
Abstract
Background
The mammalian ovary is a unique organ that displays a distinctive feature of cyclic changes throughout the entire reproductive period. The estrous/menstrual cycles are associated with drastic functional and morphological rearrangements of ovarian tissue, including follicular development and degeneration, and the formation and subsequent atrophy of the corpus luteum. The flawless execution of these reiterative processes is impossible without the involvement of programmed cell death (PCD).
Main text
PCD is crucial for efficient and careful clearance of excessive, depleted, or obsolete ovarian structures for ovarian cycling. Moreover, PCD facilitates selection of high-quality oocytes and formation of the ovarian reserve during embryonic and juvenile development. Disruption of PCD regulation can heavily impact the ovarian functions and is associated with various pathologies, from a moderate decrease in fertility to severe hormonal disturbance, complete loss of reproductive function, and tumorigenesis. This comprehensive review aims to provide updated information on the role of PCD in various processes occurring in normal and pathologic ovaries. Three major events of PCD in the ovary—progenitor germ cell depletion, follicular atresia, and corpus luteum degradation—are described, alongside the detailed information on molecular regulation of these processes, highlighting the contribution of apoptosis, autophagy, necroptosis, and ferroptosis. Ultimately, the current knowledge of PCD aberrations associated with pathologies, such as polycystic ovarian syndrome, premature ovarian insufficiency, and tumors of ovarian origin, is outlined.
Conclusion
PCD is an essential element in ovarian development, functions and pathologies. A thorough understanding of molecular mechanisms regulating PCD events is required for future advances in the diagnosis and management of various disorders of the ovary and the female reproductive system in general.
Similar content being viewed by others


Background
Regulated cell death (RCD) is an essential part оf the developmental process. The controlled elimination of unwanted cells occurs in virtually any tissue of the body, from skin to the nervous system [1]. In general, RCD might occur on three occasions: as a response to irreparable cell damage (mutations, DNA damage, metabolic impairments, viral infections, etc.), as a mechanism of tissue homeostasis (e.g., to counterbalance constant proliferation and renewal of epithelial tissues), or as an innate process of organ development and functioning (removal of superfluous cells or cells that have accomplished their task) [2]. The second and third cases are together called programmed cell death (PCD) and involve very precise and intricate regulatory mechanisms because the impairments of well-organized cell clearance in normal tissues could easily result in long list of pathologies.
In addition to well-known processes during embryogenesis, such as the development of the central nervous system [3] and finger formation [4], PCD plays a very important role during juvenile and adult life. One of the most important cases of post-embryonic PCD takes place in the ovary and is directly connected to reproductive function, pregnancy, and the estrous/menstrual cycles. The cyclic nature of reproductive processes and their absolute importance for the survival of the species require an intricate regulatory mechanism(s) that should be able to sustain the continuous cycles of the “cell proliferation-differentiation-death-recovery” process.
This review aims to provide current information on the main aspects of PCD involvement in the functions and biological processes in the normal ovary and to highlight PCD abnormalities associated with various ovarian pathologies. We performed an extensive analysis of the published information, paying special attention to the detailed description of molecular mechanisms regulating PCD and the survival of ovarian cells. The established major role of apoptosis is discussed, while also pinpointing the recent discoveries in non-apoptotic modes of PCD (autophagy, necroptosis and ferroptosis).
The mammalian ovary: from structure to functions
The first discernible germline cells in the developing embryo are called “primordial germ cells” (PGCs). During the early stages of embryogenesis, these cells migrate from the endoderm to the mesonephros, where epithelial, mesenchymal, and blood vessel cells start to form the body of the fetal ovary. PGCs actively proliferate during the migration and for some time afterward, forming multicellular clusters called “PGC cysts” (Fig. 1) [5, 6]. The cells in these cysts are connected with each other through cytoplasmic intercellular bridges [6]. At the same time, they interact with a specific subpopulation of epithelial cells called pre-granulosa cells to form the initial complexes that will later develop into ovarian follicles [5]. The proliferation of PGCs ultimately concludes in a meiotic division that generates the primary oocytes; however, meiosis is halted at prophase I due to control from the surrounding pre-granulosa cells [7, 8]. The primary oocytes reside in this state called “the dictyate” throughout the rest of embryonic development and a major part of adult life, only resuming their meiotic division in response to stimulation exerted by other elements of the reproductive system.

The essential steps of development and degeneration of follicles and the CL in a mammalian ovary. PGC cysts form in the embryonic period (blue area). After the PGC depletion before birth, the primordial follicles start to gradually develop into antral follicles. During the juvenile period (green area) all antral follicles undergo atresia. During the reproductive period (yellow area) a surge of FSH rescues the dominant follicle, while smaller antral follicles degenerate via atresia. Dominant follicles next undergo ovulation stimulated by FSH and LH and transform into CL , which subsequently involutes into corpus albicans in a PGF2α-dependent manner. Primordial follicles are repeatedly recruited until the whole ovarian reserve is depleted (pink area) and the reproductive period ends. The black arrows indicate survival and development, the red arrows indicate degradation events, and the lightning symbols indicate the crucial involvement of endocrine regulators
The first massive wave of PCD is observed in the developing ovary [9]. Up to 80% of PGCs and primary oocytes are eliminated, while only a minor part of the initial population survives [5]. Pre-granulosa cells associated with dying PGCs survive and are relocated to other, viable PGCs and primary oocytes, resulting in a drastic increase in the ratio of pre-granulosa cells to PGCs/oocytes [10]. As a result, each primary oocyte becomes surrounded by a single layer of flattened pre-granulosa cells and a thin layer of basal membrane, forming a structure known as “primordial follicle” [11]. These follicles remain dormant until birth and through the juvenile period until specific stimulation that promotes their further development [12]. In rodents, the process of primordial follicle formation and PGC depletion is extended through the first week after birth [13].
At the moment of birth, the ovary consists of three distinct compartments: the superficial epithelium (one layer of cubic epithelial cells), the cortex (a major part of the ovary that contains primordial and developing follicles), and the medulla (mesenchymal stromal tissue that houses numerous blood vessels) [14]. It had been assumed that the oocytes present at birth represent the entire reserve of potential ova available for the duration of life. However, this dogma was disproved by demonstrating that germ cells can enter meiosis and produce new oocytes and follicles during the reproductive age [15, 16].
After the formation of primordial follicles, a process termed “initial recruitment” starts and continues throughout the entire reproductive age [17]. Primordial follicles are stimulated by the local environment and transform into “primary follicles”, which are recognized by an increase in the oocyte size and the proliferation of the surrounding layer of granulosa cells. The oocyte is separated from the granulosa cells by the zona pellucida, which is filled with glycoprotein matrix, and there are long cytoplasmic protrusions connecting granulosa cells to the oocyte [18,19,20]. The ongoing growth results in development of “secondary follicle” that exhibits an even larger oocyte and two or more layers of granulosa cells surrounded by theca cells [19]. Around this point, the mature granulosa cells start to express the receptors for follicle-stimulating hormone (FSH), making primary and secondary follicles sensitive to pituitary stimuli [21]. The formation of a fluid-filled cavity marks the antral stage; the antral follicles are ready for terminal maturation—if they can survive [19].
The second crucial case of PCD occurs at this stage and is called “atresia”, a widespread degeneration of antral follicles. Follicular atresia starts with several dying granulosa cells and rapidly spreads across the entire follicle, efficiently obliterating it within several days. Atresia is a default outcome of follicle development, and most follicles degenerate without reaching maturation [17, 19]. The main factor that rescues the follicle is a surge of FSH excreted by the pituitary gland during each estrous/menstrual cycle after the onset of puberty. These repeated events are called “cyclic recruitment” and result in the selection of several dominant antral follicles based on their sensitivity to FSH and other hormones (Fig. 1). The number of dominant follicles is specific for each species and is associated with the size of the offspring (e.g., there is usually only one dominant follicle in a human ovary) [17, 19, 22].
The dominant follicle reaches the terminal maturation state, when granulosa cells start to produce high levels of estradiol; the follicle also becomes extremely responsive to FSH and luteinizing hormone (LH), another pituitary gonadotropin [22]. At some point, the inhibitory effect is exerted by the dominant follicle(s) upon the pituitary gland and results in suppression of FSH secretion. The dominant follicle develops further with support from LH and estradiol; less mature follicles are unable to survive the decline in FSH levels and undergo atretic degeneration [22, 23]. LH stimulation of granulosa cells lifts the prophase I arrest, and the first meiotic division occurs, resulting in formation of a “secondary oocyte” [24]. The final step of the follicle development is associated with a simultaneous surge in FSH, LH, and estradiol levels that promotes follicle rupture and release of the oocyte [22, 25]. The secondary oocyte subsequently interacts with a spermatozoid, undergoes the second meiotic division, develops into a mature ovum, and is fertilized to form a zygote [24].
The granulosa cells of the ruptured follicle do not simply degrade after ovulation; rather, their function is to provide a favorable environment for the embryo to develop and successfully implant into the uterus. This function is achieved through their terminal differentiation into the corpus luteum (CL), a temporary gland inside the ovarian tissue. Luteal cells produce high levels of progesterone that affect the endometrium of the uterus and promote the implantation of the blastocyst and its further development into the embryo [26].
The final fate of luteal cells is determined by the success of embryo implantation into the endometrium. If the implantation fails, then CL cells quickly die in a process called “luteolysis” that starts within several days after ovulation (Fig. 1) [26]. In many species, CL regression is primarily stimulated by prostaglandin F2α (PGF2α) secreted by the uterus. However, luteolysis in primates (including humans) is not dependent on PGF2α produced by the uterus; it is supposedly caused by a decrease in LH levels [27, 28]. If there is successful embryo implantation and pregnancy, then the lifespan of the CL is extended due to the effect of chorionic gonadotropin (CG, primates and equine) or prolactin (rodents) secreted by the implanted blastocyst or decidua, respectively [29]. The developing placenta takes over the synthesis of progesterone, rendering the CL redundant; it then undergoes prompt destruction through PCD [27]. This third major PCD event concludes the long chain of changes initiated during the cyclic recruitment of follicles and “resets” the ovary, preparing it for a new menstrual/estrous cycle.
Thus, PCD plays an essential role in both the development and normal physiology of the ovary, allowing the organism to successfully progress through multiple cycles of reproduction. Below the most common types of PCD are presented with information regarding their involvement in oocyte depletion, follicular atresia, and luteolysis, as well as various ovarian pathologies.
Common types of PCD observed in the ovary
The understanding of RCD has evolved tremendously from the most basic observations to the extremely sophisticated models describing the internal molecular cross-talk between multiple modes of cell death [2, 30]. For a long time, apoptosis had been believed to be the only type of PCD/RCD, but studies in recent decades have defined multiple other ways cells are eliminated and have brought them into the spotlight. These types of RCD are caused by different stimuli and are executed through different signaling pathways. They also differ in the effects exerted upon their environment, from relatively clean autophagy-dependent death to disruptive and inflammation-inducing pyroptosis [2]. Below is a brief description of the types of RCD that have been reported to occur in the ovarian cells.
Apoptosis
Apoptosis is the most common and best described mode of RCD that is often considered to be the “default” way for cells to die. Morphologic changes observed in apoptotic cells include chromatin condensation, fragmentation of the nucleus, membrane blebbing, reduction of the cytoplasm volume, and general cell shrinkage. The cell ultimately breaks down into small membrane-encapsulated apoptotic bodies, which undergo phagocytosis by macrophages or neighboring cells [31].
Apoptosis is orchestrated by activation of a family of proteases called caspases. These enzymes are evolutionally conserved and functionally divided into three major groups: apoptosis initiators (caspase-8, caspase-9, and caspase-10), apoptosis executioners (caspase-3, caspase-6, and caspase-7), and inflammatory caspases (caspase-1, caspase-4, caspase-5, and caspase-11) [32]. Caspase-2 stands as a unique family member as its involvement in cell death and other aspects of cell physiology is a matter of great debate [33, 34]. Various caspases facilitate the signal transduction along two major apoptosis pathways: intrinsic and extrinsic.
Intrinsic (“mitochondrial”) apoptosis activates in conditions of nutrient deprivation, growth factor shortage, detachment from the matrix and other cells, cytotoxic stress, and other non-specific factors. The subsequent overexpression and activation of BH3-only proteins belonging to the BCL-2 family (BIM, NOXA, BBC3/PUMA, and BAD) results in neutralization of pro-survival BCL-2-like proteins (BCL-2, BCL-XL, BCL-2A1, and MCL-1) and activation of the BAX/BAK complexes, which permeabilize the outer mitochondrial membrane [35]. Mitochondrial cytochrome c is released into the cytoplasm and forms (in presence of dATP) the apoptosome complex with Apoptotic Peptidase Activating Factor 1 (APAF1) and pro-caspase-9. Activated caspase-9 proteolytically cleaves and activates caspases-3 and caspase-7, which in turn cleave hundreds of substrate proteins and promote the terminal stages of apoptosis [32].
Extrinsic apoptosis is actively stimulated through “death receptor” molecules on the cell surface. These receptors belong to the tumor necrosis factor (TNF) superfamily and include CD95/Fas/Apo-1, TNFR1, TRAIL receptors, and other proteins [36]. Ligand binding results in the assembly of a protein complex that includes the receptor itself, the adaptor proteins FADD/TRADD and pro-caspase-8, and triggers caspase-8 processing [32, 36]. Active caspase-8 directly cleaves and activates executioner caspases. Additionally, caspase-8 can cleave BID and thus promote BAX/BAK activation, switching the signal transduction toward the intrinsic pathway [37].
The molecular machinery controlling apoptosis initiation and progression is very complex, and the role of its specific components can vary considerably in different cells and tissues. The development of multiple experimental models that recapitulate the inactivation or hyperactivation of individual genes and proteins in a tissue-specific manner allowed to thoroughly unravel this complicated network regarding ovary-specific processes [38].
Autophagy
Autophagy is a permanently operating process in the living cell that provides the removal of non-functional or harmful molecules and organelles, their degradation, and recycling. It is, in general, a pro-survival mechanism that sustains cell renewal and stress resistance. However, in certain conditions RCD relies on autophagic proteins and pathways and should be considered true autophagy-dependent cell death (ADCD) [2, 39].
Macroautophagy (usually referred to as “autophagy” in general) is the most common type of autophagy and is facilitated by autophagosomes, which engulf the organelles and aggregates, separating them from the cytoplasm. They later fuse with lysosomes to form autophagolysosomes for disintegration and reabsorption of useful material. Microautophagy involves direct sequestration of cytosolic content by the lysosome, and chaperone-mediated autophagy is associated with selective transfer of protein molecules into the lysosome. The affected cells appear to be filled with vesicles encapsulating fragments of other organelles [39].
Autophagy is promoted in response to a multitude of stimuli such as stress, various forms of starvation, hypoxia, infections, and intracellular disturbances [40]. Different signaling proteins including phosphatidylinositol 3-kinase (PI3K), a serine/threonine-specific protein kinase (AKT), mammalian target of rapamycin (mTORC1) kinase, and protein kinase AMP-activated catalytic subunit alpha-1 (AMPK) converge on the Unc-51-like autophagy activating kinase (ULK) protein complex. Activated ULK phosphorylates BECN1 and other targets that constitute the PI3KC3 complex associated with pre-autophagosomal membrane structures in the endoplasmic reticulum (ER) [41]. The following steps involve a large family of ATG proteins and the microtubule-associated protein 1 light chain (MAP1LC3 or LC3) protein. Pre-autophagosomal membranes bind the ATG5-ATG12-ATG16 complex and LC3 modified with phosphatidylethanolamine (also known as LC3-II) by ATG3, ATG4, and ATG7. These membranes are called “phagophores” and are ready to engulf their substrates and close into an autophagosome. The final step—fusion of an autophagosome and a lysosome—is controlled by LAMP1 and LAMP2 [40]. Lysosome-targeting compounds like bafilomycin A1 and chloroquine prevent autophagolysosome maturation and inhibit autophagy halfway through it, keeping the cellular contents sequestered but not degraded [42].
While autophagic activity is often increased in the cells undergoing RCD, it can be an attempt to attenuate the stressful conditions and rescue the cell from death. On the other hand, autophagy-related proteins interact with many complexes regulating apoptosis (FADD, caspase-8, MCL-1, BCL-XL), necroptosis (RIPK1, RIPK3), and ferroptosis (ferritin), and can actively promote either apoptotic or necrotic cell death [43]. It is very important to define ADCD as a type of cell death that essentially requires activation of autophagic machinery and can be inhibited by autophagy inactivation [2]. Up to date, ADCD in mammalian tissues has only been confirmed in several specific cases, which do not allow yet to build a general scheme of autophagy involvement into RCD or properly define its physiological impact [43]. Nevertheless, the accumulating knowledge of tight links between autophagy and RCD in various tissues and conditions invokes increasing interest in this area. This review aims to collect and organize the current evidence of how autophagy may impact death and survival of ovarian cells, highlighting the events potentially occurring through ADCD.
Necroptosis
Necrotic cell death is much more chaotic than apoptosis and was considered an uncontrolled way of accidental cell death until two seminal reports described necrosis induction in glutamate-treated neurons [44] and a switch from apoptosis to necrosis-like cell death in murine thymocytes [45]. Since then, several subtypes of regulated necrotic death have been described, with necroptosis being the first of them. Cells undergoing necroptosis display very distinct morphology: a breach of the cellular membrane results in prominent cell swelling without visible chromatin condensation in the nucleus. The content of the dying cell is released into the surrounding space and induces an inflammatory response in the adjacent cells [46].
Necroptosis progression is regulated by a pathway that relies on three key proteins: receptor interacting serine/threonine kinase 1/3 (RIPK1, RIPK3), and mixed lineage kinase domain like pseudokinase (MLKL) [47]. It is most often initiated by ligands of TNF superfamily receptors (TNF1α, FasL, and TRAIL) but can also be induced by bacterial or viral infections and chemical agents [47]. TNF-associated induction is the most well characterized, and its initial steps have significant overlap with the extrinsic apoptosis pathway. Activated TNF receptors assemble Complex I that consists of TRADD, TRAF2, RIPK1, and inhibitor of apoptosis protein (cIAP1/2) proteins and promotes survival through the NFκB pathway [48]. In the absence of cIAPs, Complex I becomes unstable, and RIPK1 binds to FADD and caspase-8 instead, forming Complex IIb that can promote caspase-8-dependent apoptosis [49]. However, if caspase-8 activity (or caspase activity in general) is inhibited, then RIPK1 undergoes autophosphorylation and assembles Complex IIc (“necroptosome”) together with RIPK3 and MLKL. Subsequent RIPK3 and MLKL phosphorylation results in MLKL oligomerization and translocation to the cytoplasmic membrane, where it forms transmembrane channels and facilitates loss of membrane integrity, condemning the cell to necroptotic death [50]. Due to its disruptive effects, necroptosis is not observed very often in normal tissues, but the aberrations of necroptotic molecular machinery can be associated with various pathologies.
Ferroptosis
Ferroptosis as a separate, iron-dependent mode of cell death was first described in 2012 and integrated earlier observations of non-apoptotic cell death associated with ions of iron, reactive oxygen species (ROS), and metabolic effects [51, 52]. The most distinct morphologic feature of ferroptosis is mitochondria shrinkage combined with cristae reduction and an increase in membrane density [51]. The plasma membrane integrity and nucleus structure are preserved until the very late stages when the dying cells acquire a necrotic morphology [46, 53].
Ferroptosis is characterized by the peroxidation of polyunsaturated fatty acids (PUFAs) incorporated into membrane phospholipids. Some monounsaturated fatty acids (MUFAs) have been shown to exert the opposite, anti-ferroptotic effects. The role of PUFAs in ferroptosis is highly dependent on their location, and the ER is the most crucial place where PUFA peroxidation occurs [52]. Peroxidation of membrane-bound PUFAs can be initiated by iron-dependent lipoxygenases and is promoted by free Fe2+ through the Fenton reaction, which produces hydroxyl radicals. These processes define the importance of iron turnover during ferroptosis initiation. Intracellular iron is deposited in complexes with ferritin or glutathione (GSH), and autophagy-dependent ferritin degradation is an important step during ferroptosis progression [52, 54]. GSH depletion also increases Fe2+ availability, but its major role in ferroptosis regulation is carried out through glutathione peroxidase 4 (GPX4), a GSH-dependent peroxidase that protects membrane lipids from oxidation by ROS [55]. GSH is synthesized from cystine imported into the cell via the xc− system, and the first described ferroptosis inductors, erastin and RAS-selective lethal (RSL3) compound, are inhibitors of the xc− system and GPX4, respectively [56, 57]. Overall, ferroptosis is a relatively new type of RCD which is being actively explored.
Regulation of PCD during embryonic and juvenile development
The loss of PGCs during the formation of primordial follicles marks the first significant PCD event in the developing ovary [5, 9]. It is observed in all studied mammals, including mice, rats, and humans [7, 58, 59]. In human ovaries, the total number of primary oocytes decreases from around 7 million cells in a 5-month-old embryo to 1–2 million cells at birth and 300,000 cells at the age of 7 years [59]. Three hypotheses explaining PGC depletion have been proposed and supported by experimental data: (I) degeneration of excess PGCs that did not attract enough pre-granulosa cells and growth factors (“Apoptosis” section [60,61,62]), (II) lethal DNA damage during crossover step in the meiosis (“Apoptosis” section [63,64,65]), and (III) sacrifice of normal PGCs to allow the surviving ones to feed on their cytoplasmic contents (“Autophagy” section [6, 66, 67]). The current knowledge of PCD regulation in the developing ovary is discussed below, and the most important regulatory pathways are summarized in Fig. 2.

