
Abstract
The major concept of "oxidative stress" is an excess elevated level of reactive oxygen species (ROS) which are generated from vigorous metabolism and consumption of oxygen. The precise harmonization of oxidative stresses between mitochondria and other organelles in the cell is absolutely vital to cell survival. Under oxidative stress, ROS produced from mitochondria and are the major mediator for tumorigenesis in different aspects, such as proliferation, migration/invasion, angiogenesis, inflammation, and immunoescape to allow cancer cells to adapt to the rigorous environment. Accordingly, the dynamic balance of oxidative stresses not only orchestrate complex cell signaling events in cancer cells but also affect other components in the tumor microenvironment (TME). Immune cells, such as M2 macrophages, dendritic cells, and T cells are the major components of the immunosuppressive TME from the ROS-induced inflammation. Based on this notion, numerous strategies to mitigate oxidative stresses in tumors have been tested for cancer prevention or therapies; however, these manipulations are devised from different sources and mechanisms without established effectiveness. Herein, we integrate current progress regarding the impact of mitochondrial ROS in the TME, not only in cancer cells but also in immune cells, and discuss the combination of emerging ROS-modulating strategies with immunotherapies to achieve antitumor effects.
Similar content being viewed by others

Background
Reactive oxygen species (ROS) are a group of highly reactive oxygen-containing molecules generated through several mechanisms in cells, such as aerobic respiration in mitochondria, metabolic enzymes, peroxisomes, and membrane-bound NADPH oxidases (NOXs) [1, 2]. Although generated via several sources, mitochondria are the major cellular organelles of ROS production; mitochondrial ROS (mtROS) are mainly produced by the electron transport chain (ETC) and oxidative phosphorylation (OXPHOS) during aerobic respiration. The superoxide (O2 –), for example, is produced from incomplete electron transfer and leakage of electrons through ETC Complexes I, III, and IV [3, 4]. Due to the multifaceted role of ROS in cell survival and function, intracellular ROS levels must be strictly controlled to maintain the equilibrium between ROS production and scavenging through multiple mechanisms. At high levels, ROS cause oxidative damage to DNA, proteins, and lipids, and become deleterious to cells. At low to medium levels, ROS also act as a cellular signaling messenger, involved in regulating several varieties of cellular functions including gene expression, proliferation, differentiation, and stress response. In other words, the imbalance of ROS level decides the severity of the oxidative stress for either the cellular compromised or survival-associated functions. Simultaneously, ROS can be regulated and controlled by their localization within the cell, i.e., cells have several protective mechanisms against ROS via compartmentalization [5]. In cancer cells, mitochondria can also be redistributed to these regions of the cell to provide energy demands for cell migration under oxidative stress [6]. Further mechanisms to localize ROS and allow for a restricted response include the control of mitochondria turnover and localization. For example, mtROS can be eliminated by mitophagy that removes damaged ROS-producing mitochondria through targeted autophagy [7]. In addition, uncoupling of OXPHOS is involved in the control of mtROS production. During aerobic respiration, mitochondrial uncoupling has been considered as a cytoprotective strategy under oxidative stress, including inflammation, aging, diabetes, or atherosclerosis [8,9,10]. However, mitochondrial uncoupling proteins (UCPs) lower the efficiency of OXPHOS and are involved in the increase of mtROS production in cancer [11]. Mitochondrial calcium ([Ca2+]m) is another factor to upregulate the entire OXPHOS machinery, resulting in faster respiratory chain activity. [Ca2+]m coupled with proton uncouplers showed significance in promoting mtROS [12, 13]. Likewise, mtROS generated by the OXPHOS uncoupler CCCP (Carbonyl cyanide m-chlorophenyl hydrazone) is important for the Peroxiredoxin 6-induced PINK/Parkin mitophagy [14].
