
Abstract
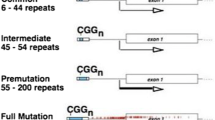
Fragile X-associated primary ovarian insufficiency (FXPOI) is among the family of disorders caused by the expansion of a CGG repeat sequence in the 5' untranslated region of the X-linked gene FMR1. About 20% of women who carry the premutation allele (55 to 200 unmethylated CGG repeats) develop hypergonadotropic hypogonadism and cease menstruating before age 40. Some proportion of those who are still cycling show hormonal profiles indicative of ovarian dysfunction. FXPOI leads to subfertility and an increased risk of medical conditions associated with early estrogen deficiency. Little progress has been made in understanding the etiology of this clinically significant disorder. Understanding the molecular mechanisms of FXPOI requires a detailed knowledge of ovarian FMR1 mRNA and FMRP’s function. In humans, non-invasive methods to discriminate the mechanisms of the premutation on ovarian function are not available, thus necessitating the development of model systems. Vertebrate (mouse and rat) and invertebrate (Drosophila melanogaster) animal studies for the FMR1 premutation and ovarian function exist and have been instrumental in advancing our understanding of the disease phenotype. For example, rodent models have shown that FMRP is highly expressed in oocytes where it is important for folliculogenesis. The two premutation mouse models studied to date show evidence of ovarian dysfunction and, together, suggest that the long repeat in the transcript itself may have some pathological effect quite apart from any effect of the toxic protein. Further, ovarian morphology in young animals appears normal and the primordial follicle pool size does not differ from that of wild-type animals. However, there is a progressive premature decline in the levels of most follicle classes. Observations also include granulosa cell abnormalities and altered gene expression patterns. Further comparisons of these models are now needed to gain insight into the etiology of the ovarian dysfunction. Premutation model systems in non-human primates and those based on induced pluripotent stem cells show particular promise and will complement current models. Here, we review the characterization of the current models and describe the development and potential of the new models. Finally, we will discuss some of the molecular mechanisms that might be responsible for FXPOI.
Similar content being viewed by others


Review
Fragile X-associated primary ovarian insufficiency (FXPOI) is among the family of disorders caused by the expansion of a CGG repeat sequence located in the 5' untranslated region (UTR) of the X-linked gene FMR1. About 20% of women who carry an allele with 55 to 200 unmethylated CGG repeats, called the premutation (PM) allele, develop hypergonadotropic hypogonadism and cease menstruating before age 40, a condition also known as premature ovarian failure (POF). The 20% contrasts with a rate of about 1% of the general population (for reviews, see [1–3]).
The term primary ovarian insufficiency (POI) encompasses both POF and occult indicators of ovarian function such as decreased levels of the anti-Müllerian hormone and increased levels of the follicle-stimulating hormone. As this entire spectrum, including the altered hormone profile is observed among women with the PM [4–9], the term ‘FXPOI’ is well suited [10]. Importantly, the proportion of women with the PM who manifest occult hormone indicators or clinical signs of ovarian dysfunction is unknown, as is the extent to which such indicators predict infertility or POF. This is a clinically significant gap as women with FXPOI can still become pregnant and may have a child with fragile X syndrome [11].
FXPOI is clinically significant. The most immediate and significant consequence of diminished ovarian function is reduced fertility [12, 13]. The state of early estrogen deficiency leads to additional clinical consequences such as an increased risk of low bone density, earlier onset osteoporosis and bone fractures [14], impaired endothelial function [15], earlier onset of coronary heart disease [16], and increased cardiovascular mortality and overall mortality (e.g., [17, 18]). In addition, women who have an early menopause are reported to suffer from more anxiety, depression, somatization, sensitivity, hostility and psychological distress than women with normal ovarian function [19].
We know very little about the mechanisms leading to FXPOI. It is well established that full mutation carriers, or those with an allele of >200 methylated repeats that leads to silencing of FMR1, do not suffer from ovarian dysfunction. Thus, the significant reduction of the FMR1 protein product, FMRP, does not appear to be the culprit. There are important molecular attributes of the PM: with increasing repeat length, there are increasing FMR1 transcript levels and decreasing FMRP levels [20–24]. As discussed in more detail below, many researchers have postulated that FMR1 mRNA gain-of-function toxicity may underlie FXPOI, as is the case for the other PM-associated disorder, fragile X-associated tremor/ataxia syndrome (FXTAS) [25].
Not all women with the PM suffer from POF or occult indicators of ovarian dysfunction. Four factors have been investigated to explain the incomplete penetrance of POF among PM carriers: repeat length, skewed X-chromosome inactivation (XCI), background genes and smoking. First, there is a strong non-linear association between the penetrance of POF and repeat number. Women with mid-range PM repeats (approximately 70 to 90 repeats) have the highest risk for POF. Carriers of both smaller and larger PM repeat lengths also have an increased risk of POF compared to the general population, but not to the same extent as mid-range repeat carriers [7, 13, 26–28]. Second, skewed XCI may play a role in modifying the risk or severity of FXPOI, as FMR1 is located on the X chromosome. However, no study has found evidence for skewed XCI based on samples from fresh blood among PM carriers with FXPOI [5, 7, 28–31]. Assuming that XCI in blood can be used as a proxy for the correct target tissue, one possible explanation for this observation is that the toxic effect of the PM acts during a stage in development when both X chromosomes are active. Third, studies have shown that the risk of POF depends not only on the PM allele, but also on other background genes [27, 32]. Finally, smoking, a known risk factor for reducing the age at menopause, has been shown to have the same effect on women with the PM as it does on non-carriers [13, 27].
In short, little is known about the etiology of FXPOI and the cause of its reduced penetrance and variable expressivity. The development and use of model systems to uncover the associated mechanism has just begun. The overall goal of this review is to describe these model systems and the initial steps taken to elucidate the mechanisms underlying the association between the FMR1 PM and ovarian function. We will begin with a description of the current rodent model systems, which are the most mature in terms of their characterization of the effect of the PM. We will then describe new models that have the potential to advance the field.
Rodent model systems: recapitulation of FXPOI
Only recently have FMR1 mutation murine models been used to study ovarian function (Table 1). Published results for two PM mouse models [33, 34] and unpublished studies for another (RKH et al., unpublished), indicate their value in studying the etiology for FXPOI. A full mutation mouse model further implicates FMR1 as having an important role in folliculogenesis [35]. Finally, the characterization of expression patterns of FMR1 in the ovary of the rat shows the potential of this model in understanding the toxic effect of the PM [36]. Here we will review these models with respect to their ovarian phenotype to underscore their importance in future research on FXPOI.