Major molecular mechanisms and regulatory elements involved in the control of PGC depletion process in the embryonic ovary. The pathways regulated by KITLG and LIF, which are produced by granulosa cells, promote PGC survival through inhibition of apoptosis and stimulation of autophagy (left side of the figure). In the absence of KITLG, the intrinsic apoptosis pathway (with potential involvement of extrinsic apoptosis regulators) induces PGC death through apoptosis (right side of the figure). Orange circles with the letter “P” indicate protein phosphorylation, red crosses indicate the lack or suppression of certain regulators, and dashed elements indicate the regulators and connections that are not definitively confirmed up to date
Apoptosis
The vast majority of PGCs and primary oocytes undergo caspase-dependent apoptosis [68, 69]. The survival and proliferation of PGCs are crucially dependent on external signaling molecules such as KIT ligand (KITLG (SCF)), Leukemia inhibitory factor (LIF), Insulin like growth factor 1 (IGF1), retinoic acid, and cAMP activators [70, 71]. The presence of KITLG and LIF is essential to prevent PGC apoptosis in vitro [60], and both factors are secreted by pre-granulosa and granulosa cells, promoting PGC survival in vivo [61, 62]. The KIT receptor (also known as c-Kit and CD117) on the surface of PGCs acts through the PI3K/AKT and MEK/ERK signaling pathways [72], and its knockout results in complete oocyte degradation by the moment of birth [73]. The exact pathways facilitating LIF signaling in PGCs have not yet been determined; however, LIF can activate the Janus kinase/Signal transducer and activator of transcription 3 (JAK/STAT3) pathway in porcine oocytes [74] and human granulosa cells [75]. Evidence from other models suggests that STAT3 activation should confer an anti-apoptotic effect [76]. Both the PI3K/AKT and STAT3 pathways may promote the expression of BCL-2, BCL-XL, and MCL-1 [76, 77]. Moreover, AKT can phosphorylate and inactivate the proapoptotic BAX and BAD proteins [77, 78] and the Forkhead box O3 (FOXO3) transcription factor, which promotes the expression of the BCL-2 inhibitor BIM in oocytes [79]. Treatment with KITLG inhibits FOXO3, BIM, and apoptosis in naked oocytes in vitro [80]. Thus, the shortage of available survival-stimulating pre-granulosa cells may translate to PGCs death until the optimal ratio between oocytes and pre-granulosa cells is achieved.
Growth factor deficiency, in general, promotes intrinsic apoptosis through the BCL-2- BAX/BAK axis. Bcl2 or Bax knockout in mice results in decrease or increase in the number of primordial follicles, respectively [81, 82], and apoptotic oocytes display a reduced Bcl2l1 to Bax transcripts ratio [83]. Independently of the PI3K/AKT and STAT3 pathways, DNA damage, ER stress, mitochondrial damage, or nutrient starvation can activate BH3-only proteins through p53 or its homolog TAp63; the latter protein is the major regulator of NOXA and BBC3 (PUMA) in oocytes [35, 63, 84]. BBC3, NOXA, and BAX are controlled by the checkpoint kinase 2 (CHK2)-p53/p63 checkpoint system and facilitate the elimination of primary oocytes with persistent DNA damage in the postnatal period [64, 65]. BBC3 seems to be more important for this process than NOXA: BBC3-only or BBC3/NOXA inactivation protects PGCs from radiation-induced apoptosis, but NOXA-only knockout does not [63]. Moreover, BBC3 is crucial for the death of PGCs during their migration to the developing ovary but not during PGC cyst breakdown; the second process seems to be BAX-independent [82, 85]. TAp63-dependent DNA damage and apoptosis in fetal oocytes are drastically promoted by the inactivation of Glycogen synthase kinase 3 beta (GSK3B) kinase [86]. This fact is surprising—as GSK3B activity is commonly suppressed by the PI3K/AKT pathway—and indicates the complexity of the interactions between pro-survival and pro-apoptotic signals in the establishment of the primordial follicle pool. DNA damage and repair are inherent to crossover recombination during meiosis, and the aforementioned information supports the idea of PGC depletion as a way to enrich the pool of competent oocytes that have not accumulated unrepaired DNA lesions.
Very little data are available regarding extrinsic apoptotic signals in the process of PGCs depletion. Primary oocytes express TNFα, TRAIL and their receptors, and treatment with TNFα or TRAIL in vitro can induce apoptosis in naked oocytes or granulosa cells, respectively [87, 88]. CD95/Fas/Apo-1 knockout increased PGC numbers in both fetal and postnatal murine ovaries [73]. Overall, this area requires further investigation.
The extrinsic and intrinsic apoptosis pathways converge on caspase machinery, and three distinct features have been reported for caspases in PGCs and primordial follicles. Caspase-3, one of essential executioners of apoptosis, seems to be completely irrelevant for oocyte pool dynamics in mice during both embryonic development and initial weeks after birth [89]. Caspase-2 is usually dispensable for apoptosis regulation [90], but Casp2 knockout in mice results in a significant increase in the number of primordial oocytes [91]. Further investigation has demonstrated that caspase-2 deficiency prevents PGC death caused by cytokine deprivation (or inactivation of interleukin processing by caspase-4/-5 or caspase-11) but does not affect apoptosis induced by meiotic defects [92]. Selective caspase-2 inhibition is sufficient to partially prevent the death of oocytes cultured in vitro [69]. Initiator caspase-9 is constitutively active in all fetal oocytes, but the progression of apoptosis is hindered by the X-linked inhibitor of apoptosis (XIAP) protein [93, 94]. This block is relieved by upregulation of LINE1 retrotransposon expression [95]. It is possible that other caspases (caspase-6, caspase-7, and caspase-8) could also be regulated in a unique way in fetal oocytes, but no reports have been published on this topic.
Surprisingly, the hallmarks of classic apoptosis are not universally observed in murine primordial follicles during early postnatal development despite the ongoing reduction in oocyte numbers [13, 96]. This fact raises many questions about the contribution of apoptosis to the depletion of oocytes during the pre-puberty period.
Autophagy
While apoptosis is the most investigated type of PCD in the embryonic ovary, some of the oocytes also display autophagic features both in vitro and in vivo [69, 97], but the current reports are as controversial as autophagy itself. A prominent increase in the number of LAMP1-labeled vesicles in oocytes soon after birth suggests that lysosomes and autophagy may be involved in the corresponding reduction of the oocyte pool [97]. However, LC3, which is essential for autophagosome maturation, is only detected in rodent PGC cysts and completely disappears from oocytes during primordial follicle formation; it is only retained in granulosa cells [98, 99].
Inactivation of autophagy by Atg7 knockout or hemizygous Becn1 deficiency in mice results in severe loss of PGCs during the neonatal period, suggesting its positive role in oocyte survival [100]. Germ cell-specific Atg7 knockout causes the same effect, ruling out the involvement of granulosa cells [101]. Autophagy and the oocyte pool size are both positively controlled by miR-378-3p (which targets pyruvate dehydrogenase kinase 1 (PDK1) and caspase-9), miR-92b-3p (which targets tuberous sclerosis complex subunit 1 (TSC1), and Sirtuin 1 (Sirt1) [99, 102]. On the other hand, the lack of MAP1LC3 indicates that canonical autophagy should not be active in primordial follicle oocytes [98]. Therefore, it is possible that ATG7 or BECN1 deficiency confers their effects through a more exotic LC3-independent autophagic process. This idea is supported by the fact that oocytes in primordial and primary follicles contain cytoplasmic autophagosomes detectable by electron microscopy [103, 104]. An experimental model demonstrated that autophagy protects PGC cysts and oocytes from the stress caused by perinatal starvation, albeit at the expense of granulosa cells [103, 105].
Despite the observations described above, the promotion of autophagic process has been also reported during attrition of primordial follicles. Degrading oocytes in ovaries of 1- to 5-day-old rats express high levels of lysosome marker LAMP1 and acid phosphatase [104]. However, LAMP1 labels all kinds of lysosomes, including those produced in an autophagy-independent manner. In addition, some autophagic oocytes simultaneously display caspase-3 activation and DNA fragmentation without clear apoptotic morphology [104]. Therefore, the causative role of autophagy in oocyte death progression remains to be confirmed. Nevertheless, the idea of “altruistic death” has been proposed. The majority of PGCs in the pre-follicular cysts are considered to be “nurse cells” that sacrifice themselves to provide nutrition to one surviving oocyte [66]. In this case, ADCD would be a fitting way for the cells to die. Recent studies in Drosophila and mice have confirmed the idea of “nurse cells” but have proposed a unique model of extrinsic cell death induction instead of common autophagy [6, 67]. During follicle formation around the PGC cyst, surrounding pre-granulosa cells generate acidic extracellular vesicles that fuse with PGCs. Pre-granulosa cells transfer cathepsins and DNases into “nurse cells”, promoting their death and relocation of their contents into the developing oocyte through intercellular cytoplasmic bridges. This unique process is inhibited by bafilomycin A1, a commonly used inhibitor of lysosome generation and autophagy that blocks the activity of vacuolar ATPase and prevents acidification, resulting in PGC survival [6]. At first glance, such effects could be interpreted as evidence that autophagy is the reason for “nurse cell” degradation, but the real mechanism is very different because it is facilitated through acidified exosome-like vesicles instead of intra-cytoplasmic lysosomes. It is also possible that autophagosome-like structures observed in primordial follicle oocytes are transferred to their cytoplasm from “nurse cells”. More studies are needed to better understand this unique way of oocyte death.
Necroptosis and ferroptosis
Necroptosis- or ferroptosis-mediated PCD in the developing ovary had not been investigated in detail until very recently. The fact that oocytes express TNFα was described back in 1994 [106], but the first evidence of necrotic cell death during the pre-puberty period was reported in 2018 [107]. Necroptotic ovarian cells comprise both PGCs and somatic cells immediately after birth, but only somatic cells still undergo necroptosis by day 21 [107, 108]. Surprisingly, the highest concentration of necroptotic events occurs in the medulla region, but its role is undefined [108].
Transferrin, a major iron carrier protein, is expressed at all stages of ovarian follicles and is used as an important component of medium for ovary cultivation conditions [109, 110]. Early stages of growing follicle atresia are associated with pro-ferroptotic changes in gene expression patterns [111], but there are currently no reports describing ferroptotic cell death events in the developing ovary. Nevertheless, a recent single-cell transcriptome study revealed that most ferroptosis-related genes are highly expressed in both oocytes and granulosa cells during the late embryonic stages and early postnatal life, sparking an increased interest in this area [112].
Regulation of PCD during follicular atresia
Out of the thousands of oocytes that survive the initial PGC depletion and form primordial follicles, only a few are able to complete maturation and successfully ovulate [113]. The vast majority of follicles undergo spontaneous atresia at the antral stage unless they are rescued by FSH secreted by the pituitary gland [19]. The initial recruitment of primordial follicles begins around birth, but follicular atresia eliminates the growing follicles throughout the entire juvenile period. After puberty onset, periodic surges of gonadotropins during each estrous/menstrual cycle rescue a number of antral follicles from degradation and allow them to develop further. During this stage, one or more dominant follicles reciprocally suppress FSH secretion and thus exert an inhibitory effect upon other maturing ones, setting them toward atretic demise [22, 23].
The major difference between PGC attrition during ovary development and atresia of antral follicles lies in the origin of degradation. While the depletion of primordial follicles is driven by oocytes themselves, follicular atresia is initiated in granulosa cells [114, 115]. Atresia can happen at any stage of follicular development, but early antral follicles are most susceptible to it [19]. Three types of atresia have been described based on where cell death is initiated: (I) “antral” atresia starts in the middle of the granulosa cell layer; (II) “basal” atresia spreads from the basal lamina and is associated with early incomplete luteinization of granulosa cells; and (III) “terminal differentiation” atresia occurs in preovulatory follicles, where globules of granulosa cells detach from the inner surface of the antral cavity and undergo apoptosis in the antral liquid [114, 116]. Despite differences in the initiation and progression details, the functional differences between these types of atresia are unclear. The initial focus of PCD quickly spreads over the granulosa cell layer and to the oocyte itself due to the presence of gap junction complexes that establish cytoplasmic bridges between cells. Such bridges may facilitate transfer of either nutrients from healthy cells or PCD mediators from atretic ones to the oocyte [20]. The most well-characterized regulators and signaling pathways of PCD involved in follicular atresia are depicted in Fig. 3.

Major molecular mechanisms and regulatory elements involved in the control of antral follicle atresia in the cycling ovary. The pathways regulated by FSH, which is secreted by the pituitary gland, promote the survival of granulosa cells and the growth and maturation of the follicle (left side of the figure). These pathways inhibit apoptosis and promote pro-survival autophagy (and mitophagy in particular) in antral and dominant follicles. When FSH levels are insufficient, extrinsic apoptosis is induced in the granulosa cell layer and results in follicular atresia (right side of the figure). Pro-ADCD additionally promotes the degeneration of pre-antral follicles under the conditions of oxidative stress. Orange circles with the letter “P” indicate protein phosphorylation, red crosses indicate the lack or suppression of certain regulators, and dashed elements indicate the regulators and connections that are not definitively confirmed up to date
Apoptosis
Apoptosis seems to be a predominant type of PCD responsible for granulosa cell death and follicular atresia, and it is easily recognized by the above-mentioned features [115]. Unlike in the case of PGC depletion which is mostly dependent on intrinsic apoptosis, both intrinsic and extrinsic pathways are significantly involved in the control of follicular atresia. The extrinsic pathway activated by adjacent granulosa cells seems to constantly push them toward apoptosis, while pituitary hormones and growth factors rescue them from demise [115].
The CD95/Fas/Apo-1–FasL system is one of the most well-characterized regulatory pathways involved in atresia initiation. Both molecules are abundantly expressed in granulosa cells of atretic follicles in a pattern that closely follows the localization of apoptotic cells [117,118,119]. The levels of CD95/Fas/Apo-1 and FasL increase during the progression of atresia and correlate with both apoptotic activity in granulosa cells and gonadotropin exposure [120]. CD95/Fas/Apo-1 and FasL are expressed during all stages of follicle development starting at the pre-antral stage, but prominent apoptosis induction is not observed until the antral stage. This may be explained by a simple increase in the expression of CD95/Fas/Apo-1, FasL, or both [120], but may also be associated with the membrane localization of both CD95/Fas/Apo-1 and FasL [36]. In the latter case, pathway activation occurs upon achieving critical concentrations of both the receptor and ligand through an increase in cell density in the multilayer walls of the antral follicle.
While the oocyte does not initiate the process of atresia, it still expresses the components of the extrinsic apoptotic pathway. The data on CD95/Fas/Apo-1 and FasL in the oocytes are controversial. Some researchers have reported the expression of CD95/Fas/Apo-1 but not FasL [117, 121], but other investigators have detected both CD95/Fas/Apo-1 and FasL in oocytes at different stages of follicle development [118, 122]. Despite expressing CD95/Fas/Apo-1, the oocytes are not susceptible to FasL-dependent apoptotic stimuli exerted by granulosa cells because they are separated by the zona pellucida [19]. Removal of the zona pellucida layer promotes apoptosis in oocytes co-incubated with FasL-positive granulosa cells in vitro [117].
Treatment with CD95/Fas/Apo-1-activating antibody alone can promote granulosa cell death in vivo but not in vitro [123,124,125,126]. However, the susceptibility of granulosa cells to CD95/Fas/Apo-1 activation in vitro is greatly increased by treatment with Interferon-γ (IFNγ), TNFα or cycloheximide, indicating that some additional mechanisms are involved in the regulation of CD95/Fas/Apo-1 system [124,125,126,127]. IFNγ is expressed in both oocytes and granulosa cells of pre-antral and early antral follicles [127] and acts through JAK/STAT pathway [128]. IFNγ treatment results in a prominent increase in CD95/Fas/Apo-1 expression in granulosa cells, elevating their sensitivity to FasL and promoting apoptosis [125,126,127].
TNFα is a dualistic regulator of cell death and survival, and its various effects are executed through two receptors: TNFR1 (pro-apoptotic) and TNFR2 (pro-survival) [129]. All three molecules show moderate-to-high expression levels in all cells of pre-antral and antral follicles [130], but TNFα predominantly causes granulosa cell apoptosis and follicle degradation in various models like human granulosa cell culture [131], isolated rat antral follicles [132], and whole bovine ovarian tissue cultured in vitro [130]. Moreover, researchers have identified the TNFR1-mediated pathway as a potential upstream regulator of atresia in porcine follicles using a transcriptomic approach [111, 133]. Like INFγ, TNFα can induce overexpression of CD95/Fas/Apo-1 [126], but its effect can also be mediated through a decrease in BCL-2 protein level [131].
TRAIL is the third major regulator of extrinsic apoptosis, but its role in follicular atresia is poorly understood. TRAIL ligand, its functional death receptor (DR5), and the decoy non-functional (DcR1 and DcR2) receptors are expressed in growing and antral follicles; apoptosis is readily initiated in TRAIL-treated granulosa cells in vitro [88]. The detailed analysis of TRAIL signaling in porcine ovaries has demonstrated that healthy developing follicles express high levels of DcR1 that are lost in atretic granulosa cells, while DR4 levels remain unchanged [134, 135]. Moreover, inactivation of DcR1 in healthy granulosa cells results in their prominent sensitization to TRAIL [136]. On the other hand, the expression of TRAIL itself is prominently increased during follicle growth [135]. Unfortunately, this promising area presently feels very under-investigated.
FasL-, TNFα-, and TRAIL-dependent cell death is additionally regulated by a specific cFLIP protein that inhibits the formation of both pro-apoptotic and necroptotic complexes, death-inducing signaling complex (DISC) and ripoptosome, respectively. Three common isoforms of cFLIP (cFLIPL, cFLIPS, and cFLIPR) have been described, and cFLIPS and cFLIPR are considered to play a more important role in RCD control due to stricter regulation of their abundance in cells [137]. The published studies on cFLIP in ovarian follicles have mostly used the model of porcine ovaries, where it is prominently expressed in granulosa cells during follicle growth [138]. Follicular atresia is associated with a reduction in cFLIPL but not cFLIPS transcription; on the other hand, cFLIPS protein is not detected in any condition [139]. Overexpression or suppression of cFLIP variants in cultured human and porcine granulosa cells renders them resistant or sensitive to CD95/Fas/Apo-1-induced apoptosis, respectively [140]. Furthermore, cFLIP controls granulosa cell death through caspase-8, confirming a unique function for cFLIP in extrinsic apoptosis [141]. Despite the well-established role of cFLIP in apoptosis, its regulation is not fully characterized. The half-life of cFLIP is very short [142], and this fact explains prominent sensitization to CD95/Fas/Apo-1-induced apoptosis by cycloheximide, as general translation inhibition quickly depletes the cFLIP supply [126]. Protein kinase C (PKC)-dependent phosphorylation of cFLIPS stabilizes the protein and prevents its rapid degradation [143], while its expression is stimulated through the AKT-FOXO3 and AKT-NFκB pathways [142, 144]. Both PKC and AKT pathways are stimulated by FSH (see below), adding cFLIP as an impactful factor in overcoming PCD in the antral follicle.
Modulation of the intrinsic pathway components in antral follicles is mostly associated with the maintenance of cell survival. Due to the developed net of blood vessels, growth factor shortage is rarely a problem in the mature ovary, but additional stimuli are required to prevent extrinsic apoptosis. FSH is the main factor that prevents follicles from inevitable apoptotic demise [19]. It is indispensable for the formation of antral follicles, as genetic knockout of either the β-subunit of FSH or the FSH receptor (FSHR) results in the complete absence of antral follicles, even atretic ones, in murine ovaries [145, 146]. It is also crucially important for the survival of granulosa cells. The peptide inhibitor of the FSH-FSHR interaction causes follicular atresia in rodents and primates [147, 148], while an excess of FSH prevents apoptosis in dominant follicles [149]. FSHR is expressed on the surface of granulosa cells in pre-antral and antral follicles [21], and its activation affects a plethora of intracellular signaling pathways through G proteins and β-arrestins, multifunctional scaffold proteins [150, 151]. The major regulatory circuits activated by FSH include the cAMP/PKA, PI3K/AKT, PLC/PKC, and MEK/ERK pathways. Current knowledge suggests that G proteins provide a rapid response via all of these pathways, while β-arrestins are involved in prolonged stimulation of only MEK/ERK signaling [150, 152]. These signaling pathways are tightly interconnected, but PI3K/AKT and cAMP/PKA seem to be the most important for granulosa cell survival and further follicle maturation, respectively [150, 153]. The experimental data indicate that cAMP/PKA-regulated granulosa cell survival may be mediated through AKT stimulation, and the differentiation-related effects of PKA are independent of PI3K/AKT [154, 155]. Differentiation and terminal maturation of antral follicles curated by cAMP/PKA are controlled through steroid hormone synthesis [156]; however, these processes lie beyond the scope of our discussion.
The PI3K/AKT pathway plays an important role in follicular atresia as it controls a multitude of apoptotic regulators [157]. It confers general anti-apoptotic effects mediated through upregulation of BCL-2 and suppression of BIM [77, 158, 159], inactivation of BAX and BAD [78, 160], and activation of the p53-inhibiting mouse double minute 2 homolog (MDM2) protein [77]. Furthermore, the FSH-stimulated AKT pathway promotes the expression of XIAP that inhibits the activity of effector caspases, preventing the induction of apoptosis through the intrinsic and extrinsic pathways [161, 162]. Atretic follicles display a decrease in XIAP compared with healthy antral follicles [163]. AKT can increase the XIAP level through NFκB-mediated stimulation of its transcription [164], and XIAP has been proposed as a major switch between follicle survival and atresia [165]. However, the pivotal role of XIAP can be challenged by FOXO transcription factors (FOXO1 and FOXO3) that are widely known as direct targets of the AKT pathway. FOXO proteins promote apoptosis through transcriptional upregulation of multiple pro-apoptotic genes. For example, FOXO1 facilitates overexpression of BBC3 and apoptosis in response to oxidative stress in murine granulosa cells [166, 167]. FOXO1 expression is drastically increased in early antral follicles; it is suppressed again in preovulatory follicles after gonadotropin stimulation but persists in atretic ones [168]. There is a similar trend, albeit with less prominent changes, for FOXO3 [169]. More importantly, FOXO transcription factors promote the expression of FasL and TRAIL [120, 169], which, as mentioned above, are directly involved in granulosa cell atresia [170]. This fact has been confirmed experimentally in porcine ovaries [169]. FOXO phosphorylation by AKT results in protein translocation from the nucleus to the cytoplasm and subsequent degradation [170]. Therefore, it is logical to assume that FSH-dependent rescue of antral follicles is mediated through the AKT-FOXO axis and results in FasL and TRAIL downregulation [155]. However, FSH treatment does not affect FasL expression in granulosa cells [149]. Surprisingly, simultaneous knockout of the Foxo1 and Foxo3 genes in granulosa cells results in a drastic reduction in the serum FSH level, indicating that there is a regulatory feedback loop between maturing ovarian follicles and the pituitary gland [171]. A recent proteomic analysis of atretic granulosa cells identified FOXO1 and AKT3 among the top regulators of follicular atresia in pigs [172]. Thus, PI3K/AKT signaling affects both extrinsic and intrinsic apoptosis in antral follicles, FSH synthesis and dominant follicle selection. Researchers should pay much attention to this pathway when exploring the molecular mechanisms of follicular atresia.