The imbalance of redox homeostasis is detrimental to biomolecules, cells, and even entire organism. It has been well known that cancer cells carry more ROS than their normal surrounding cells. Many pro-tumor events promote ROS production, including activation of oncogenes, loss of tumor suppressor function, changes in mitochondrial activity, adaptation to hypoxia, altered stromal interactions, fibrosis, and pathophysiology of inflammation. Some researchers have shown that superoxide-dependent oxidative stress may be involved in the pathophysiology of inflammation, fibrosis, and cancer [15, 16]. For instance, it is well demonstrated that ROS activate mitogen-activated protein kinase (MAPK) family comprising of JNK, p38, and ERK [17]. These MAPK family members function in a cellular context-specific manner, integrating signals that regulate proliferation, survival, apoptosis, and invasiveness [18, 19]. However, the consequences of ROS are very different, and ROS act as a double-edged sword in carcinogenesis, which both support and inhibit malignant behavior, a foe and friend [20,21,22]. The biological function and the therapeutic strategies of oxidative stress in cancer biology have been comprehensively described in other reviews [20, 23,24,25]. Under sustained ROS stress, it will potentially cause serious damage to cell structure and function, which also induces somatic mutation [26]. For example, ROS can damage both nuclear DNA (nDNA) and mitochondrial DNA (mtDNA), which leads to mutagenesis and elicits the metabolic reprogramming causing an increasing risk of carcinogenesis [27, 28]. mtROS damage mtDNA and causes adaptation of metabolic reprogramming, which are required for tumorigenesis. Therefore, to protect against ROS, cells develop antioxidant defense mechanisms for their elimination, which include endogenous and exogenous as well as enzymatic and non-enzymatic antioxidants. The superoxide dismutase (SOD) is the first antioxidant enzyme characterized, which can dismutate two superoxides (O2•–) into H2O2 and O2 [29, 30]. Catalase is then responsible for detoxifying the H2O2 into water. Glutathione (GSH), is another endogenous antioxidant mechanism within the cells [31]. Glutathione peroxidase (GPx) is a group of enzymes capable of reducing hydroperoxides using GSH as a substrate [32]. Regarding non-enzymatic mechanisms, mitophagy is an important form of autophagy for the selective removal of dysfunctional mitochondria and the elimination of mtROS [33]. These mtROS produced by dysfunctional mitochondria also can promote tumor development, possibly by perturbing the signal transduction adapter function of p62-controlled pathways [34]. Ironically, antioxidant defense mechanisms are also considered to show that control of increased ROS, which could promote tumorigenicity.
Since ROS can damage both nDNA and mtDNA, deregulated high ROS production in cancer cells may occur due to exogenous chemotherapy and radiotherapy (RT). Explicit role of high ROS level in cellular-intrinsic events of cancer leads to cell death and benefits the treatments of chemotherapy and RT. The elevated ROS levels in cancer have been shown to induce tumor cell death and increase sensitivity to anti-tumor therapy. In addition, growing evidences suggest that eliminating damaged mitochondria by selective autophagy is a powerful tool to control the inflammation in the immune system [35]. Therefore, the demand for the understanding of the complexity of ROS in malignancies will be key to exploring the potential of ROS-targeting therapies for cancer.
Recently, cancer biology is evolving from a 'cancer cell-centric' perspective to a systematical concept that considers cancer cells as a network of surrounding cells, which is called a tumor microenvironment (TME) [36]. The TME mainly includes tumor cells and their neighbor cells, including cancer‑associated fibroblasts (CAFs), vascular endothelial cells, and immune cells. By interacting with these neighbor stromal cells through soluble factors and signaling molecules, tumor cells have developed adaptive mechanisms to survive under various extreme conditions of the TME, such as hypoxia, higher ROS, and lower pH [37,38,39]. These stress phenotypes are common characteristics of many tumor types and so called the hallmarks of cancer [37, 40]. According to the hypothesis of ‘seed and soil’ first proposed by Paget in 1989, where tumor cells were known as ‘seeds’ and the surrounding microenvironment was known as ‘soil’ [41]. To survive under these environmental stresses, cancer cells in the TME activate the stress response, such as escape in apoptosis, angiogenesis that supplies their need for oxygen and nutrients, immunosuppression, invasion, and metastasis. ROS are associated with inflammation and cancer development as well as progression. This persistent inflammatory/oxidative environment leads to a vicious cycle that damages healthy adjacent epithelial and stromal cells, ultimately leading to carcinogenesis [42,43,44]. Furthermore, it is important to note that the TME significantly contributes to cancer development through creating an immunosuppressive environment that ultimately causes the suppression of cytotoxic T lymphocytes (CTL) response [45]. Similarly, ROS act as a double-edged sword and play a dual role in immune responses. One of ROS role in anti-cancer function is through the activation of T cells and NK cells to increase the ROS production, which allows the neutrophils and macrophages recruitment to kill cancer cells [46]. On the other hand, the elevated ROS can support cancer cells through promoting tumor-contributing immune cells, including myeloid-derived suppressor cells (MDSCs), tumor-associated macrophages (TAMs), and regulatory T cells (Tregs). In conclusion, the production and regulation of ROS levels in the TME-associated cancer and stromal cells play a decisive role in the progression of the disease. mtROS function is tightly controlled to maintain the balance through multiple mechanisms which are involved in inflammation and tumorigenesis [47]. However, extensive research is necessary to unveil the critical regulatory mechanisms driven by mtROS in tumor and tumor infiltrating immune cells for immune response in the TME. In this review, we will interpret how tumor cells process the mitochondrial ROS regulation to interact the components in the TME by different mechanisms and aspects: (1) The impact of mitochondrial ROS on the survival signaling in cancer cells; (2) The impact of mitochondrial ROS on inflammation and cancer immunoescape in the tumor microenvironment (TME); (3) The impact of mitochondrial ROS on immune cells in the TME; (4) The translational significance of mitochondrial ROS modulation in the prognosis and combination of cancer immunotherapy.