Model construction
The construction of each model has been reported in detail previously. Dr Usdin’s team originally constructed a knock-in (KI) murine model to study instability of the repeat sequence [37]. The approximately 130-repeat tract in the PM model was generated by serial ligation of short, stable CGG · CCG-repeat tracts, which were then used to replace the endogenous shorter murine repeat tract by homologous recombination. The KI allele had only minimal differences from the wild-type (WT) murine Fmr1 gene in the region flanking the repeat. Therefore, females have a normal mouse Fmr1 allele, and an Fmr1 allele that is almost exactly the same as the endogenous mouse allele except for the length of the repeat tract. These mice are in a C57BL/6 background. This model will be referred to as CGGnih.
Lu et al. used a transgenic model that carries a YAC with the human PM allele that includes 90 repeats [34]. The line used (YAC-TG296) includes one copy of the YAC and about 5 kb of flanking sequence and was bred to WT FVB mice for five generations [38]. These mice are homozygous for the WT Fmr1 allele. This line and several others have been used to study repeat instability [38] and the overexpression of FMRP [39].
Dr Willemsen’s team characterized their previously constructed KI mouse with an expanded CGG repeat in the PM range (CGGdut). This KI mouse model was developed by substituting the endogenous mouse 5' UTR containing the CGG repeat with the corresponding region from a human allele carrying 98 CGG repeats [40]. These mice are homozygous for the KI allele and have no WT Fmr1 allele. They are on a mixed C57BL/6 and FVB/n genetic background. This model shows instability upon transmission [41] and the biochemical, phenotypic and neuropathological characteristics of FXTAS [42]. At this time, this model provides information about FMR1 expression in ovarian tissues. Further work on the ovarian phenotype is currently being conducted.
The role of FMRP in ovarian function has also been examined in two other rodent models. Ovarian function in a mouse model for the fragile X full mutation containing a targeted disruption of the Fmr1 gene [43] has recently been described [34]. The expression of FMRP and Fmr1 during folliculogenesis has recently been evaluated in Sprague-Dawley rats [36].
Premutation leads to altered FMR1 expression levels
In all WT animals, FMRP has been identified in granulosa cells (GCs), luteal cells and most prominently in oocytes. In oocytes, expression was seen at all stages of folliculogenesis and primarily in the cytoplasm [33, 36]. Expression was not observed in the interstitial cells. For the rat model, Ferder et al. [36] found that there were changes in Fmr1 expression during follicle maturation, both at the protein and mRNA levels. FMRP levels increased with increasing follicle development. Fmr1 transcript levels were similar in pre-antral and early antral follicles, but decreased in pre-ovulatory follicles. The authors suggested that Fmr1 expression in the ovary may be regulated at different levels and these may be independently controlled. In addition, they found expression of at least four different isoforms of FMRP during all stages of follicular growth. These expression patterns differ from those observed in the brain and testis.
Increased expression of Fmr1 mRNA in the ovary has been seen in all PM mouse models. Interestingly, in the CGGnih and WT littermates, there was a non-linear age effect, where total ovary mRNA levels were higher at 12 months compared with 6 and 18 months of age. At 7 months, in situ hybridization studies of the CGGnih model showed mRNA levels to be increased in oocytes and GCs.
The expectation for relative FMRP levels differs among the PM mouse models due to their construction. No differences in FMRP levels between YAC-TG296 mice and their WT littermates were found, when measured at 6 to 8 weeks. At 1 to 2 months, the CGGnih mice showed relatively reduced levels of FMRP in GCs and luteal cells. In the CGGdut PM model, a reduction in FMRP expression was noted at 2 months. This observed decrease is similar to that found in the brain of these PM models [37, 41]. Again, the relative Fmr1 PM levels seemed more pronounced in ovaries than in the brain for both the CGGnih and the CGGdut models.
Interestingly, an abnormal distribution of FMRP when measured at 7 months has been observed in the CGGnih model: FMRP was more highly expressed in the nucleus of oocytes than in the cytoplasm. There were eight times as many oocytes with higher nuclear expression in the PM model compared with the WT.
Two phenomena considered to be a consequence of altered FMR1 expression in the brain were measured for the CGGnih model: the presence of inclusion bodies and ubiquitination. Essentially no inclusions were noted in the ovarian cells of the PM or WT mice. With respect to ubiquitination, ubiquitin in WT mice was distributed throughout the cytoplasm and nucleoplasm. The CGGnih mice showed higher levels of ubiquitin in oocytes, more oocytes with elevated ubiquitin and a pronounced nuclear/perinuclear concentration than WT mice. Also, those with the highest number of oocytes with nuclear FMRP had the highest number of oocytes with high concentrations of ubiquitin.
Morphology of the premutation ovary
Both the CGGnih and YAC-TG296 models had smaller ovaries by 4 months of age compared with WT mice, but they were grossly normal. However, in the CGGnih model from 4 to 12 months, there was no decrease in size, as would be expected with the normal decrease in the number of oocytes and corpus lutea. This might be explained by the noted interstitial hypertrophy and tubulostromal hypertrophy at 7 to 12 months. The CGGnih mouse ovaries also had more and larger non-functional ovarian cysts.
Premutation leads to a depletion of follicles in later stages of maturation
Examination of the pattern of follicle counts at all stages provides insight into the effect of the PM. The total number of primordial follicles was comparable to WT measured at PD25 (YAC-TG296) and at 4 months (CGGnih). This suggests that the establishment of the primordial pool is not affected in PM mice. At PD25 and 9 weeks in the YAC-TG296 model, the number of later subclasses of follicles was reduced, and significantly so for mature follicles, compared with WT mice. At ages over 4 months, the CGGnih mice had a significant reduction of all subclasses of follicles, with the size of the primordial pool being correlated to the number of advancing subclasses. Also, the number of corpus lutea, the bodies that result from post-ovulatory follicles, was reduced in PM mice compared with WT mice. Together, these observations suggests that the PM does not affect the establishment of the primordial follicle pool, does not block a particular stage of follicle development and does not lead to increased follicular recruitment. The fact that both follicles that depend on ovarian intrinsic factors and those that depend on the input of extrinsic factors are affected suggests that the problem may be intrinsic to the ovary.