While FSH is the main factor regulating follicle survival, other molecules contribute to this process, including IGF, the transforming growth factor (TGF) superfamily, Epidermal Growth Factor ( EGF), Fibroblast Growth Factor (FGF), estradiol, interleukins, and other regulators. Many of them (IGF, EGF, and bFGF) exert their effects through the same PI3K/AKT and MEK/ERK pathways, augmenting FSH-dependent effects [173,174,175,176]. IGF is especially interesting because it is required for proper activation of the PI3K pathway by FSH [177]. The TGF superfamily comprises a multitude of different proteins, of which activin and inhibin are the most relevant due to their ability to induce or suppress FSH secretion, respectively. The effects of TGF receptors activation are facilitated through SMAD proteins (the main signal transducers for receptors of the TGFβ superfamily) and ultimately result in increased FSHR expression [152]. The functions of estradiol and interleukins in follicular atresia are not completely understood. Studies in rodents and cattle suggest that estradiol should promote granulosa cell survival via BCL-2 regulation; however, the experiments performed on primates demonstrate that estradiol can induce atresia of dominant preovulatory follicles [178]. Interleukins control multiple regulatory circuits. Although their effects may be very controversial, the accumulated data imply that their actions are cytoprotective rather than pro-apoptotic [179].
The final stages of apoptosis in atretic follicles significantly depend on activation of caspase-3, in contrast to caspase-3-independent apoptosis in primordial follicles [89]. The pattern of caspase-3 activation in the normal ovary overlaps with the localization of atretic foci [180]. However, caspase-3 depletion is not sufficient for complete prevention of follicular degradation, as caspase-7 and caspase-9 are also involved in the process and can compensate for caspase-3 loss [89, 181]. Overall, terminal execution of apoptosis in the atretic follicles exhibits much more “canonical” mechanisms than in PGCs and primordial follicles.
Autophagy
The role of autophagy during follicle growth and atresia in the adult ovary is even more diverse and controversial than during embryonic development. The first mention of autophagy in atretic granulosa cells of quail ovaries was in 1996 [182]. However, ADCD in the follicles of human ovaries was first described in 2006, and this opened a new direction in studies of follicular atresia [183].
The first report of localization of the crucial autophagic regulator BECN1 in a normal ovary suggested its exclusive expression in theca cells, and not in granulosa cells or oocytes [184]. Nevertheless, later reports demonstrated that granulosa cells express BECN1, as well as ATG5, ATG7, MAP1LC3, and other autophagy-related proteins, which indicates that these cells are competent for autophagy [185,186,187]. While researchers initially thought that autophagy occurs in follicles in the context of the stress response to oxidized low-density lipoproteins [183], they soon discovered that antral follicle atresia is associated with active autophagy under normal physiologic conditions [188]. This finding was supported by the fact that oxidative stress is one of the factors promoting death of granulosa cells [189]. However, starting from this point, the reports describing the effects and regulation of autophagy in the follicles become very inconsistent and mainly follow one of two contradictory concepts.
The first concept regards autophagy in follicular atresia as a destructive cell death process. A comparative investigation of autophagic regulators and active caspase-3 expression in different types of follicles suggested that atresia of pre-antral follicles is executed through autophagy, while late antral follicles degrade via apoptosis [190]. Pre-antral follicles are extremely dependent on the presence of FSH, and, based on the current knowledge of FSH-dependent signaling, it should inhibit autophagy (alongside apoptosis) via the PI3K/AKT/mTORC1 pathway [41]. Several studies have confirmed this idea, reporting AKT pathway activation, LC3-II downregulation, and a reduction in the number of autophagosomes after gonadotropin treatment in vitro or in vivo, while PI3K, AKT, or mTORC1 inhibitors prevent FSH-dependent effects [188, 191]. FSH-associated inhibition of mitophagy is even able to protect granulosa cells from oxidative stress [192]. Oxidative stress can promote autophagy through JNK activation; however, the exact mechanism in granulosa cells remains vague [193, 194]. It was demonstrated that autophagy activation during oxidative stress precedes apoptosis, and ATG7 or BECN1 knockdown rescues cells, confirming that the observed phenomenon can be correctly considered ADCD [195].
On the other hand, a growing number of reports support an alternative concept concerning autophagy-mediated protection of granulosa cells from death. This idea is consistent with the common knowledge of autophagy acting as a survival mechanism in stressful conditions [39]. Epg5 gene knockout suppresses autophagy and leads to accumulation of WT1 and subsequent induction of apoptosis in granulosa cells [196]. Alternatively, the autophagic response can be induced in hypoxic conditions through Hypoxia Inducible Factor 1 Subunit Alpha (HIF1α) and is involved in a metabolic switch to glycolysis [197]. Surprisingly, both HIF1α and autophagy are stimulated in hypoxic murine granulosa cells by FSH treatment. Furthermore, FSH treatment somehow inhibits mTORC1 phosphorylation without inhibiting AKT [198]. These facts have been confirmed in porcine cells and attributed to mitophagy regulated through the HIF1α-PINK1-Parkin cascade [198, 199]. The regulatory circuits connecting FSH, the PI3K/AKT/mTORC1 pathway, and autophagy in granulosa cells may be heavily dependent on oxygen balance. FSH-PI3K/AKT signaling always acts as an anti-apoptotic factor, but it has to either compete with destructive autophagy (during oxidative stress response) or cooperate with protective mitophagy (in hypoxic conditions). Later studies revealed that HIF1α-BNIP3-stimulated autophagy promotes granulosa cell survival and differentiation into luteal cells [200, 201]. miRNA-21-3p can simultaneously inhibit VEGFA, FGF2, the AKT/mTORC1 pathway, and autophagy in bovine granulosa cells [202, 203], raising even more questions about the connections between membrane receptors, the PI3K/AKT/mTORC1 pathway, and autophagy in granulosa cells.
While it is well-established that follicular atresia begins in the granulosa, the mechanisms of oocyte degradation after breakdown of the granulosa layer are mostly unknown. The deterioration of cytoplasmic protrusions that sprout through the zona pellucida from granulosa cells should result in a drastic reduction of nutrients transported to the oocyte, and it is no surprise that the oocytes in the atretic follicles display clear signs of autophagy. Their cytoplasm is filled with a high number of autophagosomes and displays prominent LAMP1 and MAP1LC3 staining [104, 204]. Simultaneously, the same oocytes are positive for cleaved caspase-3 and DNA fragmentation, two hallmarks of apoptosis [104, 204]. The seemingly unique feature of atretic oocytes is the disturbance of cytoplasmic structures known as lamellae or lattices which could also be attributed to ongoing autophagy [205]. It is still unknown whether autophagy plays a driving role in oocyte degradation or just coincides with apoptosis in a futile attempt to rescue the stressed oocyte. The only available evidence of two processes being possibly connected is provided by protein-protein interactions between BECN1 and BAX, BCL-2, and cleaved caspase-3 [206], but no functional studies have been published.
Necroptosis and ferroptosis
The role of necroptosis and ferroptosis in follicular atresia was not properly addressed until very recently. These two types of cell death have not been detected consistently in normal ovaries in vivo, but there is recent evidence that they can contribute to granulosa and oocyte death.
The first evidence of necroptotic death in granulosa cells was reported in a study focused on a readthrough isoform of acetylcholinesterase (AChE-R) in ovarian follicles. A synthetic peptide derived from the AChE-R C-terminal region induced MLKL phosphorylation and necroptosis in cultured granulosa cells [207]. Researchers confirmed the ability of AChE to regulate MLKL phosphorylation (seemingly through RIPK1) by using a three-dimensional in vitro culture of macaque follicles, but no associations between necroptotic proteins and follicle degradation were detected [208]. The RIPK1 and RIPK3 genes are overexpressed in atretic bovine follicles, and a system biology approach suggested that necroptosis may be actively involved in atresia [209]. SIRT1 promotes granulosa cell death and regulates RIPK1 and MLKL phosphorylation; however, the mechanism behind cell death is RIPK1-dependent apoptosis rather than MLKL-dependent necroptosis because it is accompanied by caspase-3 processing and PARP cleavage [210].
Transcriptomic analysis of early atretic follicles in a pig ovary revealed that the gene expression changes during atresia initiation represent a crosstalk between apoptosis, autophagy, and ferroptosis rather than apoptosis alone (including disturbances in GSH metabolism) [111]. This is consistent with the ultrastructural changes described in early atretic oocytes that include mitochondria condensation and loss of cristae—classic signs of ferroptosis [211]. Inactivation of the basonuclin zinc finger protein 1 (BNC1) gene promotes ferroptotic oocyte death through the neurofibromin 2 (NF2)-Yes associated protein (YAP) pathway [212]. Ferroptosis contributes to granulosa cell death in patients with endometriosis through the accumulation of iron in the follicular liquid, confirming the importance of iron balance for follicle survival [213]. The circRHBG-miR-515-5p-SLC7A11 molecular axis, which is often upregulated in patients with polycystic ovary syndrome (PCOS), inhibits ferroptosis in granulosa cells [214]. More investigations are required to determine whether ferroptosis is a normal event during follicular atresia in the healthy ovary.
The available data suggests that cells of antral follicles have the required molecular machinery to undergo necroptosis or ferroptosis; however, these types of RCD seem to represent minor variants in comparison to apoptosis and autophagy. Both necroptosis and ferroptosis have been known for more than a decade, but the scarce mentions of them in the recent literature imply that their characteristic features are rarely observed outside of specific circumstances (AChE-R overexpression, BNC1 mutation, etc.). Theoretically, FasL- or TRAIL-induced necroptotic death could occur in cells with inactivated cFLIPL when apoptotic pathways are suppressed by FSH signaling, but this hypothetical event has to be confirmed experimentally in vitro and in vivo. Ferroptosis is inhibited by PI3K/AKT signaling [215] and can be promoted by autophagic degradation of ferritin [216]. Therefore, it could be prone to occur in combination with autophagy when pre-antral follicles receive insufficient FSH. While the proposed situations look rather exotic, a meticulous researcher should keep these mechanisms in mind when analyzing follicular atresia.
Regulation of PCD during luteolysis
The CL is a temporary gland that exists in a tight reciprocal conjunction with the newly formed embryo. Progesterone secreted by the CL provides a suitable environment for embryo implantation and development in the uterus. After successful implantation, the uterus and the embryo start secreting CG or prolactin that maintain the CL in a functional state until the placenta is developed enough to support the embryo on its own [26, 27, 29]. Hence, it is extremely important to have a precise regulatory system that ensures rapid CL regression in case of fertilization or implantation failure so that the ovary can enter a new menstrual/estrous cycle.
The mechanisms of luteolysis are currently less understood compared with PGC depletion and follicular atresia. One of the major reasons for this issue is that different orders of mammals have different mechanisms of luteolysis regulation. For example, the majority of animals utilize uterus-derived PGF2α as the leading stimulator of CL regression, but primates (including humans) demonstrate successful luteolysis even after hysterectomy [27]. Conversely, primates have a unique hCG hormone that is secreted by trophoblast of the developing embryo and delays luteolysis. In other animals these functions are performed by LH, prolactin, or placental lactogen [26, 29]. Nevertheless, the aggregated knowledge establishes that luteolysis is kept in the balance by external inductors of cell death and survival (Fig. 4).

Major molecular mechanisms and regulatory elements involved in the control of luteolysis in the cycling ovary. LH secreted by the pituitary gland and CG secreted by the uterus during the early pregnancy period ensure maintenance of CL and continuous synthesis of progesterone. Signaling pathways controlled by LHCGR-PKA regulatory axis suppress apoptosis and autophagy until the levels of LH and CG decrease (left side of the figure). If the pregnancy did not occur or is at late stages, then the uterus produces PGF2α, which initiates luteolysis through the promotion of extrinsic apoptosis and destructive autophagy (right side of the figure). The induction of apoptosis through death receptors is dependent on the presence of immune cells which express FasL. Orange circles with the letter “P” indicate protein phosphorylation, red crosses indicate the lack or suppression of certain regulators, and dashed elements indicate the regulators and connections that are not definitively confirmed up to date
Apoptosis
Similarly to the other cases of massive PCD in the ovary, apoptosis is the best studied mechanism of CL degeneration due to the long history of observations [217, 218]. A massive increase in apoptotic activity occurs in the CL tissue soon after ovulation if the ovum is not successfully fertilized [218]. Experimental abrogation of apoptosis, which is commonly performed by caspase-3 knockout, is complicated in the case of the CL due to prominent perinatal mortality of caspase-3-deficient mice [219]. However, in the cohort of mice that survived long enough, caspase-3 knockout prevented apoptosis progression in the ovaries and significantly hindered CL regression [220, 221]. CL development in the ovary is associated with active angiogenesis [222]; therefore, it is unlikely that the lack of nutrients or hypoxia could be the main reason for the induction of luteal cell death. It is currently considered that extrinsic apoptosis in CL is activated by a local increase in CD95/Fas/Apo-1 system components (with additional influence from IFNγ and TNFα) and by external PGF2α secretion [217].
Both CD95/Fas/Apo-1 and FasL are expressed in luteal cells in various model organisms in the case of a normal estrous/menstrual cycle or pregnancy [119, 123, 125, 223,224,225]. The levels of CD95/Fas/Apo-1 seem to increase progressively with the age of the CL [223,224,225,226,227]. CD95/Fas/Apo-1 and FasL are expressed in CL cells in a mutually exclusive way and cooperate for efficient apoptosis induction. CD95/Fas/Apo-1 is only present in steroidogenic luteal cells, while FasL is specific to immune cells (macrophages and T lymphocytes) that invade CL during the functional degradation stage and promote structural involution [228, 229]. However, this model of luteolysis needs confirmation from different sources. Activation of CD95/Fas/Apo-1 by a specific antibody or FasL results in luteal cell death in vitro and in vivo [123, 125, 224]; accordingly, mice with an inactivating FasL mutation display impaired luteolysis [123]. A specific mouse strain known as the SAMP (senescence-accelerated mouse-prone) displays many early signs of aging including a reduced reproductive potential due to the abnormal accumulation of luteal bodies in the ovaries. These CL-like formations do not undergo luteolysis and display a simultaneous reduction in CD95/Fas/Apo-1, FasL, and active caspase-3 levels [230].
The susceptibility of luteal cells to CD95/Fas/Apo-1-dependent apoptosis is further promoted by TNFα and IFNγ [125, 224, 226, 229, 231]. Studies of murine ovaries suggest that TNFα may always be expressed in CL tissue and sustains a baseline level of CD95/Fas/Apo-1-mediated death; however, a local increase in the IFNγ level drives prominent apoptosis induction [232]. On the other hand, TNFR1-deficient mice or mice treated with TNF-inactivating antibody display disturbances of the estrous cycle: they often remain in diestrus, probably due to impaired luteolysis [233, 234]. Both TNFα and IFNγ increase the expression of CD95/Fas/Apo-1, therefore, enhancing the apoptosis-inducing effects [125, 224, 232]. The exact mechanism of this regulation in luteal cells has not yet been determined, but the studies performed in other models suggest that it may be facilitated through NFκB-RelA-dependent control of transcription [235, 236]. Additional mechanisms of CD95/Fas/Apo-1-dependent regulation of apoptosis may include suppression of the soluble anti-apoptotic FasB decoy receptor and TGFβ1 [232, 237]. In general, the CD95/Fas/Apo-1-dependent mechanism of luteal cell apoptosis is very similar to the mechanism observed in atretic follicles (see “Regulation of PCD during follicular atresia - Apoptosis” section).
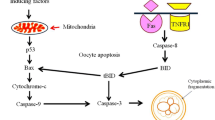
PGF2α has long been known as a luteolysis inductor that is external to the ovary itself. It is well established that PGF2a is secreted by the uterus and is crucial for CL regression in many species, with the notable exception of primates. Hysterectomy does not affect the CL lifespan in primates, but the administration of exogenous PGF2α still promotes CL degradation [27]. Many aspects of PGF2α functions remain poorly understood. For example, the ideas about the intraovarian source of PGF2α were proposed in the early 1970s [238] and were experimentally confirmed in 1976 [239], but the dynamics of prostaglandin synthesis in luteal cells were only determined in 2000 [240]. The most peculiar detail is that the regulatory mechanisms of PGF2α synthesis in the late-phase CL seem to establish a positive feedback loop, resulting in self-sustaining secretion of this major luteolytic factor [241].
The molecular pathways involved in signal transduction from the PGF2α receptor (PTGFR) to apoptotic regulators are also unclear. The only confirmed regulatory circuit associated with PTGRF is the PLC/PKC pathway, but its connection to cell death induction in the CL has to be further investigated [242, 243]. No interactions between PTGFR and extrinsic cell death regulators like FADD, TRADD, or caspase-8 have been identified; hence, it is very unlikely that PGF2α can directly induce apoptosis or necroptosis. Caspase-3-deficient mice that survive long enough to develop the CL in the ovaries are insensitive to the luteolysis-inducing effect of PGF2α, but they do demonstrate a drastic increase in caspase-8 activation upon treatment with either a Fas-activating antibody or PGF2α [221]. Moreover, PGF2α treatment increases the expression of CD95/Fas/Apo-1, FasL, accumulation of cleaved BID, and rise in BAX/BCL-2 ratio, while caspase-8 inhibition rescues cells from apoptosis, confirming that PGF2α promotes PCD by stimulating the extrinsic apoptotic pathway [244]. Injection of the PGF2α analog cloprostenol results in a significant increase in CD95/Fas/Apo-1, FasL, IFNγ, and TNFα expression within 4–12 h, suggesting strong upregulation of the extrinsic apoptotic pathway [245]. At the same time, IFNγ and TNFα reciprocally promote PGF2α secretion in luteal cells, closing the positive autoregulation circle [246]. In addition, PGF2α-induced CL regression is associated with PI3K/AKT pathway inhibition, STAT3 activation, ROS generation, and ER stress, indicating that the intrinsic pathway is at least partially involved in the sensitization of luteal cells to apoptosis [247,248,249,250]. Despite these aforementioned details, our understanding of PGF2α-dependent mechanisms of luteolysis is still far from complete. Nevertheless, we can be confident that the CD95/Fas/Apo-1-TNFα-IFNγ-PGF2α network is a cornerstone of cell death induction in CL.
In the majority of tissues, the final steps of apoptosis in the CL are mostly controlled by activation of caspase-3 [220, 221]. However, caspase-2 presents the most prominent changes in the late-stage CL, displaying a more than sevenfold increase in activity, while the relative activation of caspase-9 and caspase-3 is less prominent [251]. Unfortunately, there are no detailed reports on special features of caspase-2 in the CL, and this aspect of luteolysis regulation deserves more attention in the future.
Similarly to antral follicles that are inclined to CD95/Fas/Apo-1-dependent apoptosis, the CL seems to be predisposed toward luteolysis at the end of the menstrual/estrous cycle. Therefore, the impending apoptosis of luteal cells should be averted for successful embryo development. The onset of pregnancy is associated with the secretion of signaling factors aimed at preserving the CL and further stimulating its progesterone secretion. These factors are secreted by the developing embryo itself, associated membranes, the placenta, or the uterus at the location of embryo implantation and include CG (in primates only), prostaglandin E2 (PGE2), prolactin, and placental lactogen (in rodents). To some extent, the CL is also sustained by LH secreted by the pituitary gland [26, 252, 253]. The most important effects seem to be mediated by CG/LH and PGE2.
LH and CG are the primary stimulators of CL maintenance during pregnancy in ruminants and primates, respectively [26]. They act through the same LHCGR receptor that is expressed in the CL during all stages of the menstrual/estrous cycle or pregnancy except for the involution stage [254]. LHCGR is a G protein-coupled receptor that is capable of independent interaction with two separate G proteins thus mediating activation of either PLC/PKC, cAMP/PKA, or both pathways [255]. While the cAMP/PKA pathway is responsible for steroidogenic changes and progesterone secretion and acts through CREB- and GSK3β/β-catenin-dependent regulation of STAR enzyme expression [156, 256,257,258], PLC/PKC signaling may be associated with antisteroidogenic effects [259]. The possible effects of LHCGR-dependent PLC/PKC activation on luteal cell survival are unclear, especially considering that luteolytic PGF2α also activates this pathway [242]. A more exotic LHCGR partner is β-arrestin that may switch the downstream signaling toward AKT and MAPK cascade stimulation (including the pro-survival MEK/ERK pathway) [255, 260, 261].
LH or CG treatment promotes the viability of luteinized granulosa or luteal cells in vitro [231, 262,263,264] and protects the CL from regression in vivo [265,266,267]. The molecular changes that link LHCGR activation to anti-apoptotic effects include upregulation of BCL-2 [262, 267, 268], MCL-1 [263], survivin [269], and anti-oxidant enzymes [264] with simultaneous suppression of BAX and CD95/Fas/Apo-1 [262, 268]. The exact mechanisms facilitating these LH/CG effects in the luteal cells are still undetermined, but they may be mediated through PKA-CREB regulation of gene expression [258]. It is noteworthy that progesterone, the main product of luteal cells stimulated by LH or CG, suppresses luteolysis, reduces CD95/Fas/Apo-1 expression, and increases the BCL-2 level [270,271,272,273]. LHCGR stimulation is also associated with an increase in intraovarian PGE2 [265], the most important competitor of PGF2α in terms of defining the fate of the CL [274].
PGE2 acts through four different receptors (PTGER) that facilitate signal transduction through the cAMP/PKA pathway; PTGER2 and PTGER4, which are expressed in luteal cells, activate PKA signaling [274, 275]. PGE2 synthesis intensifies in the uterus after embryo implantation, and PGE2 is subsequently transported to the ovaries [275]. Researchers suggest that the initial PGE2 effects exerted upon the freshly developed CL define luteal cell sustenance before LH/CG influence; however, the actual relationships between PGE2 and CG/LH are unknown [276]. PGE2 on its own exerts anti-apoptotic effect [273], and its withdrawal from luteal cells results in activation of PGF2α secretion and shift toward luteolysis [277]. The current knowledge of apoptosis regulation during luteolysis is clearly incomplete, especially compared with well-defined mechanisms controlling embryonic oocyte attrition and follicular atresia. While the accumulated studies have established that the CL fate is defined by antagonism between the CD95/Fas/Apo-1-TNFα-IFNγ-PGF2α and the LH/CG-PGE2-PKA signaling networks, much more investigation is required in this area to clarify the details.
Autophagy
The CL is a relatively large structure, and its degradation should result in large amounts of cellular material being discharged. Therefore, it is no surprise that autophagy, which actively recycles cellular contents, is prominent in luteal cells, especially during the menopausal or post-pregnancy involution [278, 279]. A comparative study of CL tissue morphology during luteolysis suggested that autophagy may be the predominant mechanism during the late stages of regression [280]. Nevertheless, the authors did not use specific autophagy-associated markers to make this conclusion definitive.