The survival signaling of mitochondrial ROS stress by chaperone in cancer cells
Intracellular ROS mainly come from dysfunctional mitochondrial respiratory chain enzyme complexes [3, 4, 48] and are crucial intermediates to trigger cellular signaling promoting and suppressing tumorigenesis [17, 21, 49, 50]. Mitochondria contribute to cellular energy metabolism, redox status, calcium homeostasis, and cell death regulation in mammalian cells. Therefore, mitochondria are the sensors of environmental stresses and responders to various stresses by regulating a series of signals to communicate with the other organelles to reduce the impact of subsequent stress damages. Several factors, such as mtDNA metabolism/damage, metabolic enzyme defects, and morphology dynamic changes, contribute to mitochondrial dysfunction in cancer cells under severe stress phenotypes. Furthermore, as the center of energy metabolism and programmed cell death, the precise harmonization between mitochondria and other organelles in the cell is absolutely vital to the survival of cancer cells [51]. Here, we specifically focus on the survival strategies in cancer cells for the oxidative stress by mitochondria (Table 1 and Fig. 1).

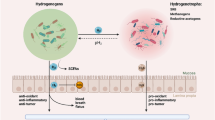
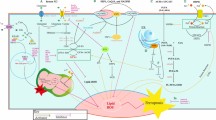
Scheme of mitochondrial ROS stress promotes cell survival and inflammation that causes an immunosuppressive tumor microenvironment (TME) to induce tumorigenesis. Mitochondria are the major cellular source of ROS generation. Mitochondrial ROS (mtROS) are mainly produced by mitochondrial aerobic respiration or as a byproduct of the activity of metabolic enzymes. Chaperone Lon is the major one of mitochondrial protein quality control system. Lon binds with NDFUS8 in the Complex I of electron transport chain and with PYCR1 reductase to up-regulate mtROS generation to promote cell proliferation and inflammation. Mitochondrial chaperone complex of HSP60-mtHSP70-Lon sequesters p53 in mitochondria matrix and stabilizes with NCLX (Na+/ Ca2+ exchanger) to restrain apoptosis and increase the cisplatin resistance under ROS stress. In addition, mtROS cause the oxidative damage on mtDNA and induce IFN signaling that upregulates PD-L1 expression to inhibit T-cell activation. Under ROS stress, cancer cells to secrete NF-κB-dependent inflammatory cytokines ( IL-6, IFN-γ, TGFβ, VEGF, IL-4, and IL-10) to cause the immunosuppressive state of macrophages, dendritic cells (DC), and T cells (Treg). Upregulation of Lon by ROS and hypoxia also induces the secretion of extracellular vehicles (EVs) that carry mtDNA and PD-L1. mtROS-induced EVs further induce the production of IFN and IL-6 from macrophages, which attenuates T-cell immunity in the TME. Macrophage-induced ROS leads to the accumulation of Treg and regDC cells. In short, mtROS cause an immunosuppressive TME to promote immunoescape, survival, and EMT/metastasis of cancer cells
Mitochondrial protein quality control in cancer cell survival
Mitochondrial protein homeostasis or protein quality control (mtPQC) is dependent on the normal function of protease and chaperone system [52]. The mtPQC system is essential for maintenance of proteostasis in mitochondria by trying to refold or by degradation of damaged proteins. Typically, the degradation of misfolded proteins is performed by ATP-dependent proteases of the AAA + (ATPases associated with a wide variety of cellular functions) family. Mitochondria contain several different AAA + proteases, LonP1, CLPP (CLP protease proteolytic subunit), and YME1L1 (ATP-dependent zinc metalloprotease) [53, 54]. The mitochondrial stress through deregulation in proteostasis generates internal imbalance which leads to mitochondrial unfolded protein response (UPRmt). UPRmt can be due to the elevated mtROS, decrease in mtDNA number or mitochondrial mass, impairment of the protein quality control system and disorder caused by oxidative phosphorylation [55, 56]. Hence, the cell activates the adaptive transcriptional regulatory response to promote the cell survival through recovery of mitochondrial function, adapting metabolism and the innate immunity. Accumulating evidence has reported that HSP60 and mtHSP70 play a chaperone role in cancer proliferation and metastasis through maintain a quality genome and assisting for refolding of unfolded and misfolded proteins [57]. The transcription factor activating transcription factor 5 (ATF5) regulates the gene expression of HSP60, GRP75 (mtHSP70) and other proteases for cancer cell survival and resistance against therapeutics and apoptosis [58, 59]. HSP60 plays an inhibitory role against cell death through its interaction with surviving and cyclophilin-D [60, 61]. Similarly, during hypoxia, mtHSP70 translocation to the outer membrane of mitochondria to interact with hypoxia-inducible factor 1 (HIF-1) leading to truncation of VDAC and thereby developing chemotherapeutic resistance by inhibiting