Zhang J et al. Efficacy and safety of ralinepag, a novel oral IP agonist, in PAH patients on mono or dual background therapy: results from a phase 2 randomised, parallel group, placebo-controlled trial. Eur Respir J. 2019;54(4).
Fischer LG, Honemann CW, Patrie JT, Durieux ME, Rich GF. Ropivacaine attenuates pulmonary vasoconstriction induced by thromboxane A2 analogue in the isolated perfused rat lung. Reg Anesth Pain Med. 2000;25(2):187–94.
Kameshima S, Nakamura Y, Uehara K, Kodama T, Yamawaki H, Nishi K et al. Effects of a Soluble Guanylate Cyclase Stimulator Riociguat on Contractility of isolated pulmonary artery and Hemodynamics of U46619-Induced Pulmonary Hypertension in Dogs. Vet Sci. 2023;10(2).
Kylhammar D, Radegran G. Cyclooxygenase-2 inhibition and thromboxane A(2) receptor antagonism attenuate hypoxic pulmonary vasoconstriction in a porcine model. Acta Physiol (Oxf). 2012;205(4):507–19.
Hirata M, Hayashi Y, Ushikubi F, Yokota Y, Kageyama R, Nakanishi S, et al. Cloning and expression of cDNA for a human thromboxane A2 receptor. Nature. 1991;349(6310):617–20.
Huang JS, Ramamurthy SK, Lin X, Le Breton GC. Cell signalling through thromboxane A2 receptors. Cell Signal. 2004;16(5):521–33.
Raychowdhury MK, Yukawa M, Collins LJ, McGrail SH, Kent KC, Ware JA. Alternative splicing produces a divergent cytoplasmic tail in the human endothelial thromboxane A2 receptor. J Biol Chem. 1994;269(30):19256–61.
Christman BW, McPherson CD, Newman JH, King GA, Bernard GR, Groves BM, et al. An imbalance between the excretion of thromboxane and prostacyclin metabolites in pulmonary Hypertension. N Engl J Med. 1992;327(2):70–5.
Dobyns EL, Wescott JY, Kennaugh JM, Ross MN, Stenmark KR. Eicosanoids decrease with successful extracorporeal membrane oxygenation therapy in neonatal pulmonary Hypertension. Am J Respir Crit Care Med. 1994;149(4 Pt 1):873–80.
Fuse S, Kamiya T. Plasma thromboxane B2 concentration in pulmonary Hypertension associated with congenital Heart Disease. Circulation. 1994;90(6):2952–5.
Katugampola SD, Davenport AP. Thromboxane receptor density is increased in human Cardiovascular Disease with evidence for inhibition at therapeutic concentrations by the AT(1) receptor antagonist losartan. Br J Pharmacol. 2001;134(7):1385–92.
Al-Naamani N, Palevsky HI, Lederer DJ, Horn EM, Mathai SC, Roberts KE, et al. Prognostic significance of biomarkers in pulmonary arterial Hypertension. Ann Am Thorac Soc. 2016;13(1):25–30.
McKenzie C, MacDonald A, Shaw AM. Mechanisms of U46619-induced contraction of rat pulmonary arteries in the presence and absence of the endothelium. Br J Pharmacol. 2009;157(4):581–96.
Mulvaney EP, Reid HM, Bialesova L, Bouchard A, Salvail D, Kinsella BT. NTP42, a novel antagonist of the thromboxane receptor, attenuates experimentally induced pulmonary arterial Hypertension. BMC Pulm Med. 2020;20(1):85.
Mulvaney EP, Renzo F, Adao R, Dupre E, Bialesova L, Salvatore V, et al. The thromboxane receptor antagonist NTP42 promotes beneficial adaptation and preserves cardiac function in experimental models of right heart overload. Front Cardiovasc Med. 2022;9:1063967.
Mulvaney EP, Reid HM, Bialesova L, Mendes-Ferreira P, Adao R, Bras-Silva C, et al. Efficacy of the thromboxane receptor antagonist NTP42 alone, or in combination with sildenafil, in the sugen/hypoxia-induced model of pulmonary arterial Hypertension. Eur J Pharmacol. 2020;889:173658.
Thompson AAR, Lawrie A. Targeting vascular remodeling to treat pulmonary arterial Hypertension. Trends Mol Med. 2017;23(1):31–45.
Lau EMT, Giannoulatou E, Celermajer DS, Humbert M. Epidemiology and treatment of pulmonary arterial Hypertension. Nat Rev Cardiol. 2017;14(10):603–14.
Qin J, Wang G, Han D. Selexipag in patients with Pulmonary Hypertension: a systematic review and Meta-analysis of Randomized controlled trials. Curr Probl Cardiol. 2023;48(2):101466.
Seyfarth HJ, Hammerschmidt S, Halank M, Neuhaus P, Wirtz HR. Everolimus in patients with severe pulmonary Hypertension: a safety and efficacy pilot trial. Pulm Circ. 2013;3(3):632–8.
Abedi F, Omidkhoda N, Arasteh O, Ghavami V, Hosseinzadeh H. The therapeutic role of rho kinase inhibitor, Fasudil, on pulmonary Hypertension; a systematic review and Meta-analysis. Drug Res (Stuttg). 2023;73(1):5–16.
Waxman A, Restrepo-Jaramillo R, Thenappan T, Ravichandran A, Engel P, Bajwa A, et al. Inhaled Treprostinil in Pulmonary Hypertension due to interstitial lung Disease. N Engl J Med. 2021;384(4):325–34.
Sadushi-Kolici R, Jansa P, Kopec G, Torbicki A, Skoro-Sajer N, Campean IA, et al. Subcutaneous treprostinil for the treatment of severe non-operable chronic thromboembolic pulmonary Hypertension (CTREPH): a double-blind, phase 3, randomised controlled trial. Lancet Respir Med. 2019;7(3):239–48.
Acknowledgements
Not applicable.
Funding
This work was supported by grants from the National Natural Science Foundation of China (82171557); Natural Science Foundation of Hunan Province (2021JJ20085); the Fundamental Research Funds for the Central Universities of Central South University (2023ZZTS0877).
Author information
Authors and Affiliations
Contributions
The review was designed by Xinqun Hu and Yuhu He. The manuscript of this review was prepared by Cheng Zeng. The draft was revised by Jing Liu and Xialei Zheng. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Zeng, C., Liu, J., Zheng, X. et al. Prostaglandin and prostaglandin receptors: present and future promising therapeutic targets for pulmonary arterial hypertension. Respir Res 24, 263 (2023). https://doi.org/10.1186/s12931-023-02559-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12931-023-02559-3