All key autophagic regulators (BECN1, MAP1LC3, ATG proteins, and LAMP1) are expressed in luteal cells [281, 282], and their abundance changes according to luteolysis dynamics. The MAP1LC3, LC3-II, LAMP1, and ATG family protein levels increase progressively during the luteal/diestrus stage and reach their maxima in the late-stage steroidogenic cells [248, 281,282,283,284]; a similar trend is observed in the CL during pregnancy [285]. However, LAMP2 levels peak at the mid-luteal stage and then decrease, indicating that autophagy during late luteolysis may be abrogated at the autophagolysosome formation step despite prominent initial induction [285]. The data on BECN1 are conflicting, as its expression seems to be the highest in the late-stage CL in pigs [281] and almost absent at the same time in humans [184]. The aggregated data suggest that moderate autophagic activity may be associated with luteal cell differentiation and survival during CL formation, but its prominent stimulation during the late luteal/diestrus phase is likely to contribute to tissue degeneration and PCD.
Luteal cells treated with PGF2α in vitro and in vivo display prominent signs of both autophagy and apoptosis [248, 283, 286]. While PGF2α-dependent apoptosis is supposedly associated with the PLC/PKC and the CD95/Fas/Apo-1–FasL pathways, autophagy stimulation may be facilitated through AMPK signaling activation in bovine [286], ER stress in goat [248], or HIF1α-BNIP3-dependent ROS generation in the rat CL [287]. The last observation is especially interesting as activation of the same pathway in granulosa follicle cells results in their survival and luteinization instead of PCD [200, 201]. Moreover, autophagy stimulation by PGF2α in rat luteal cells is independent of mTORC1 activity and is controlled through the MEK/ERK pathway; however, the possible connection between the MEK/ERK pathway and PTGFR is unknown [288]. In contrast, LH promotes PKA and mTORC1 signaling pathways and inhibits autophagy in bovine CL [286].
Autophagy activation in the late-stage CL is accompanied by caspase-3 cleavage and apoptosis [282,283,284]. Inhibition of autophagosome formation with 3-methyladenine rescues apoptotic cells, confirming that autophagy can be the driving force behind luteal cell death. Surprisingly, the administration of the common autophagy inhibitors bafilomycin A1 or chloroquine does not replicate the effects of 3-methyladenine, promoting apoptosis instead. The main difference between these treatments is that bafilomycin A1 and chloroquine do not inhibit autophagy initiation but prevent autophagosome disposal; as a result, the accumulation of immature autophagosomes may be a driving factor of ADCD during late luteolysis [248, 283, 285]. Similarly to follicular atresia, the main challenge in investigating autophagy in CL degeneration lies in discerning autophagy-driven apoptosis from the co-occurrence of apoptotic and autophagic processes.
Necroptosis
Involution of the CL is a relatively “clean” process of tissue degradation that seems to take place without pronounced inflammation, which is one of the distinct necrosis/necroptosis features. Nevertheless, luteolysis strongly depends on CD95/Fas/Apo-1-, FasL-, and TNFα-dependent signals which are also involved in necroptosis. This idea is supported by the fact that luteal cells express significant levels of AChE and phosphorylated MLKL, and MLKL phosphorylation is increased in the late-stage CL [207, 289].
Clear necroptotic events have not been described in the normal CL. Two reports refer to an increase in RIPK1 and RIPK3 expression and cell death induction in the bovine CL and cultured luteal cells in response to PGF2α, TNFα, and IFNγ treatment [290, 291]. However, neither study includes data on RIPK1 or RIPK3 phosphorylation/activation or MLKL changes. They also only utilize RIPK1 inhibitor necrostatin-1 as a functional confirmation of necroptosis, which is not sufficient [47]. Considering that the authors demonstrated caspase-3 activation in the dying cells, one would assume that in this case RIPK1-dependent apoptosis occurs instead of MLKL-dependent necroptosis; hence, confirmation of the involvement of necroptosis in luteolysis requires more in-depth experiments.
Recently, researchers described RIPK3-dependent cell death independent of MLKL activity in the murine CL [292]. They proposed a new model of RIPK3-dependent apoptosis which contributes to PGF2α-dependent luteolysis, but these data also imply that RIPK3 may switch to its canonical induction of necroptosis in some currently unknown conditions. Comparative proteomic data for several stages of CL development and regression identified necro(pto)sis among the most affected processes in the late luteal phase CL [293]. It is evident that the investigation of necroptosis in luteolysis is currently in its initial phase. Future studies should focus more on evaluating of definitive necroptosis features like MLKL activation and/or morphological changes.
Pathologies associated with PCD deregulation in the ovary
Three waves of PCD in the normal ovary converge into an intricate cascade of events tightly controlled by hormones, cytokines, and local expression of cell death proteins. Therefore, it is not surprising that aberrations in PCD regulation result in severe impairments of ovarian tissue structure, the menstrual/estrous cycle, and reproductive function. Among numerous ovarian disorders, at least three are tightly associated with PCD deregulation: PCOS, premature ovarian insufficiency (POI), and malignant tumors. While some other pathologies like stromal hyperplasia, hyperthecosis, or pregnancy luteoma display tissue homeostasis disturbances and abnormal cell propagation, there are no evidences of them being driven by PCD aberrations [294].
PCOS
PCOS is the most common endocrine/metabolic disorder diagnosed in women of reproductive age, affecting up to 20% of the population. It is a complex syndrome of unclear origin that manifests in hormonal disbalance (androgen and LH excess), obesity, diabetes, and infertility [295]. From the pathomorphological point of view, the most distinct PCOS feature is the accumulation of so-called “ovarian cysts”, which are in fact ovarian follicles at different stages of development [296]. The follicles in a polycystic ovary are unable to properly ovulate, and they do not undergo atresia, resulting in partial or complete abrogation of reproductive function.
The total amount of primary, secondary, and small antral follicles in a polycystic ovary is 2–6 times higher than in a normal one [297]. It seems logical to assume that such changes should be associated with the aberrations of either PGC depletion or antral follicle atresia. However, the events underlying PCOS development are much more controversial, and the molecular mechanisms remain elusive. Various PCOS animal models were developed; however, they often recapitulate only part of the PCOS phenotype, either in terms of morphologic ovarian changes or metabolic disturbances. The rodent models based on the administration of aromatase inhibitor letrozole or constitutive LH overexpression seem to be the most adequate up to date [298]. Nevertheless, there is a clear lack of knowledge on PCOS-related changes during PGC depletion, and the available data mostly describe the changes observed during reproductive age.
Few reports have demonstrated that granulosa cells from polycystic ovaries have a lower apoptotic rate [299, 300] and increased expression of the pro-survival regulators BIRC3, EGFR, and HSP90B1 [299,300,301]. Moreover, expression of the mitogenic factor TGFα promotes the accumulation of antral follicles and a general PCOS-like state in mice [302]. In addition, polycystic ovaries display increased LHCGR expression, which may render them hypersensitive to LH effects (which may also be upregulated [303]), including ERK1/2- and AKT-mediated survival signals [255, 304, 305].
On the other hand, a growing number of studies have demonstrated that the accumulated follicles display an increased apoptotic rate. This activation of apoptosis can be associated with overexpression of established regulators of follicular atresia, including FasL [306], TNFα [303, 307], and FOXO3 [308]. Two other recurrent events observed in patients with PCOS and animal models of this disease are FSHR suppression [305, 309] and mitochondrial dysfunction [310, 311]. Deregulation of numerous other proteins (HMGB1, SH2B3, and USP25) and non-coding RNAs promotes apoptosis in polycystic ovaries [312,313,314,315,316], but their association with the process of normal follicular atresia is unclear. Such stimulation of apoptosis in PCOS follicles is without a doubt confusing, but it plays an important role in the development of anovulation and infertility by compromising oocyte maturation [114, 314].
Opposite changes in FSHR and LHCGR expression may underpin the distinct feature of PCOS—the inability of ovarian follicles to reach their outcome, either ovulation or atresia. FSH is required for the selection of dominant follicles, which subsequently induces atresia of smaller follicles [22], so a reduction in FSH sensitivity can potentially prevent this process and suspend the follicles in the early antral state. This idea was confirmed by a study of FSHR-antagonizing peptide that promotes the accumulation of secondary and pre-antral follicles in rat ovaries and a general PCOS-like state [317, 318]. Such follicles usually display increased apoptotic activity in preparation for atresia [114]; however, the increased LH sensitivity combined with LH excess may hinder mass cell death and, therefore, prevent atretic degradation.
Mitochondrial dysfunction in polycystic ovaries is a trending research area: researchers have linked mitochondrial dysfunction to apoptosis [311], as well as autophagy and ferroptosis. Active ROS generation and mitochondrial failure in PCOS can be associated with inefficient autophagy (and mitophagy in particular) [319, 320]; however, other recent reports describe increased autophagy/mitophagy [313, 321, 322]. Overexpression of several ferroptosis-associated genes was described in samples from patients with PCOS; unfortunately, their possible role in PCOS pathogenesis is not yet clear [323]. The discovery that ferroptosis is inhibited in granulosa cells from patients with PCOS via the circRHBG-miR-515-5p-SLC7A11 axis emphasizes the importance of PCD other than apoptosis in this disease and may provide new diagnostic molecular markers or therapeutic targets [214]. A recent report described an experimental attempt to treat PCOS in a mouse model by inducing ferroptosis [324]. The administration of n-3 PUFAs in the diet to dehydroepiandrosterone-treated mice activated Hippo signaling and YAP1 exocytosis, and suppressed NRF2, a protein that regulates critical ferroptosis-related genes (GPX4, GSS, TFRC, HMOX1, and others) [325], leading to the promotion of ferroptosis in ovarian granulosa cells. Considering the increasing interest toward autophagy and ferroptosis in the ovarian follicles, much can be expected from future studies in this direction. Current approaches for PCOS management are mostly focused on the compensation of hormonal disbalance and metabolic manifestations, while the accumulation of enlarged follicles is often considered a consequence that only impacts the fertility [326]. While the potential restoration of normal atresia in polycystic ovaries through selective PCD induction is not likely to improve fertility, it may contribute to the normalization of steroid hormone levels and therefore alleviate the severity of the disease symptoms.
POI
The primordial follicles present at birth constitute the “ovarian reserve” that is supposed to be exhausted around the age of 45–50 years. If the follicles are depleted faster than usual, then this reserve can be completely exhausted at an earlier age. This condition is called POI and is diagnosed in 1–4% of women at an age of 40 years and 0.1% of women at an age of 30 years [327]. POI should be discerned from primary ovarian dysgenesis, which is caused by abnormal ovary development during embryogenesis and results in complete disruption of ovarian functions before birth [327]. POI should not be also confused with a poor ovarian response condition characterized by low sensitivity of the follicles to the hormonal signals and hindered ovulation [328]. True POI is associated with a decreased number of follicles at different stages of their development. This phenomenon can be caused by either impaired follicle formation or increased follicular degradation, with the latter being most relevant to the topic of this review.
Genome-wide studies have demonstrated a very prominent mutational heterogeneity of POI cases [329]; however, only some of the identified genes have been confirmed to be associated with PCD. The most well-established among them are FIGLA, PGRMC1, and FMR1. Folliculogenesis specific bHLH transcription factor (FIGLA) is an oocyte-specific transcription factor that regulates proper formation of the extracellular matrix around oocyte [330]. Mutations in the FIGLA gene do not clearly affect embryonic ovary development in mice but result in impaired formation of primordial follicles and drastic attrition of oocytes soon after the birth [331]. FIGLA deficiency halts meiotic progression in PGCs and promotes DNA damage and subsequent apoptosis [332]. While complete FIGLA inactivation results in sterility [331], some mutations may exert milder effects and be associated with POI, as has been reported in Chinese and Indian populations [333, 334].
Progesterone receptor membrane component 1 (PGRMC1) is a non-canonical progesterone receptor that is expressed in granulosa cells and facilitates the anti-apoptotic and antimitotic effects of progesterone [335, 336]. Conditional PGRMC1 knockout in the murine reproductive tract results in subfertility without affecting the sensitivity to gonadotropin treatment [337]. Progesterone treatment can prevent granulosa cell apoptosis in response to oxidative stress, while PGRMC1 inhibition ablates this effect [338]. PGRMC1 expression is diminished in the granulosa cells of atretic follicles [339], suggesting that the inactivating mutations of this gene may promote atresia and thus contribute to POI development; however, this phenomenon has yet to be confirmed in large patient cohorts.
The FMR1 (Fragile X messenger ribonucleoprotein 1) gene is located on the X chromosome and bears a repetitive stretch of 20–40 CGG triplets in its 5′ untranslated region. The number of these repeats can expand to 200+, result in high methylation and gene silencing, and cause mental retardation [340]. However, the carriers of a moderately increased number of CGG repeats (50–200, historically labeled as the “premutation” cohort) are prone to develop fragile X-associated POI [341]. It has no effect on the initial pool of primordial follicles [342, 343], but promotes their abnormally high recruitment and atresia rates during the pre-menopause period [341]. Mice carrying premutation FMR1 develop POI and display increased apoptosis of granulosa cells [344]. Increased atresia of pre-antral and antral follicles results in low levels of anti-Müllerian hormone produced by granulosa cells; the deficiency of this hormone lifts the arrest of primordial follicle recruitment and quickly depletes the ovarian reserve [345].
Both PGRMC1 and FMR1 genes are located on the X chromosome. Besides the single mutations in these genes, there is a much more disruptive event: monosomy of the X chromosome also known as Turner syndrome [327]. It was initially thought to cause complete gonadal dysgenesis, but histologic studies of 45 XO fetuses have demonstrated that ovaries and PGCs develop normally during the first 3 months in utero, while primary follicle formation is impaired later and results in increased PGC attrition through apoptosis [346,347,348]. It is worth noting that the X chromosome also carries the anti-apoptotic XIAP gene, which is very important for preventing embryonic PGC depletion and postnatal antral follicle atresia [94, 165]. Hence, it is clear that the loss of one X chromosome in females disrupts multiple regulators of PCD in the ovary and almost inevitably results in POI [327].
Deregulation of autophagy can affect different stages of follicle development and, therefore, cause POI. Inactivating heterozygous mutations in the ATG7 and ATG9 genes are associated with a reduced number of primordial follicles [349], a finding consistent with experimental data obtained from Atg7-deficient mice [100, 101]. Inactivation of Epg5 results in suppression of autophagy and promotion of granulosa cell apoptosis during the antral follicle stage, which may result in POI [196]. However, mutations in autophagy-related genes have not been reported among the most prevalent genetic aberrations in POI.
One more notable gene associated with POI is BNC1: researchers identified it as a potential POI determinant through genome-wide patient studies [350, 351] and functionally confirmed its role in a murine model [350]. Unlike the other genes mentioned above, BNC1 deficiency triggers not apoptosis of granulosa cells, but ferroptosis of the oocyte itself, resulting in quick depletion of the ovarian reserve via follicular atresia [212].
Most comparative POI studies have focused on genome-based approaches and the identification of POI-associated mutations, while the data on gene expression patterns are scarce due to the nature of the disease: it is commonly diagnosed when the ovarian reserve is already depleted and there are no follicles left to investigate. However, several recent studies utilizing in vitro and in vivo models have allowed researchers to identify several other PCD-related genes and regulatory pathways potentially involved in POI development. Granulosa cells subjected to serum starvation in vitro display autophagy stimulation and reduced viability, and melatonin treatment rescues them by inhibiting autophagy through the PI3K-AKT-mTORC1 pathway (similarly to FSH effects; see “Regulation of PCD during follicular atresia - Autophagy” section) [352, 353]. Treatment of rats with 4-vinylcyclohexene diepoxide results in prominent miR-144 suppression, PI3K/AKT inhibition, autophagy induction, and POI development [354]. Taken together with the data on genetic inactivation of autophagic regulators presented above, autophagy should be tightly regulated to protect follicles from premature degeneration. These reports also suggest that treatment focused on stimulating the PI3K/AKT pathway (FSH and melatonin) could be used to prevent POI development. Because POI often involves ovarian tissue fibrosis, researchers developed a model of POI induction via cisplatin administration. Cisplatin promotes ferroptosis, and the investigators detected corresponding changes in the expression of ACSI4, ALOX15, SLC7A11, and GPX4 in the ovarian tissue alongside follicle development disorders and ovarian tissue fibrosis. The resulting POI and fertility reduction could be reverted by introducing ferroptosis-inhibiting vitamin E [355]. Considering the scarcity of available information on the role of apoptosis, ferroptosis and autophagy in POI, such discoveries are very important to understand the entire complex RCD system in the ovary. It also can be used as a foundation for the development of diagnostic and therapeutic approaches for POI.
Tumors of ovarian origin
Malignant tumors represent the deadliest type of ovarian pathologies due to the lack of clear symptoms, concealed localization in the pelvic region, and the high risk of spreading to multiple adjacent organs. Like any cancer, the ovarian malignancies are tightly associated with various mechanisms of RCD suppression or evasion [356]. The entire field of RCD in ovarian cancer is enormous and lies far beyond the scope of this review. However, two specific cases should be mentioned because they are directly connected to normal PCD processes in the ovary.
Most ovarian tumors are high-grade serous carcinomas. A series of recent discoveries has demonstrated that these tumors commonly originate not from the ovary itself but rather from the fallopian tube epithelium. Therefore, they have no link to RCD events in the ovary [357]. Other ovarian cancer types are much rarer, and their cells of origin are usually unknown except for two very distinct subtypes: ovarian germ cell tumors (OGCTs) originating from PGCs and granulosa cell tumors (GCTs) [358]. Due to the very limited availability of samples, information on mechanisms underlying the development of these tumors is scarce.
OGCTs are usually diagnosed in the juvenile age and include several subtypes: dysgerminomas, immature teratomas, yolk sac tumors, and embryonal carcinomas [358]. While these malignancies differ significantly in their structure and prognosis, they share a common trait of frequent chromosome 12 amplification or isochromosome formation [359,360,361]. Chromosome 12 carries such relevant genes as KITLG and KRAS, which are crucial for PGC survival in the embryonic ovary [60, 61]. Thus, it is not surprising that their amplification promotes abnormal PGC proliferation. Moreover, OGCTs often display amplification or activating mutations of the KIT receptor and KRAS, thus further promoting PGC survival [359, 362, 363]. The tumor-driving roles of KIT, KITLG, and KRAS remain to be confirmed in animal models, but the well-established teratogenic effect of BCL-2 specifically expressed in granulosa cells [158] clearly illustrates that inhibition of apoptosis in PGCs promotes their malignisation. Unfortunately, due to the rarity of OGCTs and limited interest to their recapitulation in the laboratory animals, this intriguing model is not widely used.
GCTs arise either around menopause (45–50 years) or, in rare cases, the juvenile period (before 20 years). These two forms are histologically distinct, but they both display a common C134W mutation in the FOXL2 protein [364, 365]. Surprisingly, this mutation does not seem to have any significant impact on FOXL2 functions [366]. However, a transcriptomic study demonstrated that clinical GCT samples display changes in FOXL2 targets associated with reduced cell death and increased proliferation [367]. Wild-type FOXL2 exerts pro-apoptotic effects upon granulosa cells through caspase-8, BID, and BAK, and is associated with increased levels of TRAIL and CD95/Fas/Apo-1 expression. These associations are lost in FOXL2C134W granulosa cells [368]. FOXL2 activity is crucial for granulosa cell differentiation and secondary follicle formation, but its exact role in follicle survival/atresia remains elusive [369]. Despite the lack of mutations tightly associated with GCT cases in patients, several animal models were developed; in many of them, the promotion of tumor development is facilitated by inactivation of well-known mediators of follicular atresia (see “Regulation of PCD during follicular atresia - Apoptosis” section), such as α-inhibin, SMAD1/5, or FOXO1/3 [370]. These models additionally highlight the importance of PCD in the maintenance of normal ovarian function and hint at potential targeting of antral follicle-specific PCD regulation pathways for GCT treatment.
Conclusion
The regulation of PCD in the normal ovary is an extremely complicated process, in which the successful completion of each step is essential for the onset of the next development stage. Without proper PGC attrition, it is impossible to form stable primordial follicles; antral follicle atresia is a part of dominant follicle selection and subsequent ovulation; and CL degradation concludes each ovarian cycle and resets it. The disruption of this order of events can be devastating to the patient’s reproductive function and general health.
Over the last 50 years, the anatomical, physiological, and molecular events underlying PCD in the ovary have been established in great detail, and the new aspects related to less common RCD modes like necroptosis and ferroptosis are being reported as our knowledge expands. Table 1 provides a structured summary of numerous signaling pathways involved in PCR-dependent processes in the mammalian ovary and clearly illustrates their overwhelming complexity and interconnections. However, the available information on the possible pathologic consequences of PCD deregulation in the ovary is still incomplete and fragmentary. Most studies focused on PCOS, POI, or rare ovarian tumors are devoted to finding new diagnostic or therapeutic approaches regardless of the initial disease causes or mechanisms involved in their development. While the bias may be explained by the tremendous heterogeneity of the genetic landscape of these disorders, the diverse stimuli still converge to very distinct pathophysiological manifestations that are clearly connected to PCD impairments. It is notable that almost all studies exploring PCD aspects of ovarian disorders were published over the last decade, indicating the evident shift in the research interest. We expect that the forthcoming advances in this area may represent a new phase in managing ovarian pathologies, with a solid emphasis on PCD-associated traits instead of a symptomatic approach.
A comment from the authors
We sincerely regret that, due to the reasonable limitations of manuscript length and reference number, we could not properly cite many of the original research papers describing the facts and events discussed above. The field of PCD in the mammalian ovary is immensely vast and accumulated an overwhelming amount of information over more than half of the century. We therefore attempted to highlight the most ovary-relevant and up-to-date reports, while referring to the review articles in discussions of more general aspects.
Availability of data and materials
No unique dataset was generated during preparation of this manuscript.