apoptosis [62]. Also, mtHSP70 interaction with podoplanin (PDPN) regulates the growth and invasion of oral squamous cell carcinoma [63]. UPRmt-activated AAA proteases such as Lon and ClpP are responsible for maintaining mitochondrial homeostasis by removing harmful proteins [64]. Lon protease (LonP1) is a highly conserved, main, and abundant proteases located in the mitochondrial matrix. Mitochondrial Lon is a multi-functions protease as well as a stress protein which is induced by multiple stresses, such as starvation, ER, hypoxia, oxidative stress [65]. Elevated mtROS in depolarized mitochondria was suppressed through Lon-ClpP proteolytic quality control axis by degrading the Complex I ROS-generating domain [66]. Lon protease activity was increased in higher folds upon AKT phosphorylation of Lon. In addition, Lon interaction with FUN14 domain-containing protein 1 (FUNDC1) protects cancer cells from ROS accumulation through stabilizing ETC Complex II and Complex V [67, 68]. Lon capacity in proteostatic stress response to degrade the unfolded cytosolic proteins imported after mitochondrial FUNDC1 and cytosolic HSC70 interaction [69]. The common substrates of Lon and ClpP are involved in the regulation of metabolic functions including amino acid, oxidative phosphorylation (OXPHOS), and lipid metabolism [70].
Given that these stresses are commonly happened in various cancers, it is nothing remarkable that Lon is upregulated in fast-growing tumors and needed for cancer survival. Indeed, mitochondrial Lon protease upregulation has been found in many different human cancers, including non-small cell lung cancer [48, 71], malignant B cell lymphoma [72, 73], cervical cancer [74], bladder cancer [75], prostate cancer [76], colon cancer [77, 78], and oral squamous cell carcinoma cell lines [48]. Increasing evidence supports that downregulation of Lon impairs the structure and function of mitochondria to cause cell death [79, 80]. Mitochondrial Lon regulates the Complex I of electron transport chain and PYCR1 to up-regulate ROS generation to promote cell proliferation and transformation [48, 81]. As a cytoprotective chaperone, Lon interacts with Hsp60-mtHsp70 complex [82] and sequesters p53 [83] in mitochondria matrix to restrain apoptosis. A recent study also showed that the resistance mechanism by Lon interacting with NCLX inhibits excess mitochondrial calcium influx induced by cisplatin to trigger cell death [84]. Lon also participates in cysteine metabolism to repress lipid peroxidative in regulating ferroptosis [85]. In summary, these studies indicate that mitochondrial chaperone as a key factor to maintain sustaining proliferative signaling and resisting cell death in cancer cells (Fig. 1).
Mitochondrial ROS stress and epithelial mesenchymal transition (EMT) and metastasis
Several studies disclosed the chaperone activity of mitochondrial Lon and showed upregulation of Lon induced ROS generation playing a role in stress signaling [48, 81,82,83]. Lon can cooperate with other mitochondrial proteins such as NDFUS8 [48] or PYCR1 [81] to induce mitochondrial ROS generation; on the contrary, downregulation of Lon reduced mitochondrial ROS production [75]. High expression of Lon promotes the progression of tumorigenesis, such as metastasis and invasion were found in both cancer cell models [48, 78, 81, 86] and nude mice [77, 81]. In cancer cells, several signaling activation involved in tumorigenesis were referred to under control of Lon-induced ROS. For example, Ras-ERK(ERK1/2), MAPK(P38) [48, 81], and WNT (β-catenin) [78] signaling activation increased cell proliferation. Cell migration and invasion are cancer survival strategies to escape from lots of stresses in the TME. Increasing cell migration ability induced by EMT processes was through Lon-induced ROS MAPK or NF-κB pathways [48, 81].
Among cytokines releasing from Lon overexpression cancer cells, TGF-β upregulation appeared both in cancer cells and the microenvironment [81]. In most solid tumors, unlike early-stage cancers, TGF-β signaling promote a range of tumor-promoting effects. Even in cutaneous T cell lymphoma (CTCL), TGF-β mediated cell migration is regulated by NF-kB [87]. Many papers have proposed that TGF-β1 whereby different mechanisms stimulate mtROS production. [88,89,90,91,92]. Ishikawa et al. further showed that TGF-β1 induced mtROS production and underlay the activation of genes associated with EMT [89]. Previous studies also pointed out that Lon-induced ROS upregulates TGF-β through p38-NF-κB signaling [81, 93, 94], suggesting mitochondrial Lon contributes to the immune-suppressive microenvironment required TGF-β-mediated EMT and inflammatory response. In summary, the mitochondrial Lon in the matrix can interact with different proteins in the mitochondria under different stresses to regulate ROS generation and further activate downstream ROS-mediated signaling pathways to promote tumorigenesis and metastasis.