Premutation leads to granulosa cell abnormalities
GCs are key to the functioning of the follicle. The CGGnih mice had fewer GCs in the antral follicles than did the WT mice. Furthermore, there were significantly more antral follicles in which the GC layer was detached and the corona was partial or missing in both the CGGnih and YAC-TG296 models. Signs of atresia were also increased. In the CGGnih mice, there was a high ratio of atretic follicles to advancing follicles, irrespective of estrus cycle stage. Using terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) to analyze ovarian sections at PD35, 16 weeks and 22 weeks, the YAC-TG296 mice were seen to have increased numbers of antral follicles that appeared atretic compared with the WT mice. Thus the PM could lead to increased apoptosis in the ovaries.
Premutation leads to subfertility
Features of fertility were investigated for the YAC-TG296 mice. These mice had an increased frequency of sterility, and, among those that were fertile, reduced litter sizes and were older in having their first litter. At 9 to 22 weeks, these mice had higher follicle stimulating hormone and lower luteinizing hormone levels compared with WT mice. They also had higher levels of 17β-E2 at 10 to 12 weeks, although these levels normalized to those of WT mice at 16 to 22 weeks.
Premutation leads to altered gene expression
Expression of genes known to be involved in ovarian function was investigated for YAC-TG296 mouse ovaries at two stages: PD25 and proestrus-stage adults (8 to 14 weeks). The LH receptor (Lhr) was significantly downregulated at both stages. However, no differences in mRNA levels were found between PM and WT mice, among other major known regulators and markers of folliculogenesis. LH-induced ovulation-related genes were further investigated and found to be downregulated, specifically at the proestrus stage in adults. These findings suggest that the LH-mediated pathway could be affected in PM ovaries. The PI3K-Akt pathway, a pathway known to play a critical role in gonadotropin-mediated GC differentiation, cumulus expansion and oocyte maturation, was also investigated. YAC-TG296 mouse ovaries had a significant reduction in the levels of phosphorylated Akt, but not total Akt. Given the interaction between the Akt and mTOR pathways, the status of mTOR was also investigated. Again, there was a reduction in the levels of the phosphorylated mTOR protein but not total mTOR. Thus, the Akt-mTOR-mediated signaling cascade may be altered in PM ovaries. A role for reduced mTOR phosphorylation in FXTAS is suggested by the observation that activating mTOR ameliorates neurodegeneration in a fly model of FXTAS [44]. It will be of interest to see whether this activation improves ovarian function in flies and mouse models.
Ovarian phenotype altered in knockout model
An interesting ovarian phenotype has been observed in the full mutation knockout (KO) mouse model [35]. By 3 weeks, homozygous KO mice had an increased number of follicles compared with WT mice. By 18 weeks, the size of the ovaries in the KO mice was larger than those in the WT mice and showed prominent cysts, consistent with corpus lutea development. Lysates from 9-to-18-week-old ovaries showed increased protein levels of Tsc2, Sash1 and mTOR. The authors suggested that increased levels of these proteins seen in the absence of FMRP, may lead to precocious follicular development. Thus, this KO model may have the potential to model ovarian insufficiency; however whether the related mechanism is associated with FXPOI is an open question. Women who carry the full mutation do not show signs of POI. Whether this is due to the fact that they are heterozygous for the loss of FMRP is unknown.
Fly model: effect of modifying genes and more
In Drosophila ovaries, a small population of germ-line stem cells (GSCs) is maintained in a well-defined microenvironment. This provides an attractive system for investigating the regulatory mechanisms that determine the fate of stem cells [45, 46]. A typical Drosophila ovary is composed of 16 to 20 ovarioles. Each ovariole consists of an anterior functional unit called a germarium that houses GSCs and somatic lineages, and a linear string of differentiated egg chambers posterior to the germarium. The tip of the germarium consists of specialized cells that maintain the microenvironments called niches and these are essential for GSC proliferation and maintenance. At this tip, GSCs normally divide asymmetrically to ensure that one daughter cell remains attached to the niche cells for self-renewal, while the other is displaced from the niche, becoming a cystoblast, which initiates differentiation and sustains oogenesis [47]. Studies from multiple laboratories have identified the genes that are essential for GSC fate determination [48, 49].
Drosophila GSCs have been used as a model to show that FMRP can modulate the fate of stem cells: Yang et al. [50] found that dFmr1 is required for both maintaining GSCs and repressing differentiation. Very recently, transgenic lines have been developed that drive the expression of the PM rCGG repeat in fly ovaries and these rCGGs were found to be toxic in the germ line as well (PJ, unpublished data). These results suggest that both the reduction of FMRP and expression of PM rCGG repeats could have detrimental effects on fly ovary and stem cell maintenance.
Because of the relative ease of model construction compared with other model systems, two other important questions can be addressed at relatively low cost. First, the fly model can be used to test the effect of genetic modifiers on the ovarian phenotype. This could be valuable not only for our understanding of the pathogenic mechanism, but may also shed light on genes whose human homologs may contribute to the variable penetrance of FXPOI. Second, the Drosophila model has significant potential for increasing our understanding of the non-linear effect of repeat number by creating constructs that vary only by repeat number.
Non-human primate model: bridging the translational gap
Many genetic, cellular and physiological differences exist between the current model systems used for studying FXPOI and human females. Non-human primates (NHPs) offer a clinically relevant model system in which to explore the molecular mechanisms of the PM on ovarian function. One of the limitations in modeling FXPOI is that there are no known naturally occurring animal models with FMR1 repeat mutations, including NHPs [51, 52]. Of the species tested thus far, only NHPs have CGG repeat numbers comparable to those in humans [53]. The repeat sequence found in the great apes (Hominidae) shows striking similarity to that in humans, with CGG repeat lengths ranging from 20 to 39 interrupted by 1 to 6 AGG interruptions and the longest and most variable CGG lengths at the 3' end of the repeat [53, 54].
While it is possible that spontaneous CGG repeat expansion to the pre- and full mutation range does occur in NHP populations, screening would be expensive and unlikely to yield sufficient animal numbers for meaningful studies. Instead efforts are currently underway at the Washington National Primate Research Center to generate a NHP transgenic model of FXPOI. Using technologies based on embryonic stem cells, Dr Curnow’s team aims to introduce the human PM sequence into the macaque endogenous FMR1 gene and generate NHP females with germ-line expression of the PM. While embryonic stem cells from species other than the mouse have been historically less amenable to gene-targeting strategies, recent work on the rat, human and marmoset has shown transgene efficiencies and stability equivalent to the mouse following the refinement of the culture conditions for embryonic stem cells and gene-targeting methods [55–62]. Full-scale development of a NHP model of FXPOI necessitates a long-term approach with the generation of a self-sufficient breeding colony of FXPOI-affected females in which reproductive function related specifically to FXPOI can be studied. These studies can be done in conjunction with other pertinent aspects of fragile X-associated disorders.