Abbreviations
- RCD:
-
Regulated cell death
- PCD:
-
Programmed Cell Death
- PGCs:
-
Primordial germ cells
- FSH:
-
Follicle-stimulating hormone
- LH:
-
Luteinizing hormone
- CL:
-
Corpus luteum
- PGF2α:
-
Prostaglandin F2α
- CG/hCG:
-
Chorionic gonadotropin/human chorionic gonadotropin
- BCL-2:
-
B cell leukemia/lymphoma 2
- BIM (BCL2L11):
-
BCL2 interacting mediator (BCL2-like 11)
- NOXA (PMAIP1):
-
Phorbol-12-myristate-13-acetate-induced protein 1
- BBC3 (PUMA):
-
BCL2 binding component 3 (p53 upregulated modulator of apoptosis)
- BAD:
-
BCL2 associated agonist of cell death
- BCL-XL (BCL2L1):
-
B-cell lymphoma extra large (BCL2-like 1)
- BCL-2A1:
-
BCL2 related protein A1
- MCL-1:
-
Induced myeloid leukemia cell differentiation protein
- BAX:
-
BCL2 associated X; BCL2-like protein 4
- BAK:
-
BCL2 homologous antagonist/killer
- APAF1:
-
Apoptotic peptidase activating factor 1
- TNF:
-
Tumor necrosis factor
- CD95/Fas/Apo-1 :
-
Tumor Necrosis Factor Receptor Superfamily Member 6/ Fas cell surface death receptor
- TNFR1/2 (TNFRSF1A/1B):
-
TNF receptor superfamily member 1 A/1B
- TRAIL (TNFSF10):
-
TNF superfamily member 10
- FADD:
-
Fas associated via death domain
- TRADD:
-
TNFRSF1A associated via death domain
- BID:
-
BH3 interacting domain death agonist
- ADCD:
-
Autophagy-dependent cell death
- PI3K/PI3KC3:
-
Phosphatidylinositol 3-kinase
- AKT:
-
Serine/threoninespecific protein kinase 1
- mTOR:
-
Mammalian target of rapamycin
- AMPK (PRKAA1):
-
Protein kinase AMP-activated catalytic subunit alpha 1
- ULK:
-
Unc-51 like autophagy activating kinase
- BECN1:
-
Beclin 1
- ER:
-
Endoplasmic reticulum
- ATG3/4/5/7/9/12/16:
-
Autophagy related 3/4/5/7/9/12/16
- MAP1LC3:
-
Microtubule associated protein 1 light chain 3 (also known as LC3)
- LC3-II (MAP1LC3A):
-
Microtubule Associated Protein 1 Light Chain 3 Alpha
- LAMP1/2:
-
Lysosomal associated membrane protein 1
- RIPK1/3:
-
Receptor interacting serine/threonine kinase 1/3
- MLKL:
-
Mixed lineage kinase domain like pseudokinase
- FasL:
-
Fas ligand
- TRAF2:
-
TNF receptor associated factor 2
- cIAP1/2 (BIRC2/3):
-
Baculoviral IAP repeat containing 2/3
- IAP:
-
Inhibitor of apoptosis protein
- NFkB:
-
Nuclear factor kappa B
- ROS:
-
Reactive oxygen species
- PUFAs:
-
Polyunsaturated fatty acids
- MUFAs:
-
Monounsaturated fatty acids
- GSH:
-
Glutathione
- GPX4:
-
Glutathione peroxidase 4
- RSL3:
-
RAS-selective lethal
- KITLG (SCF):
-
KIT ligand
- LIF:
-
Leukemia inhibitory factor
- IGF1:
-
Insulin like growth factor 1
- cAMP:
-
Cyclic adenosine monophosphate
- MEK:
-
ERK kinase
- ERK (MAPK1):
-
Extracellular signal-regulated kinase (mitogen-activated protein kinase 1)
- c-KIT (KIT/CD117):
-
KIT proto-oncogene, receptor tyrosine kinase
- JAK:
-
Janus kinase
- STAT3:
-
Signal transducer and activator of transcription 3
- FOXO1/3:
-
Forkhead box O1/3
- CHK2:
-
Checkpoint kinase 2
- GSK3B:
-
Glycogen synthase kinase 3 beta
- XIAP:
-
X-linked inhibitor of apoptosis
- PDK1:
-
Pyruvate dehydrogenase kinase 1
- TSC1:
-
TSC complex subunit 1
- SIRT1:
-
Sirtuin 1
- ATPase:
-
Adenosine 5'-triphosphate
- IFNγ:
-
Interferon-γ
- DR4/5:
-
Death receptor 4/5
- DcR1/2:
-
Decoy receptor 1/2
- DISC:
-
Death-inducing signaling complex
- cFLIP (CFLAR):
-
CASP8 and FADD like apoptosis regulator
- PKC:
-
Protein kinase C
- FSHR:
-
FSH receptor
- PKA:
-
Protein kinase A, cAMP-dependent
- MDM2:
-
Mouse double minute 2 homolog
- TGFα/β1:
-
Transforming growth factor alpha/beta 1
- EGF:
-
Epidermal growth factor
- FGF:
-
Fibroblast growth factor
- JNK (MAPK8):
-
Mitogen-activated protein kinase 8
- HIF1α:
-
Hypoxia inducible factor 1 subunit alpha
- PINK1:
-
Phosphatase and tensin homolog induced kinase 1
- BNIP3:
-
BCL2 interacting protein 3
- VEGFA:
-
Vascular endothelial growth factor A
- AChE-R:
-
Acetylcholinesterase
- PARP:
-
Poly (ADP-ribose) polymerase
- BNC1:
-
Basonuclin zinc finger protein 1
- NF2:
-
Neurofibromin 2
- YAP:
-
Yes associated protein
- SLC7A11:
-
Solute carrier family 7 member 11
- PCOS:
-
Polycystic ovary syndrome
- SAMP:
-
Senescence-accelerated mouse strain
- PTGFR:
-
PGF2α receptor
- PLC:
-
Phospholipase C
- PGE2:
-
Prostaglandin E2
- MAPK:
-
Mitogen-activated protein kinase
- LHCGR:
-
LH/CG receptor
- STAR:
-
Steroidogenic Acute Regulatory Protein
- PTGER:
-
PGE2 receptors
- MAPK:
-
Mitogen-Activated Protein Kinase
- POI:
-
Premature ovarian insufficiency
- BIRC3:
-
Baculoviral IAP repeat containing 3
- EGFR:
-
EGF receptor
- HSP90B1:
-
Heat shock protein 90 beta family member 1
- HMGB1:
-
High mobility group box 1
- SH2B3:
-
SH2B adaptor protein 3
- USP25:
-
Ubiquitin specific peptidase 25
- NRF2 (NFE2L2):
-
Nuclear factor, erythroid derived 2, like 2
- FIGLA:
-
Folliculogenesis specific bHLH transcription factor
- PGRMC1:
-
Progesterone receptor membrane component 1
- FMR1:
-
Fragile X messenger ribonucleoprotein 1
- OGCTs:
-
Ovarian germ cell tumors
- GCTs:
-
Granulosa cell tumors
References
Raff MC. Social controls on cell survival and cell death. Nature. 1992;356(6368):397–400.
Galluzzi L, Vitale I, Aaronson SA, Abrams JM, Adam D, Agostinis P, et al. Molecular mechanisms of cell death: recommendations of the nomenclature committee on cell death 2018. Cell Death Differ. 2018;25(3):486–541.
Yamaguchi Y, Miura M. Programmed cell death in neurodevelopment. Dev Cell. 2015;32(4):478–90.
Jacobsen MD, Weil M, Raff MC. Role of Ced-3/ICE-family proteases in staurosporine-induced programmed cell death. J Cell Biol. 1996;133(5):1041–51.
Smith P, Wilhelm D, Rodgers RJ. Development of mammalian ovary. J Endocrinol. 2014;221(3):R145–61.
Niu W, Spradling AC. Mouse oocytes develop in cysts with the help of nurse cells. Cell. 2022;185(14):2576–90.e12.
Borum K. Oogenesis in the mouse. A study of the meiotic prophase. Exp Cell Res. 1961;24:495–507.
Jaffe LA, Egbert JR. Regulation of mammalian oocyte meiosis by intercellular communication within the ovarian follicle. Annu Rev Physiol. 2017;79:237–60.
Sarraj MA, Drummond AE. Mammalian foetal ovarian development: consequences for health and disease. Reproduction. 2012;143(2):151–63.
Sawyer HR, Smith P, Heath DA, Juengel JL, Wakefield SJ, McNatty KP. Formation of ovarian follicles during fetal development in sheep. Biol Reprod. 2002;66(4):1134–50.
Pepling ME. From primordial germ cell to primordial follicle: mammalian female germ cell development. Genesis. 2006;44(12):622–32.
Hutt KJ, McLaughlin EA, Holland MK. Primordial follicle activation and follicular development in the juvenile rabbit ovary. Cell Tissue Res. 2006;326(3):809–22.
Kerr JB, Myers M, Anderson RA. The dynamics of the primordial follicle reserve. Reproduction. 2013;146(6):R205–15.
Hoffman BL, Schorge JO, Halvorson LM, Hamid CA, Corton MM, Schaffer JI. Williams gynecology. 4th ed. New York: McGraw-Hill Education LLC; 2020.
White YA, Woods DC, Takai Y, Ishihara O, Seki H, Tilly JL. Oocyte formation by mitotically active germ cells purified from ovaries of reproductive-age women. Nat Med. 2012;18(3):413–21.
Sharma D, Bhartiya D. Stem cells in adult mice ovaries form germ cell nests, undergo meiosis, neo-oogenesis and follicle assembly on regular basis during estrus cycle. Stem Cell Rev Rep. 2021;17(5):1695–711.
McGee EA, Hsueh AJ. Initial and cyclic recruitment of ovarian follicles. Endocr Rev. 2000;21(2):200–14.
Kim J, You YJ. Oocyte quiescence: from formation to awakening. Endocrinology. 2022;163(6):1–9.
Hirshfield AN. Development of follicles in the mammalian ovary. Int Rev Cytol. 1991;124:43–101.
Kidder GM. Roles of gap junctions in ovarian folliculogenesis: implications for female infertility. In: Winterhager E, editor. Gap junctions in development and disease. Berlin: Springer; 2005. p. 223–37.
Oktay K, Briggs D, Gosden RG. Ontogeny of follicle-stimulating hormone receptor gene expression in isolated human ovarian follicles. J Clin Endocrinol Metab. 1997;82(11):3748–51.
Orisaka M, Miyazaki Y, Shirafuji A, Tamamura C, Tsuyoshi H, Tsang BK, et al. The role of pituitary gonadotropins and intraovarian regulators in follicle development: a mini-review. Reprod Med Biol. 2021;20(2):169–75.
Zeleznik AJ. The physiology of follicle selection. Reprod Biol Endocrinol. 2004;2:31.
Sanders JR, Jones KT. Regulation of the meiotic divisions of mammalian oocytes and eggs. Biochem Soc Trans. 2018;46(4):797–806.
Richards JS, Russell DL, Robker RL, Dajee M, Alliston TN. Molecular mechanisms of ovulation and luteinization. Mol Cell Endocrinol. 1998;145(1–2):47–54.
Bowen-Shauver JM, Gibori G. The corpus luteum of pregnancy. In: Leung PCK, Adashi EY, editors. The ovary. 2nd ed. San Diego: Academic; 2004. p. 201–30.
McCracken JA, Custer EE, Lamsa JC. Luteolysis: a neuroendocrine-mediated event. Physiol Rev. 1999;79(2):263–323.
Hennebold JD. Corpus luteum. In: Skinner MK, editor. Encyclopedia of reproduction. 2nd ed. Amsterdam: Elsevier; 2018. p. 99–105.
Zeleznik AJ, Pohl CR. Control of follicular development, corpus luteum function, the maternal recognition of pregnancy, and the neuroendocrine regulation of the menstrual cycle in higher primates. In: Neill JD, editor. Knobil and Neill’s physiology of reproduction. 3rd ed. Boston: Academic; 2006. p. 2449–510.
Bedoui S, Herold MJ, Strasser A. Emerging connectivity of programmed cell death pathways and its physiological implications. Nat Rev Mol Cell Biol. 2020;21(11):678–95.
Orrenius S, Nicotera P, Zhivotovsky B. Cell death mechanisms and their implications in toxicology. Toxicol Sci. 2011;119(1):3–19.
Van Opdenbosch N, Lamkanfi M. Caspases in cell death, inflammation, and disease. Immunity. 2019;50(6):1352–64.
Brown-Suedel AN, Bouchier-Hayes L. Caspase-2 substrates: to apoptosis, cell cycle control, and beyond. Front Cell Dev Biol. 2020;8:1–17.
Kopeina GS, Zhivotovsky B. Caspase-2 as a master regulator of genomic stability. Trends Cell Biol. 2021;31(9):712–20.
Singh R, Letai A, Sarosiek K. Regulation of apoptosis in health and disease: the balancing act of BCL-2 family proteins. Nat Rev Mol Cell Biol. 2019;20(3):175–93.
Guicciardi ME, Gores GJ. Life and death by death receptors. FASEB J. 2009;23(6):1625–37.
Li H, Zhu H, Xu CJ, Yuan J. Cleavage of BID by caspase 8 mediates the mitochondrial damage in the Fas pathway of apoptosis. Cell. 1998;94(4):491–501.
Pru JK, Tilly JL. Programmed cell death in the ovary: insights and future prospects using genetic technologies. Mol Endocrinol. 2001;15(6):845–53.
Galluzzi L, Baehrecke EH, Ballabio A, Boya P, Bravo-San Pedro JM, Cecconi F, et al. Molecular definitions of autophagy and related processes. Embo J. 2017;36(13):1811–36.
Khandia R, Dadar M, Munjal A, Dhama K, Karthik K, Tiwari R, et al. A comprehensive review of autophagy and its various roles in infectious, non-infectious, and lifestyle diseases: current knowledge and prospects for disease prevention, novel drug design, and therapy. Cells. 2019;8(7):674.
Hurley JH, Young LN. Mechanisms of autophagy initiation. Annu Rev Biochem. 2017;86:225–44.
Pasquier B. Autophagy inhibitors. Cell Mol Life Sci. 2016;73(5):985–1001.
Doherty J, Baehrecke EH. Life, death and autophagy. Nat Cell Biol. 2018;20(10):1110–7.
Ankarcrona M, Dypbukt JM, Bonfoco E, Zhivotovsky B, Orrenius S, Lipton SA, et al. Glutamate-induced neuronal death: a succession of necrosis or apoptosis depending on mitochondrial function. Neuron. 1995;15(4):961–73.
Hirsch T, Marchetti P, Susin SA, Dallaporta B, Zamzami N, Marzo I, et al. The apoptosis-necrosis paradox. Apoptogenic proteases activated after mitochondrial permeability transition determine the mode of cell death. Oncogene. 1997;15(13):1573–81.
Vanden Berghe T, Linkermann A, Jouan-Lanhouet S, Walczak H, Vandenabeele P. Regulated necrosis: the expanding network of non-apoptotic cell death pathways. Nat Rev Mol Cell Biol. 2014;15(2):135–47.
Khan I, Yousif A, Chesnokov M, Hong L, Chefetz I. A decade of cell death studies: breathing new life into necroptosis. Pharmacol Ther. 2021;220:107717.
Micheau O, Tschopp J. Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell. 2003;114(2):181–90.
Feoktistova M, Geserick P, Kellert B, Dimitrova DP, Langlais C, Hupe M, et al. cIAPs block ripoptosome formation, a RIP1/caspase-8 containing intracellular cell death complex differentially regulated by cFLIP isoforms. Mol Cell. 2011;43(3):449–63.
Seo J, Nam YW, Kim S, Oh DB, Song J. Necroptosis molecular mechanisms: recent findings regarding novel necroptosis regulators. Exp Mol Med. 2021;53(6):1007–17.
Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149(5):1060–72.
Stockwell BR. Ferroptosis turns 10: emerging mechanisms, physiological functions, and therapeutic applications. Cell. 2022;185(14):2401–21.
Conrad M, Angeli JP, Vandenabeele P, Stockwell BR. Regulated necrosis: disease relevance and therapeutic opportunities. Nat Rev Drug Discov. 2016;15(5):348–66.
Gao M, Monian P, Pan Q, Zhang W, Xiang J, Jiang X. Ferroptosis is an autophagic cell death process. Cell Res. 2016;26(9):1021–32.
Ursini F, Maiorino M, Valente M, Ferri L, Gregolin C. Purification from pig liver of a protein which protects liposomes and biomembranes from peroxidative degradation and exhibits glutathione peroxidase activity on phosphatidylcholine hydroperoxides. Biochim Biophys Acta. 1982;710(2):197–211.
Dixon SJ, Patel DN, Welsch M, Skouta R, Lee ED, Hayano M, et al. Pharmacological inhibition of cystine-glutamate exchange induces endoplasmic reticulum stress and ferroptosis. eLife. 2014;3:e02523.
Yang WS, SriRamaratnam R, Welsch ME, Shimada K, Skouta R, Viswanathan VS, et al. Regulation of ferroptotic cancer cell death by GPX4. Cell. 2014;156(1–2):317–31.
Beaumont HM, Mandl AM. A quantitative and cytological study of oogonia and oocytes in the foetal and neonatal rat. Proc R Soc Lond Ser B Biol Sci. 1962;155(961):557–79.
Baker TG. A quantitative and cytological study of germ cells in human ovaries. Proc R Soc Lond B Biol Sci. 1963;158:417–33.
Pesce M, Farrace MG, Piacentini M, Dolci S, De Felici M. Stem cell factor and leukemia inhibitory factor promote primordial germ cell survival by suppressing programmed cell death (apoptosis). Development. 1993;118(4):1089–94.
Carlsson IB, Laitinen MP, Scott JE, Louhio H, Velentzis L, Tuuri T, et al. Kit ligand and c-Kit are expressed during early human ovarian follicular development and their interaction is required for the survival of follicles in long-term culture. Reproduction. 2006;131(4):641–9.
Nilsson EE, Kezele P, Skinner MK. Leukemia inhibitory factor (LIF) promotes the primordial to primary follicle transition in rat ovaries. Mol Cell Endocrinol. 2002;188(1–2):65–73.
Kerr JB, Hutt KJ, Michalak EM, Cook M, Vandenberg CJ, Liew SH, et al. DNA damage-induced primordial follicle oocyte apoptosis and loss of fertility require TAp63-mediated induction of Puma and Noxa. Mol Cell. 2012;48(3):343–52.
ElInati E, Zielinska AP, McCarthy A, Kubikova N, Maciulyte V, Mahadevaiah S, et al. The BCL-2 pathway preserves mammalian genome integrity by eliminating recombination-defective oocytes. Nat Commun. 2020;11(1):2598.
Martínez-Marchal A, Huang Y, Guillot-Ferriols MT, Ferrer-Roda M, Guixé A, Garcia-Caldés M, et al. The DNA damage response is required for oocyte cyst breakdown and follicle formation in mice. PLoS Genet. 2020;16(11):e1009067.
Tilly JL. Commuting the death sentence: how oocytes strive to survive. Nat Rev Mol Cell Biol. 2001;2(11):838–48.
Mondragon AA, Yalonetskaya A, Ortega AJ, Zhang Y, Naranjo O, Elguero J, et al. Lysosomal machinery drives extracellular acidification to direct non-apoptotic cell death. Cell Rep. 2019;27(1):11–9.e3.
Stringer JM, Alesi LR, Winship AL, Hutt KJ. Beyond apoptosis: evidence of other regulated cell death pathways in the ovary throughout development and life. Hum Reprod Update. 2023;29(4):434–56.
Lobascio AM, Klinger FG, Scaldaferri ML, Farini D, De Felici M. Analysis of programmed cell death in mouse fetal oocytes. Reproduction. 2007;134(2):241–52.
De Felici M. Regulation of primordial germ cell development in the mouse. Int J Dev Biol. 2000;44(6):575–80.
Kordowitzki P, Krajnik K, Skowronska A, Skowronski MT. Pleiotropic effects of IGF1 on the oocyte. Cells. 2022;11(10):1610.
Farini D, La Sala G, Tedesco M, De Felici M. Chemoattractant action and molecular signaling pathways of kit ligand on mouse primordial germ cells. Dev Biol. 2007;306(2):572–83.
Moniruzzaman M, Sakamaki K, Akazawa Y, Miyano T. Oocyte growth and follicular development in KIT-deficient Fas-knockout mice. Reproduction. 2007;133(1):117–25.
Dang-Nguyen TQ, Haraguchi S, Kikuchi K, Somfai T, Bodo S, Nagai T. Leukemia inhibitory factor promotes porcine oocyte maturation and is accompanied by activation of signal transducer and activator of transcription 3. Mol Reprod Dev. 2014;81(3):230–9.
Pastuschek J, Poetzsch J, Morales-Prieto DM, Schleußner E, Markert UR, Georgiev G. Stimulation of the JAK/STAT pathway by LIF and OSM in the human granulosa cell line COV434. J Reprod Immunol. 2015;108:48–55.
Gu Y, Mohammad IS, Liu Z. Overview of the STAT-3 signaling pathway in cancer and the development of specific inhibitors. Oncol Lett. 2020;19(4):2585–94.
Downward J. PI 3-kinase, Akt and cell survival. Semin Cell Dev Biol. 2004;15(2):177–82.
Yamaguchi H, Wang HG. The protein kinase PKB/Akt regulates cell survival and apoptosis by inhibiting Bax conformational change. Oncogene. 2001;20(53):7779–86.
Sui XX, Luo LL, Xu JJ, Fu YC. Evidence that FOXO3a is involved in oocyte apoptosis in the neonatal rat ovary. Biochem Cell Biol. 2010;88(4):621–8.
Liu H, Luo LL, Qian YS, Fu YC, Sui XX, Geng YJ, et al. FOXO3a is involved in the apoptosis of naked oocytes and oocytes of primordial follicles from neonatal rat ovaries. Biochem Biophys Res Commun. 2009;381(4):722–7.
Ratts VS, Flaws JA, Kolp R, Sorenson CM, Tilly JL. Ablation of bcl-2 gene expression decreases the numbers of oocytes and primordial follicles established in the post-natal female mouse gonad. Endocrinology. 1995;136(8):3665–8.
Alton M, Taketo T. Switch from BAX-dependent to BAX-independent germ cell loss during the development of fetal mouse ovaries. J Cell Sci. 2007;120(Pt 3):417–24.
Lobascio AM, Klinger FG, De Felici M. Isolation of apoptotic mouse fetal oocytes by AnnexinV assay. Int J Dev Biol. 2007;51(2):157–60.
Lena AM, Rossi V, Osterburg S, Smirnov A, Osterburg C, Tuppi M, et al. The p63 C-terminus is essential for murine oocyte integrity. Nat Commun. 2021;12(1):383.
Myers M, Morgan FH, Liew SH, Zerafa N, Gamage TU, Sarraj M, et al. PUMA regulates germ cell loss and primordial follicle endowment in mice. Reproduction. 2014;148(2):211–9.
Wen J, Yan H, He M, Zhang T, Mu X, Wang H, et al. GSK-3β protects fetal oocytes from premature death via modulating TAp63 expression in mice. BMC Biol. 2019;17(1):23.