The interplay between calcium and ROS in cancer cell survival
Many physiological and pathophysiological processes were associated with calcium (Ca2+) and ROS, resourceful signaling molecules, and their mutual interplay can regulate the dysfunctional mitochondria and maintain mitochondrial bioenergetics. The relationship between calcium and ROS is mutual. Calcium is a secondary messenger that controls various cellular functions from cell signaling, secretion, metastasis, autophagy, and cell death, and Ca2+ also communicates with other systems particularly ROS [95]. Calcium regulates oxidative phosphorylation by activating enzymes isocitrate dehydrogenases, pyruvate dehydrogenases, α-ketoglutarate dehydrogenases, and ATP synthesis in mitochondria, which increases metabolic rate and thereby leaks respiratory chain electrons producing mitochondrial ROS [12, 95]. Studies on ovarian cancer denoted that intracellular ROS levels were modulated by calcium in cytosol and mitochondria under cisplatin treatment in cisplatin sensitive and resistant SKOV3 cells. Treatment of BAPTA-AM (a Ca2+ chelator) or 2-APB (an IP3R inhibitor) decreased intracellular ROS in SKOV3 cisplatin sensitive cells and protected cancer cells from apoptosis. Therefore ROS and Ca2+ mutual interplay in chemotherapy decide the fate of cells [96]. Calcium channels like voltage dependent Ca2+ channels (VDCC), Store operated channels, TRP channels, and IP3R are redox regulated because of the presence of cysteine residue in their domains [95, 97,98,99,100]. Takahashi et al., found that ROS-activated TRPA1 calcium channel increased intracellular calcium and activated calcium mediated pro-survival pathways PI3K/AKT, mTOR, RAS-ERK [101, 102].
Mitochondrial Ca2+ uptake influences cellular Ca2+ signals to generate ATP synthesis through Complex V, the F0F1 ATPase activity. Mitochondrial calcium uniporter (MCU) is the selective channel responsible for mitochondrial Ca2+ uptake leading to mtROS generation and HIF1α signaling events for breast cancer progression [103, 104]. The mtROS and total ROS generated after MCU mediated calcium uptake leading to trigger signaling events by inhibiting the NAD + /SIRT3/SOD2 pathway [105]. Under hypoxia-induced oxidative stress, mtROS generation upon mtCa2+ uptake is dependent on S-glutathionylation of MCU cysteine 97 (Cys-97) residues without any involvement of MCU regulators [106]. Like MCU, the efflux channel of mitochondria also plays a role in generating mtROS for cellular activity under hypoxia. Acute hypoxia causes the activation of NCLX through Complex I inactivation and allows the mitochondrial Na+ import/ mtCa2+ export to cytosol. This leads to a consequence to increases superoxide production at Complex III, generating a redox signal [107]. Impact of acute hypoxia-induced mitochondrial ROS activates STIM1 puncta formation and SOCE activation through HIF1α and subsequent Ca2+ signal benefits tumor cell proliferation [108].
The physical interactions between ER and mitochondria called as mitochondria associated membranes (MAMs) are hotspots of calcium regulation, which accumulate calcium into mitochondria in chemotherapy leading to cancer cell death [109]. Redox nanodomains at ER-mitochondria contact sites increase calcium influx into mitochondria and regulate calcium oscillations by IP3R channels and metabolic activities of cells [110]. ER-mitochondria contact (MAM) are enriched with the proteins responsible for Ca2+ and ROS transport between mitochondria and ER. MAMs contains ER-localized IP3R/RyR receptors, SERCA pumps, mitochondrial Voltage-dependent anion channel (VDAC), and the mitochondrial Ca2+ uniporter (MCU) in the outer and inner mitochondrial membrane [111]. To avoid the lethal cell death triggered by mtROS, cells set up the preventive mechanism to neutralize the mitochondria generated ROS through activation of mitophagy, release of mitochondrial calcium through NCLX efflux channel [84], MnSOD enzymatic dismutation [112, 113]. Taken together, it is important that, for the normal function of mitochondria Ca2+ and ROS homeostasis are very signification, a little dynamic alterations or imbalance in calcium leads to different consequences on cellular function especially during pathophysiological process. Thus understanding these interconnecting molecules related pathways would pave for discovery of novel drug therapies in diseases.