Induced pluripotent stem cell model: examination of affected tissues
Findings from the PM mouse models noted above suggest that GC function is involved in the cellular cause of FXPOI, as is true for other forms of POI [63–65]. It is difficult to study GC function in women, as the procedure to obtain follicles with GCs is quite invasive and, thus, patient material is scarce. Patient-specific induced pluripotent stem cells (iPSCs) derived from adult somatic cells and which have differentiated into GC-like cells represent one novel possible option for generating an abundance of material for research purposes without any invasive procedures.
Work by Kang et al. has shown the ability of mouse iPSCs to differentiate into GC-like cells that express FSHR and secrete estradiol after co-culture with mouse GCs isolated from stimulated follicles [64]. However, this research has not been extended to human iPSCs. Adapting this protocol would enable researchers to investigate repeat length instability, cellular and signaling defects and cell viability in in vitro GCs derived from patient-specific human iPSCs. These types of in vitro studies could elucidate novel defects in somatic cells that support follicle survival and maturation that contribute to POI.
Recently, Hayashi et al. showed that functional oocytes could be derived from mouse iPSCs [66]. While this differentiation method relied on in vivo co-culture with normal mouse GCs transplanted under the ovarian bursa, the technique showed the feasibility of reconstituting a follicle and generating a functional oocyte from mouse iPSCs. If this system could be adapted to human female iPSCs using a wholly in vitro methodology in combination with a GC differentiation protocol, researchers could study how signaling defects in GCs contribute to oocyte death in POI. For treatment of infertility related to POI, patient-specific iPSCs could be differentiated into functional oocytes with GCs from non-POI patients to allow POI women to produce their own genetic offspring. Although these types of experiments and clinical implications are years away from being realized, human POI iPSC studies are a novel way for furthering understanding of FXPOI and its consequences.
Possible mechanisms of FXPOI: what have we learned from model systems?
As emphasized above, we know little about the disease pathology underlying FXPOI. A number of lines of evidence suggest that the pathology is not related to an FMRP deficit. Firstly, FXPOI is seen in women who have repeat numbers that are not associated with an FMRP deficit, at least in peripheral blood. Secondly, women who carry the completely silenced full mutation, and thus do not express FMRP in, on average, half their cells, do not show symptoms of FXPOI. Thirdly, the YAC-TG296 mouse model is homozygous for the WT Fmr1 allele, yet shows signs of ovarian dysfunction. Thus FXPOI, like FXTAS, is not likely to be the result of the loss of FMRP. However, how this relates to the observation that Fmr1 KO mice also show ovarian dysfunction is unclear.
In terms of the molecular mechanism, there may be parallels to FXTAS. Work with tissue cultures has shown that expression of mRNA from the PM allele is deleterious to a wide variety of cell types; thus it is reasonable to think that FXTAS and FXPOI may share a common pathological basis. A variety of models have been proposed to explain the pathology of FXTAS and support for these comes from various model systems. The RNA gain-of-function model predicts that the long rCGG track sequesters specific CGG-binding proteins, resulting in the loss of normal cell function. Various proteins have been identified that directly bind to CGG-RNA and whose sequestration may affect cell viability, including: hnRNP A2/B1, a protein involved in pre-mRNA processing [67, 68]; Pur α, a protein that has been implicated in transcription regulation and neuronal development [67]; and the miRNA-processing complex, DROSHA-DGCR8 [69]. These proteins in turn are able to recruit additional proteins like CUGBP1 in the case of hnRNP A2/B1 [70] and the RNA helicase, Rm62, in the case of Purα/DDX5 [71]. Overexpression of DROSHA [69], hnRNP A2/B1, CUGBP1 [72], Pur α [67] and Rm62 [71] rescue neurodegeneration in a fly model of FXTAS, but whether they rescue the mammalian phenotype remains to be seen.
Transcripts from the FMR1 locus may be deleterious in other ways. For example, the rCGG forms stable secondary structures including hairpins [73, 74], which are substrates for the human enzyme Dicer [74]. Dicer is responsible for generating small RNAs that can act via the RNA interference pathway to reduce post-transcriptionally the expression of genes containing similar repeat tracts. Expression of RNA with 80 CAG repeats, which also forms hairpins, generates Dicer-dependent small RNAs that are toxic to neuronal cells in culture [75]. It remains to be seen whether rCGGs could be toxic in similar ways.
Various antisense transcripts are also made from the human FMR1 gene and these potentially contribute to disease pathology in different ways. While the expression of some of these transcripts is low in normal cells, in PM carriers some of these transcripts are present at levels comparable to that of the sense transcript [76]. Some double-stranded RNAs, including rCUG.rCAG, can activate the innate immune response in Drosophila in a Dicer-dependent manner [75]. Such double-stranded RNAs could be generated via the annealing of sense and antisense transcripts produced from the FMR1 gene. The antisense rCGGs may also sequester proteins, as proposed for the sense transcript. Furthermore, since the repeat is located in a putative open reading frame on some of the antisense transcripts, it could produce a repeat-containing protein, in this case a polyproline-containing protein, which could contribute to disease pathology [76].
Another protein-based model arises from the observation that repeated sequences can increase the frequency at which translation initiates at non-ATG codons, a process known as repeat-associated non-ATG (RAN) translation [77]. In humans, and in mice and flies containing the human 5' UTR, there is evidence to suggest that such start sites are used to make polyglycine and/or polyalanine-containing proteins that are neurotoxic [78]. Such proteins can be detected in the brains of individuals with FXTAS.
The YAC-TG296 and the CGGdut mouse models do have the human FMR1 5' UTR upstream of the repeat and thus can make the polyglycine and polyalanine proteins. In contrast, the CGGnih mouse retains the murine 5' UTR and thus has a stop codon immediately upstream of the repeat. The fact that the CGGnih mouse does exhibit signs of ovarian dysfunction suggests that at least some pathology may arise independently of RAN translation. However, the relative contribution of RNA-based pathology and protein-based pathology to the overall phenotype remains to be determined.