Morrison LJ, Marcinkiewicz JL. Tumor necrosis factor alpha enhances oocyte/follicle apoptosis in the neonatal rat ovary. Biol Reprod. 2002;66(2):450–7.
Jääskeläinen M, Kyrönlahti A, Anttonen M, Nishi Y, Yanase T, Secchiero P, et al. TRAIL pathway components and their putative role in granulosa cell apoptosis in the human ovary. Differentiation. 2009;77(4):369–76.
Matikainen T, Perez GI, Zheng TS, Kluzak TR, Rueda BR, Flavell RA, et al. Caspase-3 gene knockout defines cell lineage specificity for programmed cell death signaling in the ovary. Endocrinology. 2001;142(6):2468–80.
Vakifahmetoglu-Norberg H, Zhivotovsky B. The unpredictable caspase-2: what can it do? Trends Cell Biol. 2010;20(3):150–9.
Bergeron L, Perez GI, Macdonald G, Shi L, Sun Y, Jurisicova A, et al. Defects in regulation of apoptosis in caspase-2-deficient mice. Genes Dev. 1998;12(9):1304–14.
Morita Y, Maravei DV, Bergeron L, Wang S, Perez GI, Tsutsumi O, et al. Caspase-2 deficiency prevents programmed germ cell death resulting from cytokine insufficiency but not meiotic defects caused by loss of ataxia telangiectasia-mutated (atm) gene function. Cell Death Differ. 2001;8(6):614–20.
Ene AC, Park S, Edelmann W, Taketo T. Caspase 9 is constitutively activated in mouse oocytes and plays a key role in oocyte elimination during meiotic prophase progression. Dev Biol. 2013;377(1):213–23.
Liu X, Castle V, Taketo T. Interplay between caspase 9 and X-linked inhibitor of apoptosis protein (XIAP) in the oocyte elimination during fetal mouse development. Cell Death Dis. 2019;10(11):790.
Malki S, van der Heijden GW, O’Donnell KA, Martin SL, Bortvin A. A role for retrotransposon LINE-1 in fetal oocyte attrition in mice. Dev Cell. 2014;29(5):521–33.
Tingen CM, Bristol-Gould SK, Kiesewetter SE, Wellington JT, Shea L, Woodruff TK. Prepubertal primordial follicle loss in mice is not due to classical apoptotic pathways. Biol Reprod. 2009;81(1):16–25.
Rodrigues P, Limback D, McGinnis LK, Plancha CE, Albertini DF. Multiple mechanisms of germ cell loss in the perinatal mouse ovary. Reproduction. 2009;137(4):709–20.
Choi JY, Jo MW, Lee EY, Yoon BK, Choi DS. The role of autophagy in follicular development and atresia in rat granulosa cells. Fertil Steril. 2010;93(8):2532–7.
Sun YC, Wang YY, Sun XF, Cheng SF, Li L, Zhao Y, et al. The role of autophagy during murine primordial follicle assembly. Aging. 2018;10(2):197–211.
Gawriluk TR, Hale AN, Flaws JA, Dillon CP, Green DR, Rucker EB 3rd. Autophagy is a cell survival program for female germ cells in the murine ovary. Reproduction. 2011;141(6):759–65.
Song ZH, Yu HY, Wang P, Mao GK, Liu WX, Li MN, et al. Germ cell-specific Atg7 knockout results in primary ovarian insufficiency in female mice. Cell Death Dis. 2015;6(1):e1589.
Sun X, Klinger FG, Liu J, De Felici M, Shen W, Sun X. Mir-378-3p maintains the size of mouse primordial follicle pool by regulating cell autophagy and apoptosis. Cell Death Dis. 2020;11(9):737.
Wang YY, Sun YC, Sun XF, Cheng SF, Li B, Zhang XF, et al. Starvation at birth impairs germ cell cyst breakdown and increases autophagy and apoptosis in mouse oocytes. Cell Death Dis. 2017;8(2):e2613.
Escobar ML, Echeverria OM, Ortiz R, Vazquez-Nin GH. Combined apoptosis and autophagy, the process that eliminates the oocytes of atretic follicles in immature rats. Apoptosis. 2008;13(10):1253–66.
Watanabe R, Kimura N. Non-suckling starvation of neonatal mice promotes primordial follicle formation with activation of ovarian autophagy. J Reprod Dev. 2018;64(1):89–94.
Marcinkiewicz JL, Krishna A, Cheung CM, Terranova PF. Oocytic tumor necrosis factor alpha: localization in the neonatal ovary and throughout follicular development in the adult rat. Biol Reprod. 1994;50(6):1251–60.
Pajokh M, Mesbah F, Bordbar H, Talaei-Khozani T. Different cell death types determination in juvenile mice ovarian follicles. Iran J Vet Res. 2018;19(4):298–303.
Pajokh M, Talaei-Khozani T, Bordbar H, Mesbah F. Apoptosis, autophagy, and necrosis in murine embryonic gonadal ridges and neonatal ovaries: an animal model. Iran J Med Sci. 2019;44(1):35–43.
Aleshire SL, Osteen KG, Maxson WS, Entman SS, Bradley CA, Parl FF. Localization of transferrin and its receptor in ovarian follicular cells: morphologic studies in relation to follicular development. Fertil Steril. 1989;51(3):444–9.
Wright CS, Hovatta O, Margara R, Trew G, Winston RM, Franks S, et al. Effects of follicle-stimulating hormone and serum substitution on the in-vitro growth of human ovarian follicles. Hum Reprod. 1999;14(6):1555–62.
Zhang J, Liu Y, Yao W, Li Q, Liu H, Pan Z. Initiation of follicular atresia: gene networks during early atresia in pig ovaries. Reproduction. 2018;156(1):23–33.
Wang JJ, Ge W, Zhai QY, Liu JC, Sun XW, Liu WX, et al. Single-cell transcriptome landscape of ovarian cells during primordial follicle assembly in mice. PLoS Biol. 2020;18(12):e3001025.
Ford EA, Beckett EL, Roman SD, McLaughlin EA, Sutherland JM. Advances in human primordial follicle activation and premature ovarian insufficiency. Reproduction. 2020;159(1):R15–R29.
Regan SLP, Knight PG, Yovich JL, Leung Y, Arfuso F, Dharmarajan A. Granulosa cell apoptosis in the ovarian Follicle-A changing view. Front Endocrinol (Lausanne). 2018;9:61.
Matsuda F, Inoue N, Manabe N, Ohkura S. Follicular growth and atresia in mammalian ovaries: regulation by survival and death of granulosa cells. J Reprod Dev. 2012;58(1):44–50.
Irving-Rodgers HF, Krupa M, Rodgers RJ. Cholesterol side-chain cleavage cytochrome P450 and 3beta-hydroxysteroid dehydrogenase expression and the concentrations of steroid hormones in the follicular fluids of different phenotypes of healthy and atretic bovine ovarian follicles. Biol Reprod. 2003;69(6):2022–8.
Mori T, Xu JP, Mori E, Sato E, Saito S, Guo MW. Expression of Fas–Fas ligand system associated with atresia through apoptosis in murine ovary. Horm Res. 1997;48(Suppl 3):11–9.
Hakuno N, Koji T, Yano T, Kobayashi N, Tsutsumi O, Taketani Y, et al. Fas/APO-1/CD95 system as a mediator of granulosa cell apoptosis in ovarian follicle atresia. Endocrinology. 1996;137(5):1938–48.
Kondo H, Maruo T, Peng X, Mochizuki M. Immunological evidence for the expression of the Fas antigen in the infant and adult human ovary during follicular regression and atresia. J Clin Endocrinol Metab. 1996;81(7):2702–10.
Lin F, Fu YH, Han J, Shen M, Du CW, Li R, et al. Changes in the expression of Fox O1 and death ligand genes during follicular atresia in porcine ovary. Genet Mol Res. 2014;13(3):6638–45.
Dharma SJ, Kelkar RL, Nandedkar TD. Fas and Fas ligand protein and mRNA in normal and atretic mouse ovarian follicles. Reproduction. 2003;126(6):783–9.
Guan S, Guo L, Zhang T, Zhu B, Wang X, Zhang C. Effects of gonadotropin on Fas and/or FasL expression and proliferation in rat ovary. Theriogenology. 2015;83(1):21–9.
Sakamaki K, Yoshida H, Nishimura Y, Nishikawa S, Manabe N, Yonehara S. Involvement of Fas antigen in ovarian follicular atresia and luteolysis. Mol Reprod Dev. 1997;47(1):11–8.
Porter DA, Vickers SL, Cowan RG, Huber SC, Quirk SM. Expression and function of Fas antigen vary in bovine granulosa and theca cells during ovarian follicular development and atresia. Biol Reprod. 2000;62(1):62–6.
Quirk SM, Cowan RG, Joshi SG, Henrikson KP. Fas antigen-mediated apoptosis in human granulosa/luteal cells. Biol Reprod. 1995;52(2):279–87.
Quirk SM, Porter DA, Huber SC, Cowan RG. Potentiation of Fas-mediated apoptosis of murine granulosa cells by interferon-gamma, tumor necrosis factor-alpha, and cycloheximide. Endocrinology. 1998;139(12):4860–9.
Lee HJ, Kim JY, Park JE, Yoon YD, Tsang BK, Kim JM. Induction of Fas-mediated apoptosis by interferon-γ is dependent on granulosa cell differentiation and follicular maturation in the rat ovary. Dev Reprod. 2016;20(4):315–29.
Xu X, Fu XY, Plate J, Chong AS. IFN-gamma induces cell growth inhibition by Fas-mediated apoptosis: requirement of STAT1 protein for up-regulation of Fas and FasL expression. Cancer Res. 1998;58(13):2832–7.
Baud V, Karin M. Signal transduction by tumor necrosis factor and its relatives. Trends Cell Biol. 2001;11(9):372–7.
Silva AWB, Ribeiro RP, Menezes VG, Barberino RS, Passos JRS, Dau AMP, et al. Expression of TNF-α system members in bovine ovarian follicles and the effects of TNF-α or dexamethasone on preantral follicle survival, development and ultrastructure in vitro. Anim Reprod Sci. 2017;182:56–68.
Sasson R, Winder N, Kees S, Amsterdam A. Induction of apoptosis in granulosa cells by TNF alpha and its attenuation by glucocorticoids involve modulation of Bcl-2. Biochem Biophys Res Commun. 2002;294(1):51–9.
Kaipia A, Chun SY, Eisenhauer K, Hsueh AJ. Tumor necrosis factor-alpha and its second messenger, ceramide, stimulate apoptosis in cultured ovarian follicles. Endocrinology. 1996;137(11):4864–70.
Terenina E, Fabre S, Bonnet A, Monniaux D, Robert-Granié C, SanCristobal M, et al. Differentially expressed genes and gene networks involved in pig ovarian follicular atresia. Physiol Genom. 2017;49(2):67–80.
Wada S, Manabe N, Inoue N, Nakayama M, Matsui T, Miyamoto H. TRAIL-decoy receptor-1 disappears in granulosa cells of atretic follicles in porcine ovaries. J Reprod Dev. 2002;48(2):167–73.
Inoue N, Manabe N, Matsui T, Maeda A, Nakagawa S, Wada S, et al. Roles of tumor necrosis factor-related apoptosis-inducing ligand signaling pathway in granulosa cell apoptosis during atresia in pig ovaries. J Reprod Dev. 2003;49(4):313–21.
Wada S, Manabe N, Nakayama M, Inou N, Matsui T, Miyamoto H. TRAIL-decoy receptor 1 plays inhibitory role in apoptosis of granulosa cells from pig ovarian follicles. J Vet Med Sci. 2002;64(5):435–9.
Ivanisenko NV, Seyrek K, Hillert-Richter LK, König C, Espe J, Bose K, et al. Regulation of extrinsic apoptotic signaling by c-FLIP: towards targeting cancer networks. Trends Cancer. 2022;8(3):190–209.
Goto Y, Matsuda-Minehata F, Inoue N, Matsui T, Maeda A, Manabe N. Porcine (Sus scrofa) cellular FLICE-like inhibitory protein (cFLIP): molecular cloning and comparison with the human and murine cFLIP. J Reprod Dev. 2004;50(5):549–55.
Matsuda-Minehata F, Goto Y, Inoue N, Manabe N. Changes in expression of anti-apoptotic protein, cFLIP, in granulosa cells during follicular atresia in porcine ovaries. Mol Reprod Dev. 2005;72(2):145–51.
Matsuda-Minehata F, Goto Y, Inoue N, Sakamaki K, Chedrese PJ, Manabe N. Anti-apoptotic activity of porcine cFLIP in ovarian granulosa cell lines. Mol Reprod Dev. 2007;74(9):1165–70.
Matsuda F, Inoue N, Goto Y, Maeda A, Cheng Y, Sakamaki K, et al. cFLIP regulates death receptor-mediated apoptosis in an ovarian granulosa cell line by inhibiting procaspase-8 cleavage. J Reprod Dev. 2008;54(5):314–20.
Chang L, Kamata H, Solinas G, Luo JL, Maeda S, Venuprasad K, et al. The E3 ubiquitin ligase itch couples JNK activation to TNFalpha-induced cell death by inducing c-FLIP(L) turnover. Cell. 2006;124(3):601–13.
Kaunisto A, Kochin V, Asaoka T, Mikhailov A, Poukkula M, Meinander A, et al. PKC-mediated phosphorylation regulates c-FLIP ubiquitylation and stability. Cell Death Differ. 2009;16(9):1215–26.
Skurk C, Maatz H, Kim HS, Yang J, Abid MR, Aird WC, et al. The akt-regulated forkhead transcription factor FOXO3a controls endothelial cell viability through modulation of the caspase-8 inhibitor FLIP. J Biol Chem. 2004;279(2):1513–25.
Kumar TR, Wang Y, Lu N, Matzuk MM. Follicle stimulating hormone is required for ovarian follicle maturation but not male fertility. Nat Genet. 1997;15(2):201–4.
Dierich A, Sairam MR, Monaco L, Fimia GM, Gansmuller A, LeMeur M, et al. Impairing follicle-stimulating hormone (FSH) signaling in vivo: targeted disruption of the FSH receptor leads to aberrant gametogenesis and hormonal imbalance. Proc Natl Acad Sci USA. 1998;95(23):13612–7.
Chitnis SS, Navlakhe RM, Shinde GC, Barve SJ, D’Souza S, Mahale SD, et al. Granulosa cell apoptosis induced by a novel FSH binding inhibitory peptide from human ovarian follicular fluid. J Histochem Cytochem. 2008;56(11):961–8.
Chitnis SS, Selvaakumar C, Jagtap DD, Barnwal RP, Chary KV, Mahale SD, et al. Interaction of follicle-stimulating hormone (FSH) receptor binding inhibitor-8: a novel FSH-binding inhibitor, with FSH and its receptor. Chem Biol Drug Des. 2009;73(6):637–43.
Zhou XL, Teng Y, Cao R, Fu H, Xiong K, Sun WX, et al. Rescue from dominant follicle atresia by follicle-stimulating hormone in mice. Genet Mol Res. 2013;12(3):2945–52.
Casarini L, Crepieux P. Molecular mechanisms of action of FSH. Front Endocrinol (Lausanne). 2019;10:305.
Shukla AK, Dwivedi-Agnihotri H. Structure and function of β-arrestins, their emerging role in breast cancer, and potential opportunities for therapeutic manipulation. Adv Cancer Res. 2020;145:139–56.
Chu YL, Xu YR, Yang WX, Sun Y. The role of FSH and TGF-beta superfamily in follicle atresia. Aging. 2018;10(3):305–21.
Hunzicker-Dunn M, Maizels ET. FSH signaling pathways in immature granulosa cells that regulate target gene expression: branching out from protein kinase A. Cell Signal. 2006;18(9):1351–9.
Puri P, Little-Ihrig L, Chandran U, Law NC, Hunzicker-Dunn M, Zeleznik AJ. Protein kinase A: a master kinase of granulosa cell differentiation. Sci Rep. 2016;6:28132.
Shen M, Liu Z, Li B, Teng Y, Zhang J, Tang Y, et al. Involvement of FoxO1 in the effects of follicle-stimulating hormone on inhibition of apoptosis in mouse granulosa cells. Cell Death Dis. 2014;5(10):e1475.
Liu YX, Zhang Y, Li YY, Liu XM, Wang XX, Zhang CL, et al. Regulation of follicular development and differentiation by intra-ovarian factors and endocrine hormones. Front Biosci (Landmark Ed). 2019;24(5):983–93.
Hemmings BA, Restuccia DF. PI3K-PKB/Akt pathway. Cold Spring Harb Perspect Biol. 2012;4(9):a011189.
Hsu SY, Lai RJ, Finegold M, Hsueh AJ. Targeted overexpression of Bcl-2 in ovaries of transgenic mice leads to decreased follicle apoptosis, enhanced folliculogenesis, and increased germ cell tumorigenesis. Endocrinology. 1996;137(11):4837–43.
Wang XL, Wu Y, Tan LB, Tian Z, Liu JH, Zhu DS, et al. Follicle-stimulating hormone regulates pro-apoptotic protein bcl-2-interacting mediator of cell death-extra long (BimEL)-induced porcine granulosa cell apoptosis. J Biol Chem. 2012;287(13):10166–77.
del Peso L, Gonzalez-Garcia M, Page C, Herrera R, Nunez G. Interleukin-3-induced phosphorylation of BAD through the protein kinase Akt. Science. 1997;278(5338):687–9.
Asselin E, Wang Y, Tsang BK. X-linked inhibitor of apoptosis protein activates the phosphatidylinositol 3-kinase/Akt pathway in rat granulosa cells during follicular development. Endocrinology. 2001;142(6):2451–7.
Wang Y, Asselin E, Tsang BK. Involvement of transforming growth factor alpha in the regulation of rat ovarian X-linked inhibitor of apoptosis protein expression and follicular growth by follicle-stimulating hormone. Biol Reprod. 2002;66(6):1672–80.
Li J, Kim JM, Liston P, Li M, Miyazaki T, Mackenzie AE, et al. Expression of inhibitor of apoptosis proteins (IAPs) in rat granulosa cells during ovarian follicular development and atresia. Endocrinology. 1998;139(3):1321–8.
Wang Y, Chan S, Tsang BK. Involvement of inhibitory nuclear factor-kappab (NFkappaB)-independent NFkappaB activation in the gonadotropic regulation of X-linked inhibitor of apoptosis expression during ovarian follicular development in vitro. Endocrinology. 2002;143(7):2732–40.
Phillipps HR, Hurst PR. XIAP: a potential determinant of ovarian follicular fate. Reproduction. 2012;144(2):165–76.
Shen M, Lin F, Zhang J, Tang Y, Chen WK, Liu H. Involvement of the up-regulated FoxO1 expression in follicular granulosa cell apoptosis induced by oxidative stress. J Biol Chem. 2012;287(31):25727–40.
Liu ZQ, Shen M, Wu WJ, Li BJ, Weng QN, Li M, et al. Expression of PUMA in follicular granulosa cells regulated by FoxO1 activation during oxidative stress. Reprod Sci. 2015;22(6):696–705.
Shi F, LaPolt PS. Relationship between FoxO1 protein levels and follicular development, atresia, and luteinization in the rat ovary. J Endocrinol. 2003;179(2):195–203.
Matsuda F, Inoue N, Maeda A, Cheng Y, Sai T, Gonda H, et al. Expression and function of apoptosis initiator FOXO3 in granulosa cells during follicular atresia in pig ovaries. J Reprod Dev. 2011;57(1):151–8.
Zhang X, Tang N, Hadden TJ, Rishi AK, Akt. FoxO and regulation of apoptosis. Biochim Biophys Acta. 2011;1813(11):1978–86.
Liu Z, Castrillon DH, Zhou W, Richards JS. FOXO1/3 depletion in granulosa cells alters follicle growth, death and regulation of pituitary FSH. Mol Endocrinol. 2013;27(2):238–52.
Yang F, Liu Q, Chen Y, Ye H, Wang H, Zeng S. Integrative proteomic and phosphoproteomic analyses of granulosa cells during follicular atresia in porcine. Front Cell Dev Biol. 2020;8:624985.
Kwintkiewicz J, Giudice LC. The interplay of insulin-like growth factors, gonadotropins, and endocrine disruptors in ovarian follicular development and function. Semin Reprod Med. 2009;27(1):43–51.
Tilly JL, Billig H, Kowalski KI, Hsueh AJ. Epidermal growth factor and basic fibroblast growth factor suppress the spontaneous onset of apoptosis in cultured rat ovarian granulosa cells and follicles by a tyrosine kinase-dependent mechanism. Mol Endocrinol. 1992;6(11):1942–50.
Price CA. Mechanisms of fibroblast growth factor signaling in the ovarian follicle. J Endocrinol. 2016;228(2):R31–R43.
Celestino JJH, Chaves RN, Matos MHT, Saraiv MVA, Bruno JB, Maia-Júnior JE, et al. Mechanisms of atresia in ovarian follicles. Anim Reprod. 2009;6:495–508.
Zhou P, Baumgarten SC, Wu Y, Bennett J, Winston N, Hirshfeld-Cytron J, et al. IGF-I signaling is essential for FSH stimulation of AKT and steroidogenic genes in granulosa cells. Mol Endocrinol. 2013;27(3):511–23.
Chauvin S, Cohen-Tannoudji J, Guigon CJ. Estradiol signaling at the heart of folliculogenesis: its potential deregulation in human ovarian pathologies. Int J Mol Sci. 2022;23(1):512.
Smolikova K, Mlynarcikova A, Scsukova S. Role of interleukins in the regulation of ovarian functions. Endocr Regul. 2012;46(4):237–53.
Boone DL, Tsang BK. Caspase-3 in the rat ovary: localization and possible role in follicular atresia and luteal regression. Biol Reprod. 1998;58(6):1533–9.
Matsui T, Manabe N, Goto Y, Inoue N, Nishihara S, Miyamoto H. Expression and activity of Apaf1 and caspase-9 in granulosa cells during follicular atresia in pig ovaries. Reproduction. 2003;126(1):113–20.
D’Herde K, De Prest B, Roels F. Subtypes of active cell death in the granulosa of ovarian atretic follicles in the quail (Coturnix coturnix japonica). Reprod Nutr Dev. 1996;36(2):175–89.
Duerrschmidt N, Zabirnyk O, Nowicki M, Ricken A, Hmeidan FA, Blumenauer V, et al. Lectin-like oxidized low-density lipoprotein receptor-1-mediated autophagy in human granulosa cells as an alternative of programmed cell death. Endocrinology. 2006;147(8):3851–60.