Mitochondrial ROS-induced mitophagy under hypoxic resistance
The mitophagy activation and sustainability are important for the cancer cells to develop hypoxic resistance property among other alternative pathways is depending on the severity and duration of hypoxia. This balancing act during hypoxia is strongly dependent on HIF1α, mTOR and UPR [114]. Hypoxia drives the receptor mediated mitophagy through participation of several protein components in conjunction with key regulatory receptor molecule like Bnip3-like/NIP3-like protein X (BNIP3L/NIX), Bcl-2/Adenovirus E1B 19 kDa-interacting protein 3 (BNIP3), and FUNDC1 [115,116,117]. mtROS plays a very prominent role during hypoxia for the activation of mitophagy receptor, Recently, in glioblastoma cells, hypoxia induced NIX promotes mitophagy and regulates mtROS on associating with GTPase RHEB (controls OXPHOS activity), and further regulating the mTOR/AKT/ HIF1α signaling axis [118, 119]. Contrarily, it is also suggested that hypoxic cell survival majorly relies on receptor mediated mitophagy/independent of PINK-Parkin mediated mitophagy in cancer cells depending on the behaviour of mitochondria during hypoxia. In argument to this, HEY1 and PINK1 expressions were reciprocal to each other and has shown poor clinical outcomes. HIF1/HEY axis overcomes oxidative stress through repressing PINK1 which is responsible for mitochondrial biogenesis and suppresses ROS level to make mitochondria less reliable for HCC survival [120, 121]. FUNDC1, a hypoxia specific mitophagy receptor, conducts mitophagy independent of PINK/parkin function and regulates mitochondrial homeostasis. Mitochondrial dysfunction under hypoxia allows increase in MAM formation and FUNDC1 also reported to regulate mitochondrial dynamics at the MAM region by regulating the dynamic related proteins DNM1L/Drp1 and OPA1 [122]. Recently, FUNDC1 dependent MAM formation promotes the angiogenesis in endothelial cells [123]. The hypoxia dependent receptor mediated mitophagy activation is completely dependent on the tumor heterogeneity and the resistance development property by mitophagy is dependent on the severity of mitochondrial damage pattern caused in hypoxic TME.
The impact of mitochondrial ROS stress on inflammation and cancer immunoescape in the tumor microenvironment (TME)
To survive under the ROS stress in the TME, cancer cells activate the stress response of escape in apoptosis, metastasis, and immunosuppression/immunoevasion. Cancer immunoevasion is still a great barrier in the current immunotherapy treatment. The host tumor response in context to control tumor development and progression is through the immunoediting process which are majorly staged into three steps (1) elimination, (2) equilibrium, and (3) escape [124, 125]. The cancer immunoescape gains advantage in immunosuppressive TME through developing abnormalities in antigen presenting and anti-tumor cells (T cells, Dendritic cells), developing immune resistant tumor cells or posting a blockade during T cells trafficking [126]. Emerging evidence showed that ROS are not only mediators of oxidative stress but also players of immune regulation in tumor development [127]. In this review, we will discuss how ROS and hypoxia modulate tumor and immune cells in the TME, which regulates inflammation and causes immunoescape (Table 1 and Fig. 1).
Mitochondrial ROS stress in the TME
It is generally accepted that the TME is a chronic inflammatory environment that contributes the development and progression of most tumors. There is growing evidence that the mitochondrial ROS play a "central" role in inflammatory TME that ultimately exacerbates cancer [47, 128]. Within the TME, active oncogenic signaling promotes cancer cells to autocrine and paracrine small molecular or cytokines to surrounding cells for tumor promotion. In cancer cells, elevated ROS have been shown to contribute to metastasis and angiogenesis through the secretion of inflammatory cytokines, the stabilization of HIF, and activation of AMPK signaling networks to enhance NADPH production. The ROS property in TME has been implicated with immune cell activation and suppression determining the cancer status. TME influences the PGC1α expression, an important contributor in mitochondrial biogenesis, to promote the accumulation of tumor-infiltrating T cells to resume anti-cancer activity. On the other hand, high ROS inhibits T cell responses by suppressing the formation of TCR and MHC antigen complex, which promotes cancer progression through evading immune response [129]. Previously, the mechanisms of how oxidative stress modulates chronic inflammation-induced carcinogenesis from the TME point of view described in other reviews [5, 47, 130]. In this review, we mainly discuss the impact of mtROS stress on the TME, including cancer cells and various types of immune myeloid cells (Fig. 1).