Many diseases caused by expansion of different repeats are associated with the formation of intranuclear inclusions in patients, in cells in tissue culture as well as in the brains of mice and fly models. While intranuclear inclusions in the brain are a hallmark of FXTAS, very few inclusions are seen in the ovaries of PM mouse models and humans. More data are necessary to establish definitively whether inclusions are a feature of FXPOI. The few inclusions noted in stromal cells of grossly normal appearing ovaries from humans [79] may suggest an underlying toxic gain-of-function related to protein degradation. The absence of inclusions in follicles may be the result of the rapid loss of affected follicles, too fast for inclusions to be observed [79]. This would be similar to that seen in Purkinje cells in FXTAS [80]. However, it is unclear whether intranuclear inclusions are protective, pathogenic or neutral markers of disease pathology.
Lastly, more work needs to focus on altered gene expression in PM models. Data from the YAC-TG296 model shows reduced expression of phosphorylated Akt and mTOR, while the KO model showed elevated mTOR levels. mTOR dysregulation in these animals is of interest since both underexpression and overexpression of mTOR [81, 82] can result in ovarian dysfunction. It has been shown that mTOR inhibition results in reduced GC proliferation [83], a significant phenotype in the PM mouse models. We speculate that a role for reduced mTOR phosphorylation in FXPOI could also account for the non-linear relation between CGG repeat number and the risk of FXPOI. As the repeat number increases, FMRP levels are predicted to decrease, because of the difficulties associated with the translation of large PM alleles. This decrease in FMRP could in turn lead to increased levels of mTOR phosphorylation, which could offset the loss of mTOR resulting from the consequences of expression of the PM rCGGs. However, whether mTOR dysregulation is the proximal cause of the ovarian dysfunction seen in FXPOI still remains to be determined and studies to address mTOR levels in human female PM carriers are sorely needed. Also, some phenotype differences observed between models may be related to the different background strains. Although this can complicate comparisons, it also points to the importance of modifying genes to explain the variable presentation of FXPOI.
Conclusions
Clearly, the value of model systems in determining the underlying cause of FXPOI cannot be overstated. Each system has its advantages. The fly model will be valuable for determining the non-linear effect of increasing repeat number on the ovary in a cost-efficient way. It will also be the model of choice for identifying modifier genes using effective genetic screens. As always, caution must be taken in ascribing phenotype outcomes in the fly to mammals. In particular, the development and aging processes occurring within ovaries differ between the fly model and mammalian systems. Thus, disease progression and histological studies of FXPOI will need to be studied in mammalian models. Already rodent models have shown their potential and will continue to help in elucidating mechanisms and identifying potential treatments. Still, the genetic, cellular and physiological differences between current vertebrate model systems and humans suggest that additional model systems should be developed and tested in parallel to expedite translational research efforts. The translational bridge between current animal models and humans can best be met by NHP studies. Finally, patient-specific iPSCs derived from adult somatic cells and differentiated into GC-like cells represent a viable option for generating material needed for research without invasive procedures that minimizes the excessive use of animals. Thus, the combined use of model systems promises to elucidate the underlying mechanisms of FXPOI and the associated risk factors.
Abbreviations
- FXPOI:
-
fragile X-associated primary ovarian insufficiency
- FXTAS:
-
fragile X-associated tremor/ataxia syndrome
- GC:
-
granulosa cell
- GSC:
-
germ-line stem cell
- iPSC:
-
induced pluripotent stem cell
- kb:
-
kilobase
- KI:
-
knock-in
- KO:
-
knockout
- miRNA:
-
microRNA
- NHP:
-
non-human primate
- PM:
-
premutation
- POF:
-
premature ovarian failure
- POI:
-
primary ovarian insufficiency
- RAN:
-
repeat-associated non-ATG
- UTR:
-
untranslated region
- XCI:
-
X-chromosome inactivation
- WT:
-
wild type
- YAC:
-
yeast artificial chromosome.
References
Sherman SL: Seminars in genetics: premature ovarian failure in the fragile X syndrome. Am J Med Genet. 2000, 97: 189-195.
De Caro JJ, Dominguez C, Sherman SL: Reproductive health of adolescent girls who carry the FMR1 premutation: expected phenotype based on current knowledge of fragile X-associated primary ovarian insufficiency. Ann N Y Acad Sci. 2008, 1135: 99-111.
Sullivan SD, Welt C, Sherman S: FMR1 and the continuum of primary ovarian insufficiency. Semin Reprod Med. 2011, 29: 299-307.
Welt CK, Smith PC, Taylor AE: Evidence of early ovarian aging in fragile X premutation carriers. J Clin Endocrinol Metab. 2004, 89: 4569-4574.
Murray A, Ennis S, MacSwiney F, Webb J, Morton NE: Reproductive and menstrual history of females with fragile X expansions. Eur J Hum Genet. 2000, 8: 247-252.
Rohr J, Allen EG, Charen K, Giles J, He W, Dominguez C, Sherman SL: Anti-Mullerian hormone indicates early ovarian decline in fragile X mental retardation (FMR1) premutation carriers: a preliminary study. Hum Reprod. 2008, 23: 1220-1225.
Sullivan AK, Marcus M, Epstein MP, Allen EG, Anido AE, Paquin JJ, Yadav-Shah M, Sherman SL: Association of FMR1 repeat size with ovarian dysfunction. Hum Reprod. 2005, 20: 402-412.
Spath MA, Feuth TB, Allen EG, Smits AP, Yntema HG, van Kessel AG, Braat DD, Sherman SL, Thomas CM: Intra-individual stability over time of standardized anti-Müllerian hormone in FMR1 premutation carriers. Hum Reprod. 2011, 26: 2185-2191.
Hundscheid RD, Braat DD, Kiemeney LA, Smits AP, Thomas CM: Increased serum FSH in female fragile X premutation carriers with either regular menstrual cycles or on oral contraceptives. Hum Reprod. 2001, 16: 457-462.
Welt CK: Primary ovarian insufficiency: a more accurate term for premature ovarian failure. Clin Endocrinol (Oxf). 2008, 68: 499-509.
Wittenberger MD, Hagerman RJ, Sherman SL, McConkie-Rosell A, Welt CK, Rebar RW, Corrigan EC, Simpson JL, Nelson LM: The FMR1 premutation and reproduction. Fertil Steril. 2007, 87: 456-465.