Gaytan M, Morales C, Sanchez-Criado JE, Gaytan F. Immunolocalization of beclin 1, a bcl-2-binding, autophagy-related protein, in the human ovary: possible relation to life span of corpus luteum. Cell Tissue Res. 2008;331(2):509–17.
Liu Q, Gao H, Yang F, Zhang H, Zeng S. FSH promotes progesterone synthesis by enhancing autophagy to accelerate lipid droplet degradation in porcine granulosa cells. Front Cell Dev Biol. 2021;9:626927.
Zheng Y, Ma L, Liu N, Tang X, Guo S, Zhang B, et al. Autophagy and apoptosis of porcine ovarian granulosa cells during follicular development. Animal (Basel). 2019;9(12):1111.
Ding Y, Zhu Q, He Y, Lu Y, Wang Y, Qi J, et al. Induction of autophagy by Beclin-1 in granulosa cells contributes to follicular progesterone elevation in ovarian endometriosis. Transl Res. 2021;227:15–29.
Choi J, Jo M, Lee E, Choi D. AKT is involved in granulosa cell autophagy regulation via mTOR signaling during rat follicular development and atresia. Reproduction. 2014;147(1):73–80.
Murdoch WJ. Inhibition by oestradiol of oxidative stress-induced apoptosis in pig ovarian tissues. J Reprod Fertil. 1998;114(1):127–30.
Meng L, Jan SZ, Hamer G, van Pelt AM, van der Stelt I, Keijer J, et al. Preantral follicular atresia occurs mainly through autophagy, while antral follicles degenerate mostly through apoptosis. Biol Reprod. 2018;99(4):853–63.
Shen M, Jiang Y, Guan Z, Cao Y, Li L, Liu H, et al. Protective mechanism of FSH against oxidative damage in mouse ovarian granulosa cells by repressing autophagy. Autophagy. 2017;13(8):1364–85.
Shen M, Jiang Y, Guan Z, Cao Y, Sun SC, Liu H. FSH protects mouse granulosa cells from oxidative damage by repressing mitophagy. Sci Rep. 2016;6:38090.
Gao H, Lin L, Haq IU, Zeng SM. Inhibition of NF-kappaB promotes autophagy via JNK signaling pathway in porcine granulosa cells. Biochem Biophys Res Commun. 2016;473(1):311–6.
Cao Y, Shen M, Jiang Y, Sun SC, Liu H. Melatonin reduces oxidative damage in mouse granulosa cells via restraining JNK-dependent autophagy. Reproduction. 2018;155(3):307–19.
Zhang JQ, Ren QL, Chen JF, Gao BW, Wang XW, Zhang ZJ, et al. Autophagy contributes to oxidative stress-induced apoptosis in porcine granulosa cells. Reprod Sci. 2021;28(8):2147–60.
Liu W, Chen M, Liu C, Wang L, Wei H, Zhang R, et al. Epg5 deficiency leads to primary ovarian insufficiency due to WT1 accumulation in mouse granulosa cells. Autophagy. 2023;19(2):644–59.
Zhou J, Li C, Yao W, Alsiddig MC, Huo L, Liu H, et al. Hypoxia-inducible factor-1α-dependent autophagy plays a role in glycolysis switch in mouse granulosa cells. Biol Reprod. 2018;99(2):308–18.
Zhou J, Yao W, Li C, Wu W, Li Q, Liu H. Administration of follicle-stimulating hormone induces autophagy via upregulation of HIF-1α in mouse granulosa cells. Cell Death Dis. 2017;8(8):e3001.
Li C, Zhou J, Liu Z, Zhou J, Yao W, Tao J, et al. FSH prevents porcine granulosa cells from hypoxia-induced apoptosis via activating mitophagy through the HIF-1α-PINK1-Parkin pathway. FASEB J. 2020;34(3):3631–45.
Tang Z, Zhang Z, Lin Q, Xu R, Chen J, Wang Y, et al. HIF-1α/BNIP3-mediated autophagy contributes to the luteinization of granulosa cells during the formation of corpus luteum. Front Cell Dev Biol. 2020;8:619924.
Tang Z, Xu R, Zhang Z, Shi C, Zhang Y, Yang H, et al. HIF-1α protects granulosa cells from hypoxia-induced apoptosis during follicular development by inducing autophagy. Front Cell Dev Biol. 2021;9:631016.
Ma L, Tang X, Guo S, Liang M, Zhang B, Jiang Z. miRNA-21-3p targeting of FGF2 suppresses autophagy of bovine ovarian granulosa cells through AKT/mTOR pathway. Theriogenology. 2020;157:226–37.
Ma L, Zheng Y, Tang X, Gao H, Liu N, Gao Y, et al. Mir-21-3p inhibits autophagy of bovine granulosa cells by targeting VEGFA via PI3K/AKT signaling. Reproduction. 2019;158(5):441–52.
Escobar Sánchez ML, Echeverría Martínez OM, Vázquez-Nin GH. Immunohistochemical and ultrastructural visualization of different routes of oocyte elimination in adult rats. Eur J Histochem. 2012;56(2):e17.
Escobar ML, Echeverría OM, García G, Ortíz R, Vázquez-Nin GH. Immunohistochemical and ultrastructural study of the lamellae of oocytes in atretic follicles in relation to different processes of cell death. Eur J Histochem. 2015;59(3):2535.
Escobar ML, Echeverria OM, Palacios-Martínez S, Juárez-Chavero S, Sánchez-Sánchez L, Vázquez-Nin GH. Beclin 1 interacts with active Caspase-3 and Bax in oocytes from atretic follicles in the rat ovary. J Histochem Cytochem. 2019;67(12):873–89.
Blohberger J, Kunz L, Einwang D, Berg U, Berg D, Ojeda SR, et al. Readthrough acetylcholinesterase (AChE-R) and regulated necrosis: pharmacological targets for the regulation of ovarian functions? Cell Death Dis. 2015;6(3):e1685.
Du Y, Bagnjuk K, Lawson MS, Xu J, Mayerhofer A. Acetylcholine and necroptosis are players in follicular development in primates. Sci Rep. 2018;8(1):6166.
McEvoy MJ, Sinderewicz E, Creedon L, McAfee M, Jonczyk AW, Piotrowska-Tomala KK, et al. Death processes in bovine theca and granulosa cells modelled and analysed using a systems biology approach. Int J Mol Sci. 2021;22(9):4888.
Sapuleni J, Szymanska M, Meidan R. Diverse actions of sirtuin-1 on ovulatory genes and cell death pathways in human granulosa cells. Reprod Biol Endocrinol. 2022;20(1):104.
Devine PJ, Payne CM, McCuskey MK, Hoyer PB. Ultrastructural evaluation of oocytes during atresia in rat ovarian follicles. Biol Reprod. 2000;63(5):1245–52.
Wang F, Liu Y, Ni F, Jin J, Wu Y, Huang Y, et al. BNC1 deficiency-triggered ferroptosis through the NF2-YAP pathway induces primary ovarian insufficiency. Nat Commun. 2022;13(1):5871.
Ni Z, Li Y, Song D, Ding J, Mei S, Sun S, et al. Iron-overloaded follicular fluid increases the risk of endometriosis-related infertility by triggering granulosa cell ferroptosis and oocyte dysmaturity. Cell Death Dis. 2022;13(7):579.
Zhang D, Yi S, Cai B, Wang Z, Chen M, Zheng Z, et al. Involvement of ferroptosis in the granulosa cells proliferation of PCOS through the circRHBG/miR-515/SLC7A11 axis. Ann Transl Med. 2021;9(16):1348.
Tan S, Kong Y, Xian Y, Gao P, Xu Y, Wei C, et al. The mechanisms of ferroptosis and the applications in tumor treatment: enemies or friends? Front Mol Biosci. 2022;9:938677.
Zhou Y, Shen Y, Chen C, Sui X, Yang J, Wang L, et al. The crosstalk between autophagy and ferroptosis: what can we learn to target drug resistance in cancer? Cancer Biol Med. 2019;16(4):630–46.
Stocco C, Telleria C, Gibori G. The molecular control of corpus luteum formation, function, and regression. Endocr Rev. 2007;28(1):117–49.
Vaskivuo TE, Ottander U, Oduwole O, Isomaa V, Vihko P, Olofsson JI, et al. Role of apoptosis, apoptosis-related factors and 17beta-hydroxysteroid dehydrogenases in human corpus luteum regression. Mol Cell Endocrinol. 2002;194(1–2):191–200.
Kuida K, Zheng TS, Na S, Kuan C, Yang D, Karasuyama H, et al. Decreased apoptosis in the brain and premature lethality in CPP32-deficient mice. Nature. 1996;384(6607):368–72.
Carambula SF, Matikainen T, Lynch MP, Flavell RA, Gonçalves PB, Tilly JL, et al. Caspase-3 is a pivotal mediator of apoptosis during regression of the ovarian corpus luteum. Endocrinology. 2002;143(4):1495–501.
Carambula SF, Pru JK, Lynch MP, Matikainen T, Gonçalves PB, Flavell RA, et al. Prostaglandin F2alpha- and FAS-activating antibody-induced regression of the corpus luteum involves caspase-8 and is defective in caspase-3 deficient mice. Reprod Biol Endocrinol. 2003;1:15.
Fraser HM, Wulff C. Angiogenesis in the corpus luteum. Reprod Biol Endocrinol. 2003;1:88.
Roughton SA, Lareu RR, Bittles AH, Dharmarajan AM. Fas and Fas ligand messenger ribonucleic acid and protein expression in the rat corpus luteum during apoptosis-mediated luteolysis. Biol Reprod. 1999;60(4):797–804.
Taniguchi H, Yokomizo Y, Okuda K. Fas–Fas ligand system mediates luteal cell death in bovine corpus luteum. Biol Reprod. 2002;66(3):754–9.
Slot KA, Voorendt M, de Boer-Brouwer M, van Vugt HH, Teerds KJ. Estrous cycle dependent changes in expression and distribution of Fas, Fas ligand, Bcl-2, Bax, and pro- and active caspase-3 in the rat ovary. J Endocrinol. 2006;188(2):179–92.
Przygrodzka E, Witek KJ, Kaczmarek MM, Andronowska A, Ziecik AJ. Expression of factors associated with apoptosis in the porcine corpus luteum throughout the luteal phase of the estrous cycle and early pregnancy: their possible involvement in acquisition of luteolytic sensitivity. Theriogenology. 2015;83(4):535–45.
Peluffo MC, Young KA, Hennebold JD, Stouffer RL. Expression and regulation of tumor necrosis factor (TNF) and TNF-receptor family members in the macaque corpus luteum during the menstrual cycle. Mol Reprod Dev. 2009;76(4):367–78.
Kuranaga E, Kanuka H, Furuhata Y, Yonezawa T, Suzuki M, Nishihara M, et al. Requirement of the Fas ligand-expressing luteal immune cells for regression of corpus luteum. FEBS Lett. 2000;472(1):137–42.
Komatsu K, Manabe N, Kiso M, Shimabe M, Miyamoto H. Changes in localization of immune cells and cytokines in corpora lutea during luteolysis in murine ovaries. J Exp Zool Comp Exp Biol. 2003;296(2):152–9.
Kiso M, Manabe N, Komatsu K, Shimabe M, Miyamoto H. Abnormal structural luteolysis in ovaries of the senescence accelerated mouse (SAM): expression of Fas ligand/Fas-mediated apoptosis signaling molecules in luteal cells. J Reprod Dev. 2003;49(6):457–63.
Matsubara H, Ikuta K, Ozaki Y, Suzuki Y, Suzuki N, Sato T, et al. Gonadotropins and cytokines affect luteal function through control of apoptosis in human luteinized granulosa cells. J Clin Endocrinol Metab. 2000;85(4):1620–6.
Komatsu K, Manabe N, Kiso M, Shimabe M, Miyamoto H. Soluble Fas (FasB) regulates luteal cell apoptosis during luteolysis in murine ovaries. Mol Reprod Dev. 2003;65(4):345–52.
Roby KF, Son DS, Terranova PF. Alterations of events related to ovarian function in tumor necrosis factor receptor type I knockout mice. Biol Reprod. 1999;61(6):1616–21.
Henkes LE, Sullivan BT, Lynch MP, Kolesnick R, Arsenault D, Puder M, et al. Acid sphingomyelinase involvement in tumor necrosis factor alpha-regulated vascular and steroid disruption during luteolysis in vivo. Proc Natl Acad Sci USA. 2008;105(22):7670–5.
Kimura M, Haisa M, Uetsuka H, Takaoka M, Ohkawa T, Kawashima R, et al. TNF combined with IFN-alpha accelerates NF-kappaB-mediated apoptosis through enhancement of Fas expression in colon cancer cells. Cell Death Differ. 2003;10(6):718–28.
Zheng Y, Ouaaz F, Bruzzo P, Singh V, Gerondakis S, Beg AA. NF-kappa B RelA (p65) is essential for TNF-alpha-induced fas expression but dispensable for both TCR-induced expression and activation-induced cell death. J Immunol. 2001;166(8):4949–57.
Gao H, Chen XL, Zhang ZH, Song XX, Hu ZY, Liu YX. IFN-gamma and TNF-alpha inhibit expression of TGF-beta-1, its receptors TBETAR-I and TBETAR-II in the corpus luteum of PMSG/hCG treated rhesus monkey. Front Biosci. 2005;10:2496–503.
Powell WS, Hammarström S, Samuelsson B, Sjöberg B, Letter. Prostaglandin-F2alpha receptor in human corpora lutea. Lancet. 1974;1(7866):1120.
Challis JR, Calder AA, Dilley S, Forster CS, Hillier K, Hunter DJ, et al. Production of prostaglandins E and falpha by corpora lutea, corpora albicantes and stroma from the human ovary. J Endocrinol. 1976;68(3):401–8.
Fridén BE, Wallin A, Brännström M. Phase-dependent influence of nonsteroidogenic cells on steroidogenesis and prostaglandin production by the human corpus luteum. Fertil Steril. 2000;73(2):359–65.
Wiltbank MC, Ottobre JS. Regulation of intraluteal production of prostaglandins. Reprod Biol Endocrinol. 2003;1:91.
Jimenez de Asua L, Goin M. Prostaglandin F2 alpha (PGF2 alpha) triggers protein kinase C (PKC) and tyrosine kinase activity in cultured mammalian cells. Adv Exp Med Biol. 1997;400A:531–8.
Sales KJ, Maldonado-Pérez D, Grant V, Catalano RD, Wilson MR, Brown P, et al. Prostaglandin F(2alpha)-F-prostanoid receptor regulates CXCL8 expression in endometrial adenocarcinoma cells via the calcium-calcineurin-NFAT pathway. Biochim Biophys Acta. 2009;1793(12):1917–28.
Yadav VK, Lakshmi G, Medhamurthy R. Prostaglandin F2alpha-mediated activation of apoptotic signaling cascades in the corpus luteum during apoptosis: involvement of caspase-activated DNase. J Biol Chem. 2005;280(11):10357–67.
Kliem H, Berisha B, Meyer HH, Schams D. Regulatory changes of apoptotic factors in the bovine corpus luteum after induced luteolysis. Mol Reprod Dev. 2009;76(3):220–30.
Galvão A, Skarzynski DJ, Szóstek A, Silva E, Tramontano A, Mollo A, et al. Cytokines tumor necrosis factor-α and interferon-γ participate in modulation of the equine corpus luteum as autocrine and paracrine factors. J Reprod Immunol. 2012;93(1):28–37.
Rovani MT, Ilha GF, Gasperin BG, Nóbrega JE Jr, Siddappa D, Glanzner WG, et al. Prostaglandin F2α-induced luteolysis involves activation of signal transducer and activator of transcription 3 and inhibition of AKT signaling in cattle. Mol Reprod Dev. 2017;84(6):486–94.
Wen X, Liu L, Li S, Lin P, Chen H, Zhou D, et al. Prostaglandin F2α induces goat corpus luteum regression via endoplasmic reticulum stress and autophagy. Front Physiol. 2020;11:868.
Yang Y, Sun M, Shan Y, Zheng X, Ma H, Ma W, et al. Endoplasmic reticulum stress-mediated apoptotic pathway is involved in corpus luteum regression in rats. Reprod Sci. 2015;22(5):572–84.
Tanaka M, Miyazaki T, Tanigaki S, Kasai K, Minegishi K, Miyakoshi K, et al. Participation of reactive oxygen species in PGF2alpha-induced apoptosis in rat luteal cells. J Reprod Fertil. 2000;120(2):239–45.
Peluffo MC, Bussmann L, Stouffer RL, Tesone M. Expression of caspase-2, -3, -8 and -9 proteins and enzyme activity in the corpus luteum of the rat at different stages during the natural estrous cycle. Reproduction. 2006;132(3):465–75.
Bazer FW. History of maternal recognition of pregnancy. Adv Anat Embryol Cell Biol. 2015;216:5–25.
Niringiyumukiza JD, Cai H, Xiang W. Prostaglandin E2 involvement in mammalian female fertility: ovulation, fertilization, embryo development and early implantation. Reprod Biol Endocrinol. 2018;16(1):43.
Takao Y, Honda T, Ueda M, Hattori N, Yamada S, Maeda M, et al. Immunohistochemical localization of the LH/HCG receptor in human ovary: HCG enhances cell surface expression of LH/HCG receptor on luteinizing granulosa cells in vitro. Mol Hum Reprod. 1997;3(7):569–78.
Choi J, Smitz J. Luteinizing hormone and human chorionic gonadotropin: origins of difference. Mol Cell Endocrinol. 2014;383(1–2):203–13.
Chin EC, Harris TE, Abayasekara DR. Changes in cAMP-dependent protein kinase (PKA) and progesterone secretion in luteinizing human granulosa cells. J Endocrinol. 2004;183(1):39–50.
Roy L, McDonald CA, Jiang C, Maroni D, Zeleznik AJ, Wyatt TA, et al. Convergence of 3′,5′-cyclic adenosine 5′-monophosphate/protein kinase A and glycogen synthase kinase-3beta/beta-catenin signaling in corpus luteum progesterone synthesis. Endocrinology. 2009;150(11):5036–45.
Priyanka S, Medhamurthy R. Characterization of cAMP/PKA/CREB signaling cascade in the bonnet monkey corpus luteum: expressions of inhibin-alpha and StAR during different functional status. Mol Hum Reprod. 2007;13(6):381–90.
Wiltbank MC, Diskin MG, Flores JA, Niswender GD. Regulation of the corpus luteum by protein kinase C. II. Inhibition of lipoprotein-stimulated steroidogenesis by prostaglandin F2 alpha. Biol Reprod. 1990;42(2):239–45.
Reiter E, Ahn S, Shukla AK, Lefkowitz RJ. Molecular mechanism of β-arrestin-biased agonism at seven-transmembrane receptors. Annu Rev Pharmacol Toxicol. 2012;52:179–97.
Casarini L, Reiter E, Simoni M. β-Arrestins regulate gonadotropin receptor-mediated cell proliferation and apoptosis by controlling different FSHR or LHCGR intracellular signaling in the hGL5 cell line. Mol Cell Endocrinol. 2016;437:11–21.
Bowolaksono A, Fauzi M, Sundari AM, Pustimbara A, Lestari R, Abinawanto, et al. The effects of luteinizing hormone as a suppression factor for apoptosis in bovine luteal cells in vitro. Reprod Domest Anim. 2021;56(5):744–53.
Chen SU, Chen RJ, Shieh JY, Chou CH, Lin CW, Lu HF, et al. Human chorionic gonadotropin up-regulates expression of myeloid cell leukemia-1 protein in human granulosa-lutein cells: implication of corpus luteum rescue and ovarian hyperstimulation syndrome. J Clin Endocrinol Metab. 2010;95(8):3982–92.
Kawaguchi S, Sakumoto R, Okuda K. Induction of the expressions of antioxidant enzymes by luteinizing hormone in the bovine corpus luteum. J Reprod Dev. 2013;59(3):219–24.
Bołzan E, Andronowska A, Bodek G, Morawska-Pucińska E, Krawczyński K, Dabrowski A, et al. The novel effect of hCG administration on luteal function maintenance during the estrous cycle/pregnancy and early embryo development in the pig. Pol J Vet Sci. 2013;16(2):323–32.
Kunal SB, Killivalavan A, Medhamurthy R. Involvement of src family of kinases and cAMP phosphodiesterase in the luteinizing hormone/chorionic gonadotropin receptor-mediated signaling in the corpus luteum of monkey. Reprod Biol Endocrinol. 2012;10:25.
Del Canto F, Sierralta W, Kohen P, Muñoz A, Strauss JF 3rd, Devoto L. Features of natural and gonadotropin-releasing hormone antagonist-induced corpus luteum regression and effects of in vivo human chorionic gonadotropin. J Clin Endocrinol Metab. 2007;92(11):4436–43.
Sugino N, Suzuki T, Kashida S, Karube A, Takiguchi S, Kato H. Expression of Bcl-2 and Bax in the human corpus luteum during the menstrual cycle and in early pregnancy: regulation by human chorionic gonadotropin. J Clin Endocrinol Metab. 2000;85(11):4379–86.
Kumazawa Y, Kawamura K, Sato T, Sato N, Konishi Y, Shimizu Y, et al. HCG up-regulates survivin mRNA in human granulosa cells. Mol Hum Reprod. 2005;11(3):161–6.
Telleria CM, Goyeneche AA, Cavicchia JC, Stati AO, Deis RP. Apoptosis induced by antigestagen RU486 in rat corpus luteum of pregnancy. Endocrine. 2001;15(2):147–55.
Kuranaga E, Kanuka H, Hirabayashi K, Suzuki M, Nishihara M, Takahashi M. Progesterone is a cell death suppressor that downregulates Fas expression in rat corpus luteum. FEBS Lett. 2000;466(2–3):279–82.
Okuda K, Korzekwa A, Shibaya M, Murakami S, Nishimura R, Tsubouchi M, et al. Progesterone is a suppressor of apoptosis in bovine luteal cells. Biol Reprod. 2004;71(6):2065–71.
Liszewska E, Rekawiecki R, Kotwica J. Effect of progesterone on the expression of bax and bcl-2 and on caspase activity in bovine luteal cells. Prostaglandins Other Lipid Mediat. 2005;78(1–4):67–81.
Arosh JA, Banu SK, McCracken JA. Novel concepts on the role of prostaglandins on luteal maintenance and maternal recognition and establishment of pregnancy in ruminants. J Dairy Sci. 2016;99(7):5926–40.
Lee J, McCracken JA, Stanley JA, Nithy TK, Banu SK, Arosh JA. Intraluteal prostaglandin biosynthesis and signaling are selectively directed towards PGF2alpha during luteolysis but towards PGE2 during the establishment of pregnancy in sheep. Biol Reprod. 2012;87(4):97.