The cancer cells attempt to evade the anti-tumor surveillance system which are termed as adaptive immune resistance. To avoid immune destruction, tumor cells hijack the physiological immune response caused by the activated T cells. mtROS are used by cancer cells and immunosuppressive immune cells to create immune tolerance to tumors [69,70,71,72,73,74,75,76]. The mitochondrial Lon has been shown to regulate the balance of mtROS production through cooperation with different proteins in the mitochondria [48, 66, 81,82,83, 131, 132]. Moreover, the mitochondrial Lon-induced mtROS-NF-κB axis stimulates inflammatory cytokines releasing from cancer cells to establish immune suppression of the TME [81]. Interestingly, mitochondrial Lon promotes tumorigenesis in an NF-κB-dependent manner, but Lon expression is also repressed by IκB kinase (IKK) inhibitor VII (IKKi7) [81]. Among ROS-induced inflammatory signaling, NF-κB is constitutive activation in many different types of cancer and promotes a variety of inflammatory factors [133, 134]. Another well-known signaling that responds to ROS induction in mediating tumorigenesis is MAPK cascades [135]. ROS-induced MAPK activation can also regulate NF-κB signaling to promote inflammatory factors secretion, such as IL-1β, IL-6, and TNFα [133, 136, 137]. Kuo et al., also especially pointed out that NF-κB and MAPK promoting inflammatory cytokines section, such as IL-6 and VEGF are regulated by mitochondrial Lon-induced ROS [81]. Another hypoxic factor, HIF1α can assist a series of kinase cascades activation leading to STAT3 to promote IL-6 secretion [138]. In addition, HIF1α is a key stimulator to induce upregulation of mitochondrial Lon that generates ROS [48]. A recent study also indicated that under cisplatin treatment, IL-6 secretion promoted by STAT3 signaling is dependent on Lon-induced increase in intracellular ROS and calcium [84]. Therefore, the positive feedback of Lon-induced ROS via NF-kB axis enhanced downstream signaling to promote tumor progression.
ROS exert a significant impact on the expression of programmed cell death protein 1 and programmed cell death-ligand 1 (PD-1 and PD-L1). The variable PD-L1 response to ROS modulation reflects the complexity of ROS biology in the TME. Through binding with PD-1, an inhibitory immune checkpoint receptor expressed on activated immune cells, PD-L1 (B7-H1) on tumor cells attenuates the effector function in dendritic cells [139] and macrophages [140]. In addition, HIF-1α contribution to positive PD-L1 expression is ROS dependent and this accompanies the infiltration of tumor supportive immune cells, myeloid-derived suppressor cells (MDSCs), regulatory T cells (Treg), and tumor-associated macrophages (TAMs) [141]. TAMs integrate the multiple molecular links between ROS and PD-L1. Elimination of ROS through redox-active drug MnTE-2-PyP5 + selectively inhibits M2 macrophage polarization and pro-tumorigenic function [142, 143]. Treg cell apoptosis triggered by oxidative stress is a novel tumor immune-evasion mechanism in the TME. Apoptotic Treg cells efficiently convert ATP into immunosuppressive adenosine via CD39 and CD73 in vitro and in vivo [144, 145]. MDSCs often represent the major producer of oxidizing species in the TME which undergoes massive expansion during tumor progression. ROS through MDSCs shows immune suppression capacity through modifying TCR and CD8 channels, leading to the antigen specific tolerance of peripheral CD8 + T cells upon CD8 + T cells losing their ability to bind phosphorylated MHC [146]. MDSCs suppress T cell proliferation also through increasing the production of Arginase-1, nitric oxide (NO), peroxynitrite (cytotoxic to T cells) and by inducing Treg cells and TGF-β secretion [147,148,149]. By focusing on how the interplays between cancer cells and immune cells influence the redox status in the TME, we will highlight the therapeutic potential of the rational combination of mtROS-modulating agents with cancer immunotherapies.
Hypoxia-induced mitochondrial ROS stress in the TME
Hypoxia is a prominent feature of the TME of solid tumor and is considered a major factor driving adaptation toward host immunosurveillance evasion. The key molecular mechanism by which cells adapt to hypoxia is through transcription factor, HIF [150]. HIF transcriptionally activates a wide repertoire of genes that promote tumor growth and metastasis. HIF-1 is particularly crucial for shifting the metabolic program of cancer cells from oxidative phosphorylation to glycolysis. Hypoxic tumor, in contrast to non-malignant tissue, to support their energy demands depends more on the anaerobic glycolysis where the final product pyruvate metabolized to lactate to restrict the OXPHOS activity [151].