Streuli I, Fraisse T, Ibecheole V, Moix I, Morris MA, de Ziegler D: Intermediate and premutation FMR1 alleles in women with occult primary ovarian insufficiency. Fertil Steril. 2009, 92: 464-470.
Allen EG, Sullivan AK, Marcus M, Small C, Dominguez C, Epstein MP, Charen K, He W, Taylor K, Sherman SL: Examination of reproductive aging milestones among women who carry the FMR1 premutation. Hum Reprod. 2007, 22: 2142-2152.
Gallagher JC: Effect of early menopause on bone mineral density and fractures. Menopause. 2007, 14: 567-571.
Kalantaridou SN, Naka KK, Papanikolaou E, Kazakos N, Kravariti M, Calis KA, Paraskevaidis EA, Sideris DA, Tsatsoulis A, Chrousos GP, Michalis LK: Impaired endothelial function in young women with premature ovarian failure: normalization with hormone therapy. J Clin Endocrinol Metab. 2004, 89: 3907-3913.
Atsma F, Bartelink ML, Grobbee DE, van der Schouw YT: Postmenopausal status and early menopause as independent risk factors for cardiovascular disease: a meta-analysis. Menopause. 2006, 13: 265-279.
Jacobsen BK, Heuch I, Kvale G: Age at natural menopause and all-cause mortality: a 37-year follow-up of 19,731 Norwegian women. Am J Epidemiol. 2003, 157: 923-929.
Mondul AM, Rodriguez C, Jacobs EJ, Calle EE: Age at natural menopause and cause-specific mortality. Am J Epidemiol. 2005, 162: 1089-1097.
van der Stege JG, Groen H, van Zadelhoff SJ, Lambalk CB, Braat DD, van Kasteren YM, van Santbrink EJ, Apperloo MJ, Weijmar Schultz WC, Hoek A: Decreased androgen concentrations and diminished general and sexual well-being in women with premature ovarian failure. Menopause. 2008, 15: 23-31.
Tassone F, Beilina A, Carosi C, Albertosi S, Bagni C, Li L, Glover K, Bentley D, Hagerman PJ: Elevated FMR1 mRNA in premutation carriers is due to increased transcription. RNA. 2007, 13: 555-562.
Kenneson A, Zhang F, Hagedorn CH, Warren ST: Reduced FMRP and increased FMR1 transcription is proportionally associated with CGG repeat number in intermediate-length and premutation carriers. Hum Mol Genet. 2001, 10: 1449-1454.
Allen EG, He W, Yadav-Shah M, Sherman SL: A study of the distributional characteristics of FMR1 transcript levels in 238 individuals. Hum Genet. 2004, 114: 439-447.
Peprah E, He W, Allen E, Oliver T, Boyne A, Sherman SL: Examination of FMR1 transcript and protein levels among 74 premutation carriers. J Hum Genet. 2010, 55: 66-68.
Garcia-Alegria E, Ibanez B, Minguez M, Poch M, Valiente A, Sanz-Parra A, Martinez-Bouzas C, Beristain E, Tejada MI: Analysis of FMR1 gene expression in female premutation carriers using robust segmented linear regression models. RNA. 2007, 13: 756-762.
Hagerman RJ, Leehey M, Heinrichs W, Tassone F, Wilson R, Hills J, Grigsby J, Gage B, Hagerman PJ: Intention tremor, parkinsonism, and generalized brain atrophy in male carriers of fragile X. Neurology. 2001, 57: 127-130.
Ennis S, Ward D, Murray A: Nonlinear association between CGG repeat number and age of menopause in FMR1 premutation carriers. EurJ Hum Genet. 2006, 14: 253-255.
Spath MA, Feuth TB, Smits AP, Yntema HG, Braat DD, Thomas CM, van Kessel AG, Sherman SL, Allen EG: Predictors and risk model development for menopausal age in fragile X premutation carriers. Genet Med. 2011, 13: 643-650.
Tejada MI, Garcia-Alegria E, Bilbao A, Martinez-Bouzas C, Beristain E, Poch M, Ramos-Arroyo MA, Lopez B, Fernandez Carvajal I, Ribate MP, Ramos F: Analysis of the molecular parameters that could predict the risk of manifesting premature ovarian failure in female premutation carriers of fragile X syndrome. Menopause. 2008, 15: 945-949.
Bione S, Benedetti S, Goegan M, Menditto I, Marozzi A, Ferrari M, Toniolo D: Skewed X-chromosome inactivation is not associated with premature ovarian failure in a large cohort of Italian patients. Am J Med Genet A. 2006, 140: 1349-1351.
Rodriguez-Revenga L, Madrigal I, Badenas C, Xuncla M, Jimenez L, Mila M: Premature ovarian failure and fragile X female premutation carriers: no evidences for a skewed X-chromosome inactivation pattern. Menopause. 2009, 16: 944-949.
Spath MA, Nillesen WN, Smits AP, Feuth TB, Braat DD, van Kessel AG, Yntema HG: X chromosome inactivation does not define the development of premature ovarian failure in fragile X premutation carriers. Am J Med Genet A. 2010, 152A: 387-393.
Hunter JE, Epstein MP, Tinker SW, Charen KH, Sherman SL: Fragile X-associated primary ovarian insufficiency: evidence for additional genetic contributions to severity. Genet Epidemiol. 2008, 32: 553-559.
Hoffman GE, Le WW, Entezam A, Otsuka N, Tong ZB, Nelson L, Flaws JA, McDonald JH, Jafar S, Usdin K: Ovarian abnormalities in a mouse model of fragile X primary ovarian insufficiency. J Histochem Cytochem. 2012, 60: 439-456.
Lu C, Lin L, Tan H, Wu H, Sherman SL, Gao F, Jin P, Chen D: Fragile X premutation RNA is sufficient to cause primary ovarian insufficiency in mice. Hum Mol Genet. 2012, 21: 5039-5047.
Ascano M, Mukherjee N, Bandaru P, Miller JB, Nusbaum JD, Corcoran DL, Langlois C, Munschauer M, Dewell S, Hafner M, Williams Z, Ohler U, Tuschl T: FMRP targets distinct mRNA sequence elements to regulate protein expression. Nature. 2012, 492: 382-386.
Ferder I, Parborell F, Sundblad V, Chiauzzi V, Gomez K, Charreau EH, Tesone M, Dain L: Expression of fragile X mental retardation protein and Fmr1 mRNA during folliculogenesis in the rat. Reprod Abstr Ser. 2013, 145: 335-343.