Ziecik AJ, Przygrodzka E, Jalali BM, Kaczmarek MM. Regulation of the porcine corpus luteum during pregnancy. Reproduction. 2018;156(3):R57–R67.
Nio-Kobayashi J, Kudo M, Sakuragi N, Iwanaga T, Duncan WC. Loss of luteotropic prostaglandin E plays an important role in the regulation of luteolysis in women. Mol Hum Reprod. 2017;23(5):271–81.
Stacy BD, Gemmell RT, Thorburn GD. Morphology of the corpus luteum in the sheep during regression induced by prostaglandin F2alpha. Biol Reprod. 1976;14(3):280–91.
Jiang YF, Hsu MC, Cheng CH, Tsui KH, Chiu CH. Ultrastructural changes of goat corpus luteum during the estrous cycle. Anim Reprod Sci. 2016;170:38–50.
Morales C, García-Pardo L, Reymundo C, Bellido C, Sánchez-Criado JE, Gaytán F. Different patterns of structural luteolysis in the human corpus luteum of menstruation. Hum Reprod. 2000;15(10):2119–28.
Grzesiak M, Michalik A, Rak A, Knapczyk-Stwora K, Pieczonka A. The expression of autophagy-related proteins within the corpus luteum lifespan in pigs. Domest Anim Endocrinol. 2018;64:9–16.
Aboelenain M, Kawahara M, Balboula AZ, Montasser Ael M, Zaabel SM, Okuda K, et al. Status of autophagy, lysosome activity and apoptosis during corpus luteum regression in cattle. J Reprod Dev. 2015;61(3):229–36.
Choi J, Jo M, Lee E, Choi D. The role of autophagy in corpus luteum regression in the rat. Biol Reprod. 2011;85(3):465–72.
Leopardo NP, Velazquez ME, Cortasa S, González CR, Vitullo AD. A dual death/survival role of autophagy in the adult ovary of Lagostomus maximus (Mammalia-Rodentia). PLoS ONE. 2020;15(5):e0232819.
Tang Z, Huang Y, Zhang Z, Tang Y, Chen J, Sun F, et al. Accumulated autophagosomes and excessive apoptosis during the luteal development of pregnant rats. Int J Clin Exp Pathol. 2017;10(12):11384–92.
Przygrodzka E, Monaco CF, Plewes MR, Li G, Wood JR, Cupp AS, et al. Protein kinase A and 5′ AMP-activated protein kinase signaling pathways exert opposite effects on induction of autophagy in luteal cells. Front Cell Dev Biol. 2021;9:723563.
Tang Z, Chen J, Zhang Z, Bi J, Xu R, Lin Q, et al. HIF-1α activation promotes luteolysis by enhancing ROS levels in the corpus luteum of pseudopregnant rats. Oxid Med Cell Longev. 2021;2021:1764929.
Choi J, Jo M, Lee E, Choi D. ERK1/2 is involved in luteal cell autophagy regulation during corpus luteum regression via an mTOR-independent pathway. Mol Hum Reprod. 2014;20(10):972–80.
Bagnjuk K, Stöckl JB, Fröhlich T, Arnold GJ, Behr R, Berg U, et al. Necroptosis in primate luteolysis: a role for ceramide. Cell Death Discov. 2019;5:67.
Hojo T, Piotrowska-Tomala KK, Jonczyk AW, Lukasik K, Jankowska K, Okuda K, et al. Receptor interacting protein kinases-dependent necroptosis as a new, potent mechanism for elimination of the endothelial cells during luteolysis in cow. Theriogenology. 2019;128:193–200.
Hojo T, Siemieniuch MJ, Lukasik K, Piotrowska-Tomala KK, Jonczyk AW, Okuda K, et al. Programmed necrosis—a new mechanism of steroidogenic luteal cell death and elimination during luteolysis in cows. Sci Rep. 2016;6:38211.
Li D, Chen J, Guo J, Li L, Cai G, Chen S, et al. A phosphorylation of RIPK3 kinase initiates an intracellular apoptotic pathway that promotes prostaglandin(2α)-induced corpus luteum regression. eLife. 2021;10:e67409.
Likszo P, Skarzynski DJ, Moza Jalali B. Changes in porcine corpus luteum proteome associated with development, maintenance, regression, and rescue during estrous cycle and early pregnancy. Int J Mol Sci. 2021;22(21):11740.
D’Angelo E, Prat J. Non-neoplastic and tumor-like conditions of the ovary. In: Lopez-Beltran A, Raspollini MR, editors. Gynecologic and urologic pathology: similarities, differences and challenges. Cambridge: Cambridge University Press; 2019. p. 13–23.
Escobar-Morreale HF. Polycystic ovary syndrome: definition, aetiology, diagnosis and treatment. Nat Rev Endocrinol. 2018;14(5):270–84.
Dewailly D, Lujan ME, Carmina E, Cedars MI, Laven J, Norman RJ, et al. Definition and significance of polycystic ovarian morphology: a task force report from the androgen excess and polycystic ovary syndrome society. Hum Reprod Update. 2014;20(3):334–52.
Norman RJ, Dewailly D, Legro RS, Hickey TE. Polycystic ovary syndrome. Lancet. 2007;370(9588):685–97.
Dunn AJ. Animal models in polycystic ovarian syndrome. Clin Obstet Gynecol. 2021;64(1):126–33.
Das M, Djahanbakhch O, Hacihanefioglu B, Saridogan E, Ikram M, Ghali L, et al. Granulosa cell survival and proliferation are altered in polycystic ovary syndrome. J Clin Endocrinol Metab. 2008;93(3):881–7.
Li L, Mo H, Zhang J, Zhou Y, Peng X, Luo X. The role of heat shock protein 90B1 in patients with polycystic ovary syndrome. PLoS ONE. 2016;11(4):e0152837.
Almahbobi G, Misajon A, Hutchinson P, Lolatgis N, Trounson AO. Hyperexpression of epidermal growth factor receptors in granulosa cells from women with polycystic ovary syndrome. Fertil Steril. 1998;70(4):750–8.
Ma YJ, Dissen GA, Merlino G, Coquelin A, Ojeda SR. Overexpression of a human transforming growth factor-alpha (TGF alpha) transgene reveals a dual antagonistic role of TGF alpha in female sexual development. Endocrinology. 1994;135(4):1392–400.
Wu G, Hu X, Ding J, Yang J. Abnormal expression of HSP70 may contribute to PCOS pathology. J Ovarian Res. 2019;12(1):74.
Comim FV, Teerds K, Hardy K, Franks S. Increased protein expression of LHCG receptor and 17α-hydroxylase/17-20-lyase in human polycystic ovaries. Hum Reprod. 2013;28(11):3086–92.
Wang H, Feng X, Wang T, Pan J, Zheng Z, Su Y, et al. Role and mechanism of the p-JAK2/p-STAT3 signaling pathway in follicular development in PCOS rats. Gen Comp Endocrinol. 2023;330:114138.
Honnma H, Endo T, Henmi H, Nagasawa K, Baba T, Yamazaki K, et al. Altered expression of Fas/Fas ligand/caspase 8 and membrane type 1-matrix metalloproteinase in atretic follicles within dehydroepiandrosterone-induced polycystic ovaries in rats. Apoptosis. 2006;11(9):1525–33.
Mohammadi S, Kayedpoor P, Karimzadeh-Bardei L, Nabiuni M. The effect of curcumin on TNF-α, IL-6 and CRP expression in a model of polycystic ovary syndrome as an inflammation state. J Reprod Infertil. 2017;18(4):352–60.
Mikaeili S, Rashidi BH, Safa M, Najafi A, Sobhani A, Asadi E, et al. Altered FoxO3 expression and apoptosis in granulosa cells of women with polycystic ovary syndrome. Arch Gynecol Obstet. 2016;294(1):185–92.
Owens LA, Kristensen SG, Lerner A, Christopoulos G, Lavery S, Hanyaloglu AC, et al. Gene expression in granulosa cells from small antral follicles from women with or without polycystic ovaries. J Clin Endocrinol Metab. 2019;104(12):6182–92.
Cozzolino M, Seli E. Mitochondrial function in women with polycystic ovary syndrome. Curr Opin Obstet Gynecol. 2020;32(3):205–12.
Lai Q, Xiang W, Li Q, Zhang H, Li Y, Zhu G, et al. Oxidative stress in granulosa cells contributes to poor oocyte quality and IVF-ET outcomes in women with polycystic ovary syndrome. Front Med. 2018;12(5):518–24.
Zhu HL, Chen YQ, Zhang ZF. Downregulation of lncRNA ZFAS1 and upregulation of microRNA-129 repress endocrine disturbance, increase proliferation and inhibit apoptosis of ovarian granulosa cells in polycystic ovarian syndrome by downregulating HMGB1. Genomics. 2020;112(5):3597–608.
Zhang C, Hu J, Wang W, Sun Y, Sun K. HMGB1-induced aberrant autophagy contributes to insulin resistance in granulosa cells in PCOS. Faseb j. 2020;34(7):9563–74.
Tan M, Cheng Y, Zhong X, Yang D, Jiang S, Ye Y, et al. LNK promotes granulosa cell apoptosis in PCOS via negatively regulating insulin-stimulated AKT-FOXO3 pathway. Aging. 2021;13(3):4617–33.
Gao Y, Chen J, Ji R, Ding J, Zhang Y, Yang J. USP25 regulates the proliferation and apoptosis of ovarian granulosa cells in polycystic ovary syndrome by modulating the PI3K/AKT pathway via deubiquitinating PTEN. Front Cell Dev Biol. 2021;9:779718.
Tamaddon M, Azimzadeh M, Tavangar SM. microRNAs and long non-coding RNAs as biomarkers for polycystic ovary syndrome. J Cell Mol Med. 2022;26(3):654–70.
Prabhudesai KS, Raje S, Dhamanaskar A, Modi D, Dighe V, Contini A, et al. Identification and in vivo validation of a 9-mer peptide derived from FSHβ with FSHR antagonist activity. Peptides. 2020;132:170367.
Waghu FH, Desai K, Srinivasan S, Prabhudesai KS, Dighe V, Venkatesh KV, et al. FSHR antagonists can trigger a PCOS-like state. Syst Biol Reprod Med. 2022;68(2):129–37.
Kobayashi M, Yoshino O, Nakashima A, Ito M, Nishio K, Ono Y, et al. Inhibition of autophagy in theca cells induces CYP17A1 and PAI-1 expression via ROS/p38 and JNK signalling during the development of polycystic ovary syndrome. Mol Cell Endocrinol. 2020;508:110792.
Wang F, Han J, Wang X, Liu Y, Zhang Z. Roles of HIF-1α/BNIP3 mediated mitophagy in mitochondrial dysfunction of letrozole-induced PCOS rats. J Mol Histol. 2022;53(5):833–42.
Li D, You Y, Bi FF, Zhang TN, Jiao J, Wang TR, et al. Autophagy is activated in the ovarian tissue of polycystic ovary syndrome. Reproduction. 2018;155(1):85–92.
Yi S, Zheng B, Zhu Y, Cai Y, Sun H, Zhou J. Melatonin ameliorates excessive PINK1/Parkin-mediated mitophagy by enhancing SIRT1 expression in granulosa cells of PCOS. Am J Physiol Endocrinol Metab. 2020;319(1):E91–E101.
Lin S, Jin X, Gu H, Bi F. Relationships of ferroptosis-related genes with the pathogenesis in polycystic ovary syndrome. Front Med (Lausanne). 2023;10:1120693.
Zhang P, Pan Y, Wu S, He Y, Wang J, Chen L, et al. n-3 PUFA promotes ferroptosis in PCOS GCs by inhibiting YAP1 through activation of the Hippo pathway. Nutrients. 2023;15(8):1927.
Song X, Long D. Nrf2 and ferroptosis: a new research direction for neurodegenerative diseases. Front Neurosci. 2020;14:267.
Glendining KA, Campbell RE. Recent advances in emerging PCOS therapies. Curr Opin Pharmacol. 2023;68:102345.
Man L, Lustgarten Guahmich N, Vyas N, Tsai S, Arazi L, Lilienthal D, et al. Ovarian reserve disorders, can we prevent them? A review. Int J Mol Sci. 2022;23(23):15426.
Ishizuka B. Current understanding of the etiology, symptomatology, and treatment options in premature ovarian insufficiency (POI). Front Endocrinol (Lausanne). 2021;12:626924.
França MM, Mendonca BB. Genetics of ovarian insufficiency and defects of folliculogenesis. Best Pract Res Clin Endocrinol Metab. 2022;36(1):101594.
Liang L, Soyal SM, Dean J. FIGalpha, a germ cell specific transcription factor involved in the coordinate expression of the zona pellucida genes. Development. 1997;124(24):4939–47.
Soyal SM, Amleh A, Dean J. FIGalpha, a germ cell-specific transcription factor required for ovarian follicle formation. Development. 2000;127(21):4645–54.
Wang Z, Liu CY, Zhao Y, Dean J, FIGLA. LHX8 and SOHLH1 transcription factor networks regulate mouse oocyte growth and differentiation. Nucleic Acids Res. 2020;48(7):3525–41.
Tosh D, Rani HS, Murty US, Deenadayal A, Grover P. Mutational analysis of the FIGLA gene in women with idiopathic premature ovarian failure. Menopause. 2015;22(5):520–6.
Zhao H, Chen ZJ, Qin Y, Shi Y, Wang S, Choi Y, et al. Transcription factor FIGLA is mutated in patients with premature ovarian failure. Am J Hum Genet. 2008;82(6):1342–8.
Peluso JJ. Progesterone receptor membrane component 1 and its role in ovarian follicle growth. Front Neurosci. 2013;7:99.
Yuan XH, Yang CR, Wang XN, Zhang LL, Gao XR, Shi ZY. Progesterone maintains the status of granulosa cells and slows follicle development partly through PGRMC1. J Cell Physiol. 2018;234(1):709–20.
McCallum ML, Pru CA, Niikura Y, Yee SP, Lydon JP, Peluso JJ, et al. Conditional ablation of progesterone receptor membrane component 1 results in subfertility in the female and development of endometrial cysts. Endocrinology. 2016;157(9):3309–19.
Will EA, Liu X, Peluso JJ. AG 205, a progesterone receptor membrane component 1 antagonist, ablates progesterone’s ability to block oxidative stress-induced apoptosis of human granulosa/luteal cells†. Biol Reprod. 2017;96(4):843–54.
Peluso JJ, Pru CA, Liu X, Kelp NC, Pru JK. Progesterone receptor membrane component 1 and 2 regulate granulosa cell mitosis and survival through a NFΚB-dependent mechanism†. Biol Reprod. 2019;100(6):1571–80.
Murray A. Premature ovarian failure and the FMR1 gene. Semin Reprod Med. 2000;18(1):59–66.
Rose BI, Brown SE. An explanation of the mechanisms underlying fragile X-associated premature ovarian insufficiency. J Assist Reprod Genet. 2020;37(6):1313–22.
Hoffman GE, Le WW, Entezam A, Otsuka N, Tong ZB, Nelson L, et al. Ovarian abnormalities in a mouse model of fragile X primary ovarian insufficiency. J Histochem Cytochem. 2012;60(6):439–56.
Sherman SL, Curnow EC, Easley CA, Jin P, Hukema RK, Tejada MI, et al. Use of model systems to understand the etiology of fragile X-associated primary ovarian insufficiency (FXPOI). J Neurodev Disord. 2014;6(1):26.
Lu C, Lin L, Tan H, Wu H, Sherman SL, Gao F, et al. Fragile X premutation RNA is sufficient to cause primary ovarian insufficiency in mice. Hum Mol Genet. 2012;21(23):5039–47.
Pankhurst MW. A putative role for anti-Müllerian hormone (AMH) in optimising ovarian reserve expenditure. J Endocrinol. 2017;233(1):R1–R13.
Singh RP, Carr DH. The anatomy and histology of XO human embryos and fetuses. Anat Rec. 1966;155(3):369–83.
Reynaud K, Cortvrindt R, Verlinde F, De Schepper J, Bourgain C, Smitz J. Number of ovarian follicles in human fetuses with the 45,X karyotype. Fertil Steril. 2004;81(4):1112–9.
Modi DN, Sane S, Bhartiya D. Accelerated germ cell apoptosis in sex chromosome aneuploid fetal human gonads. Mol Hum Reprod. 2003;9(4):219–25.
Delcour C, Amazit L, Patino LC, Magnin F, Fagart J, Delemer B, et al. ATG7 and ATG9A loss-of-function variants trigger autophagy impairment and ovarian failure. Genet Med. 2019;21(4):930–8.
Zhang D, Liu Y, Zhang Z, Lv P, Liu Y, Li J, et al. Basonuclin 1 deficiency is a cause of primary ovarian insufficiency. Hum Mol Genet. 2018;27(21):3787–800.
Liu H, Wei X, Sha Y, Liu W, Gao H, Lin J, et al. Whole-exome sequencing in patients with premature ovarian insufficiency: early detection and early intervention. J Ovarian Res. 2020;13(1):114.
Wu D, Zhao W, Xu C, Zhou X, Leng X, Li Y. Melatonin suppresses serum starvation-induced autophagy of ovarian granulosa cells in premature ovarian insufficiency. BMC Womens Health. 2022;22(1):474.
Liu Y, Fang Y, Wei J, Zhang C, Wu D, Li Y. Melatonin protects against primary ovarian insufficiency by activating the PI3K/Akt/mTOR pathway and inhibiting autophagy. Ann Clin Lab Sci. 2022;52(6):895–903.
Zhou Q, Jin X, Wang J, Li H, Yang L, Wu W, et al. 4-vinylcyclohexene diepoxide induces premature ovarian insufficiency in rats by triggering the autophagy of granule cells through regulating miR-144. J Reprod Immunol. 2023;157:103928.
Du R, Cheng X, Ji J, Lu Y, Xie Y, Wang W, et al. Mechanism of ferroptosis in a rat model of premature ovarian insufficiency induced by cisplatin. Sci Rep. 2023;13(1):4463.
Al-Alem LF, Baker AT, Pandya UM, Eisenhauer EL, Rueda BR. Understanding and targeting apoptotic pathways in ovarian cancer. Cancers (Basel). 2019;11(11):1631.
Shih IM, Wang Y, Wang TL. The origin of ovarian cancer species and precancerous landscape. Am J Pathol. 2021;191(1):26–39.
Frumovitz M, Leitao MM, Ramalingam P. Diagnosis and treatment of rare gynecologic cancers. Amsterdam, Netherlands: Elsevier; 2022.
Cheng L, Roth LM, Zhang S, Wang M, Morton MJ, Zheng W, et al. KIT gene mutation and amplification in dysgerminoma of the ovary. Cancer. 2011;117(10):2096–103.
Palmer RD, Foster NA, Vowler SL, Roberts I, Thornton CM, Hale JP, et al. Malignant germ cell tumours of childhood: new associations of genomic imbalance. Br J Cancer. 2007;96(4):667–76.
Cheng L, Zhang S, Talerman A, Roth LM. Morphologic, immunohistochemical, and fluorescence in situ hybridization study of ovarian embryonal carcinoma with comparison to solid variant of yolk sac tumor and immature teratoma. Hum Pathol. 2010;41(5):716–23.
Zong X, Zhang Y, Peng X, Cao D, Yu M, Wang J, et al. Analysis of the genomic landscape of yolk sac tumors reveals mechanisms of evolution and chemoresistance. Nat Commun. 2021;12(1):3579.
Hoei-Hansen CE, Kraggerud SM, Abeler VM, Kaern J, Rajpert-De Meyts E, Lothe RA. Ovarian dysgerminomas are characterised by frequent KIT mutations and abundant expression of pluripotency markers. Mol Cancer. 2007;6:12.
Shah SP, Köbel M, Senz J, Morin RD, Clarke BA, Wiegand KC, et al. Mutation of FOXL2 in granulosa-cell tumors of the ovary. N Engl J Med. 2009;360(26):2719–29.
Jamieson S, Butzow R, Andersson N, Alexiadis M, Unkila-Kallio L, Heikinheimo M, et al. The FOXL2 C134W mutation is characteristic of adult granulosa cell tumors of the ovary. Mod Pathol. 2010;23(11):1477–85.
Benayoun BA, Caburet S, Dipietromaria A, Georges A, D’Haene B, Pandaranayaka PJ, et al. Functional exploration of the adult ovarian granulosa cell tumor-associated somatic FOXL2 mutation p.Cys134Trp (c.402C>G). PLoS ONE. 2010;5(1):e8789.
Benayoun BA, Anttonen M, L’Hôte D, Bailly-Bechet M, Andersson N, Heikinheimo M, et al. Adult ovarian granulosa cell tumor transcriptomics: prevalence of FOXL2 target genes misregulation gives insights into the pathogenic mechanism of the p.Cys134Trp somatic mutation. Oncogene. 2013;32(22):2739–46.
Kim JH, Yoon S, Park M, Park HO, Ko JJ, Lee K, et al. Differential apoptotic activities of wild-type FOXL2 and the adult-type granulosa cell tumor-associated mutant FOXL2 (C134W). Oncogene. 2011;30(14):1653–63.
Schmidt D, Ovitt CE, Anlag K, Fehsenfeld S, Gredsted L, Treier AC, et al. The murine winged-helix transcription factor Foxl2 is required for granulosa cell differentiation and ovary maintenance. Development. 2004;131(4):933–42.
Kim SY. Insights into granulosa cell tumors using spontaneous or genetically engineered mouse models. Clin Exp Reprod Med. 2016;43(1):1–8.
Acknowledgements
The figures were prepared using template images from Servier Medical Art collection provided by Servier (https://smart.servier.com) and licensed under a Creative Commons Attribution 3.0 unported license.
Funding
This work was supported by a grant from the non-commercial organization “The Russian Science Foundation” (23-74-30006). The work in the authors’ laboratories is supported (to BZ) by grants from the Swedish (22 2013) and Stockholm (181301) Cancer Societies.
Author information
Authors and Affiliations
Contributions
MSC performed the major part of literature search, structured the information, wrote the main part of the text, prepared the figures; ARM performed literature search for “Common types of PCD observed in the ovary” section and contributed to writing of “Apoptosis” sections; BZ conceptualized the review, structured the information, edited the main text, supervised the manuscript preparation; GSK conceptualized the review, structured the information, edited the main text, supervised the manuscript preparation. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Chesnokov, M.S., Mamedova, A.R., Zhivotovsky, B. et al. A matter of new life and cell death: programmed cell death in the mammalian ovary. J Biomed Sci 31, 31 (2024). https://doi.org/10.1186/s12929-024-01017-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12929-024-01017-6