Entezam A, Biacsi R, Orrison B, Saha T, Hoffman GE, Grabczyk E, Nussbaum RL, Usdin K: Regional FMRP deficits and large repeat expansions into the full mutation range in a new fragile X premutation mouse model. Gene. 2007, 395: 125-134.
Peier AM, Nelson DL: Instability of a premutation-sized CGG repeat in FMR1 YAC transgenic mice. Genomics. 2002, 80: 423-432.
Peier AM, McIlwain KL, Kenneson A, Warren ST, Paylor R, Nelson DL: (Over)correction of FMR1 deficiency with YAC transgenics: behavioral and physical features. Hum Mol Genet. 2000, 9: 1145-1159.
Bontekoe CJ, Bakker CE, Nieuwenhuizen IM, van der Linde H, Lans H, De LD, Hirst MC, Oostra BA: Instability of a (CGG)98 repeat in the Fmr1 promoter. Hum Mol Genet. 2001, 10: 1693-1699.
Brouwer JR, Huizer K, Severijnen LA, Hukema RK, Berman RF, Oostra BA, Willemsen R: CGG-repeat length and neuropathological and molecular correlates in a mouse model for fragile X-associated tremor/ataxia syndrome. J Neurochem. 2008, 107: 1671-1682.
Willemsen R, Hoogeveen-Westerveld M, Reis S, Holstege J, Severijnen LA, Nieuwenhuizen IM, Schrier M, van Unen L, Tassone F, Hoogeveen AT, Hagerman PJ, Mientjes EJ, Oostra BA: The FMR1 CGG repeat mouse displays ubiquitin-positive intranuclear neuronal inclusions; implications for the cerebellar tremor/ataxia syndrome. Hum Mol Genet. 2003, 12: 949-959.
The Dutch-Belgian Fragile X Consortium: Fmr1 knockout mice: a model to study fragile X mental retardation. Cell. 1994, 78: 23-33.
Lin Y, Tang C, He H, Duan R: Activation of mTOR ameliorates fragile X premutation rCGG repeat-mediated neurodegeneration. PloS one. 2013, 8: e62572-
Spradling A, Drummond-Barbosa D, Kai T: Stem cells find their niche. Nature. 2001, 414: 98-104.
Lin H: Stem cells: to be and not to be. Nature. 2003, 425: 353-355.
Spradling AC, de Cuevas M, Drummond-Barbosa D, Keyes L, Lilly M, Pepling M, Xie T: The Drosophila germarium: stem cells, germ line cysts, and oocytes. Cold Spring Harb Symp Quant Biol. 1997, 62: 25-34.
Wong MD, Jin Z, Xie T: Molecular mechanisms of germline stem cell regulation. Annu Rev Genet. 2005, 39: 173-195.
Lin H: Cell biology of stem cells: an enigma of asymmetry and self-renewal. J Cell Biol. 2008, 180: 257-260.
Yang L, Duan R, Chen D, Wang J, Jin P: Fragile X mental retardation protein modulates the fate of germline stem cells in Drosophila. Hum Mol Genet. 1814–1820, 2007: 16-
Garcia Arocena D, Breece KE, Hagerman PJ: Distribution of CGG repeat sizes within the fragile X mental retardation 1 (FMR1) homologue in a non-human primate population. Hum Genet. 2003, 113: 371-376.
Deelen W, Bakker C, Halley DJ, Oostra BA: Conservation of CGG region in FMR1 gene in mammals. Am J Med Genet. 1994, 51: 513-516.
Eichler EE, Hammond HA, Macpherson JN, Ward PA, Nelson DL: Population survey of the human FMR1 CGG repeat substructure suggests biased polarity for the loss of AGG interruptions. Hum Mol Genet. 1995, 4: 2199-2208.
Zhong N, Yang W, Dobkin C, Brown WT: Fragile X gene instability: anchoring AGGs and linked microsatellites. Am J Hum Genet. 1995, 57: 351-361.
Zwaka TP, Thomson JA: Homologous recombination in human embryonic stem cells. Nat Biotechnol. 2003, 21: 319-321.
Urbach A, Schuldiner M, Benvenisty N: Modeling for Lesch-Nyhan disease by gene targeting in human embryonic stem cells. Stem Cells. 2004, 22: 635-641.
Ying QL, Wray J, Nichols J, Batlle-Morera L, Doble B, Woodgett J, Cohen P, Smith A: The ground state of embryonic stem cell self-renewal. Nature. 2008, 453: 519-523.
Shiozawa S, Kawai K, Okada Y, Tomioka I, Maeda T, Kanda A, Shinohara H, Suemizu H, James Okano H, Sotomaru Y, Sasaki E, Okano H: Gene targeting and subsequent site-specific transgenesis at the beta-actin (ACTB) locus in common marmoset embryonic stem cells. Stem Cells Dev. 2011, 20: 1587-1599.
Tong C, Li P, Wu NL, Yan Y, Ying QL: Production of p53 gene knockout rats by homologous recombination in embryonic stem cells. Nature. 2010, 467: 211-213.
Khan IF, Hirata RK, Russell DW: AAV-mediated gene targeting methods for human cells. Nat Protoc. 2011, 6: 482-501.
Khan IF, Hirata RK, Wang PR, Li Y, Kho J, Nelson A, Huo Y, Zavaljevski M, Ware C, Russell DW: Engineering of human pluripotent stem cells by AAV-mediated gene targeting. Mol Ther. 2010, 18: 1192-1199.
Asuri P, Bartel MA, Vazin T, Jang JH, Wong TB, Schaffer DV: Directed evolution of adeno-associated virus for enhanced gene delivery and gene targeting in human pluripotent stem cells. Mol Ther. 2012, 20: 329-338.
Bouhali K, Dipietromaria A, Fontaine A, Caburet S, Barbieri O, Bellessort B, Fellous M, Veitia RA, Levi G: Allelic reduction of Dlx5 and Dlx6 results in early follicular depletion: a new mouse model of primary ovarian insufficiency. Hum Mol Genet. 2011, 20: 2642-2650.
Kang Y, Cheng MJ, Xu CJ: Secretion of oestrogen from murine-induced pluripotent stem cells co-cultured with ovarian granulosa cells in vitro. Cell Biol Int. 2011, 35: 871-874.
Yang X, Zhou Y, Peng S, Wu L, Lin HY, Wang S, Wang H: Differentially expressed plasma microRNAs in premature ovarian failure patients and the potential regulatory function of mir-23a in granulosa cell apoptosis. Reproduction. 2012, 144: 235-244.
Hayashi K, Ogushi S, Kurimoto K, Shimamoto S, Ohta H, Saitou M: Offspring from oocytes derived from in vitro primordial germ cell-like cells in mice. Science. 2012, 338: 971-975.
Jin P, Duan R, Qurashi A, Qin Y, Tian D, Rosser TC, Liu H, Feng Y, Warren ST: Pur alpha binds to rCGG repeats and modulates repeat-mediated neurodegeneration in a Drosophila model of fragile X tremor/ataxia syndrome. Neuron. 2007, 55: 556-564.
Sellier C, Rau F, Liu Y, Tassone F, Hukema RK, Gattoni R, Schneider A, Richard S, Willemsen R, Elliott DJ, Hagerman PJ, Charlet-Berguerand N: Sam68 sequestration and partial loss of function are associated with splicing alterations in FXTAS patients. EMBO J. 2010, 29: 1248-1261.
Sellier C, Freyermuth F, Tabet R, Tran T, He F, Ruffenach F, Alunni V, Moine H, Thibault C, Page A, Tassone F, Willemsen R, Disney MD, Hagerman PJ, Todd PK, Charlet-Berguerand N: Sequestration of DROSHA and DGCR8 by expanded CGG RNA repeats alters microRNA processing in fragile X-associated tremor/ataxia syndrome. Cell Rep. 2013, 3: 869-880.
Kuyumcu-Martinez NM, Wang GS, Cooper TA: Increased steady-state levels of CUGBP1 in myotonic dystrophy 1 are due to PKC-mediated hyperphosphorylation. Mol Cell. 2007, 28: 68-78.
Qurashi A, Li W, Zhou JY, Peng J, Jin P: Nuclear accumulation of stress response mRNAs contributes to the neurodegeneration caused by fragile X premutation rCGG repeats. PLoS Genet. 2011, 7: e1002102-
Sofola OA, Jin P, Qin Y, Duan R, Liu H, de Haro M, Nelson DL, Botas J: RNA-binding proteins hnRNP A2/B1 and CUGBP1 suppress fragile X CGG premutation repeat-induced neurodegeneration in a Drosophila model of FXTAS. Neuron. 2007, 55: 565-571.
Tian B, White RJ, Xia T, Welle S, Turner DH, Mathews MB, Thornton CA: Expanded CUG repeat RNAs form hairpins that activate the double-stranded RNA-dependent protein kinase PKR. RNA (New York, NY). 2000, 6: 79-87.
Handa V, Saha T, Usdin K: The fragile X syndrome repeats form RNA hairpins that do not activate the interferon-inducible protein kinase, PKR, but are cut by Dicer. Nucleic Acids Res. 2003, 31: 6243-6248.
Samaraweera SE, O'Keefe LV, van Eyk CL, Lawlor KT, Humphreys DT, Suter CM, Richards RI: Modeling and analysis of repeat RNA toxicity in Drosophila. Methods Mol Biol. 2013, 1017: 173-192.
Ladd PD, Smith LE, Rabaia NA, Moore JM, Georges SA, Hansen RS, Hagerman RJ, Tassone F, Tapscott SJ, Filippova GN: An antisense transcript spanning the CGG repeat region of FMR1 is upregulated in premutation carriers but silenced in full mutation individuals. Hum Mol Genet. 2007, 16: 3174-3187.
Zu T, Gibbens B, Doty NS, Gomes-Pereira M, Huguet A, Stone MD, Margolis J, Peterson M, Markowski TW, Ingram MA, Nan Z, Forster C, Low WC, Schoser B, Somia NV, Clark HB, Schmechel S, Bitterman PB, Gourdon G, Swanson MS, Moseley M, Ranum LP: Non-ATG-initiated translation directed by microsatellite expansions. Proc Natl Acad Sci USA. 2011, 108: 260-265.
Todd PK, Oh SY, Krans A, He F, Sellier C, Frazer M, Renoux AJ, Chen KC, Scaglione KM, Basrur V, Elenitoba-Johnson K, Vonsattel JP, Louis ED, Sutton MA, Taylor JP, Mills RE, Charlet-Berguerand N, Paulson HL: CGG repeat-associated translation mediates neurodegeneration in fragile X tremor ataxia syndrome. Neuron. 2013, 78: 440-455.
Chang MC, DeCaro JJ, Zheng M, Gearing M, Shubeck L, Sherman SL, Welt CK: Ovarian histopathological and ubiquitin-immunophenotypic features in fragile X-associated primary ovarian insufficiency: a study of five cases and selected controls. Histopathology. 2011, 59: 1018-1023.
Greco CM, Hagerman RJ, Tassone F, Chudley AE, Del Bigio MR, Jacquemont S, Leehey M, Hagerman PJ: Neuronal intranuclear inclusions in a new cerebellar tremor/ataxia syndrome among fragile X carriers. Brain. 2002, 125: 1760-1771.
Adhikari D, Flohr G, Gorre N, Shen Y, Yang H, Lundin E, Lan Z, Gambello MJ, Liu K: Disruption of Tsc2 in oocytes leads to overactivation of the entire pool of primordial follicles. Mol Hum Reprod. 2009, 15: 765-770.
Adhikari D, Liu K: mTOR signaling in the control of activation of primordial follicles. Cell Cycle. 2010, 9: 1673-1674.
Yu J, Yaba A, Kasiman C, Thomson T, Johnson J: mTOR controls ovarian follicle growth by regulating granulosa cell proliferation. PloS one. 2011, 6: e21415-
Acknowledgements
The authors would like to thank the following funding sources: Emory's Genetics Discovery Fund (SLS); the Intramural Program of the National Institute of Diabetes and Digestive and Kidney Diseases at the National Institutes of Health (KU); National Institute of Child Health and Development and National Institute of Mental Health at the National Institutes of Health R21HD071876 for support of NHP studies (ECC) and the National Fragile X Foundation (RKH, RW).
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
Each author contributed sections of the review that relate to their specific expertise in model systems and/or FXPOI. All were involved in the writing and editing of the final manuscript. All authors read and approved the final manuscript.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Sherman, S.L., Curnow, E.C., Easley, C.A. et al. Use of model systems to understand the etiology of fragile X-associated primary ovarian insufficiency (FXPOI). J Neurodevelop Disord 6, 26 (2014). https://doi.org/10.1186/1866-1955-6-26
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1866-1955-6-26