Contrarily, Gisbergen et al. showed that decrease in OXPHOS results in reduced stabilization of HIF-1α and its downstream targets including carbonic anhydrase IX (CAIX], VEGF [152]. Similarly, there are lot of hypoxic factors influences mitochondria stress phenotype activation and contributes to cancer adaptation and resistance. Developing evidences indicate that ROS produce oxidative stress and regulates immune response for tumor development. [153]. Hypoxia induces production of mtROS through the mitochondrial complex I dysfunction in the inner mitochondrial membrane and the concomitant activation of the mitochondrial Na + /Ca2 + exchanger, NCLX [154]. Hypoxia stabilizes HIF-1α by forming a dimer with HIF-1β which are supported by mtROS production triggering hypoxia-responsive genes to increase angiogenesis [123]. On the other hand, the existence of mtDNA was found to be a mediator of HIF1α and DRP1 relationship under hypoxia in eliciting metabolic reprogramming, and mitochondrial biogenesis in neuroblastoma cells additionally influenced by ROS generated in cytosol [155]. Similarly, in transitional cell carcinoma (TCC), mtDNA amplification under hypoxia alleviates cisplatin induced mitochondrial oxidative stress damage to mtDNA by lowering the mitochondria ROS level and improved the mitochondrial ultra-structural changes resulted in cisplatin resistance [156]. Interestingly, the serum of rectal cancer patients also showed the lower ratio of ROS (reflecting hypoxic tumor) to high cell-free mtDNA damage contributes to systemic inflammation and poor histologic tumor response to neoadjuvant radiotherapy [157]. The intracellular/extracellular transport of the mtDNA is reportedly possible through the extracellular vesicles (EVs) [158] which helps the mtDNA to trigger pro-inflammatory cytokines leading to its own degradation [159]. mtDNA release into cytosol upon elevated ROS also reported to trigger T cell inhibitory function for cancer progression. Hypoxia triggers abundant EV secretions and hypoxia tumor cells derived exosomes contains many mitochondria derived immunosuppressive components and chemokines contributing to tumor progression through macrophage differentiation [160].
The impact of mitochondrial ROS on T cell, macrophage, and dendritic cell (DC) in the TME
Excessive ROS production leads to chronic inflammation, which is one of the environmental factors that help tumor immunosuppression [161]. Antagonism between immune cells and ROS requires tightly controlled feedback mechanisms to avoid excessive ROS formation [162]. For instance, the ROS levels in NK and T cells need to be delicately controlled to avoid the hazardous effects of high levels of ROS. Yang et al. report that NK cells primed by IL-15 acquire resistance against oxidative stress through the thioredoxin system, and have benefit in protecting other lymphocytes within the TME [163]. It has been well-studied that mild ROS levels are required for proper T cell activation and differentiation, but high and excessive level of ROS upregulates Fas expression and downregulates anti-apoptotic Bcl2 expression to promote T cell apoptosis [164]. On the other hand, proper levels of ROS are needed for the function of antigen-presenting cells. It has been reported extracellular ROS alter the immunogenicity of antigenic peptides, altering T cell priming [165]. Immunogenic cell death (ICD) leads to exhibition and secretion of damage-associated molecular patens (DAMPs), including adenosine triphosphate (ATP), ER protein calreticulin, and nuclear heat-shock protein high mobility group box 1 (HMGB1). These DAMPs interact with their receptors on DCs, leading to DC activation and ultimately antitumor T cell responses [166, 167]. A study shows that scavenging of extracellular ROS using tumor ECM-targeted nanomaterials preserves the stimulatory activity of HMGB1 and restores ICD-induced antitumor immunity [168]. Furthermore, neutrophils, macrophages and MDSCs are known to produce high amounts of ROS to kill tumor [169, 170]. These findings suggest that the level and duration of ROS determine whether ICD occurs and leads to effective antitumor immunity [171, 172]. This is further supported by a study demonstrating that GSH-deficiency in Tregs leads to increased serine metabolism, mTOR activation, and proliferation, resulting in diminished Treg suppressive function in vitro and in vivo [173].
Excluding cancer cells, macrophages are the most immune cells population circulating in the TME that maintain immune homeostasis. In TME, cancer cells remodel the peripheral and distant microenvironment by secreting tumor-derived factors which can stimulate local and circulating monocytes and macrophages to activate and accelerate tumor progression. Macrophages are stimulated by the cytokines secreted by cancer cells, which polarize macrophages with different functions [174]. Macrophages are broadly divided into two categories: classical M1 and alternative M2 macrophages [175,176,177]. ROS are involved in pro- and anti-inflammatory macrophage phenotypes by contextual fashion [178]. ROS induce some macrophage programming signaling pathway to affect macrophage polarization [179]. For example, M1 macrophage through Nox2 produces ROS to activate NF-κB stimulating phagocytosis [180], but the high level of ROS was harmful to macrophage [181]. The macrophages exhibit similar functions as M2 macrophages that secrete many cytokines, chemokines, and proteases to promote tumor growth, metastasis, angiogenesis, and immunosuppression and they are commonly termed tumor-associated macrophages (TAMs) [182,183,184]. ROS-induced signaling pathways that promote inflammatory factors secretion in macrophages are well-validated, but little literature mentions the role of mitochondrial chaperone in mtROS induction and macrophages. A recent study showed that mitochondrial Lon is upregulated in differentiation macrophages compared with monocytes and preferential higher in M2-like differentiation macrophages both in vitro and in vivo [81]. This result provides evidence that ROS-induced by Lon in macrophage may play an important role in TAMs differentiation. These results indicated several signaling pathways response to mitochondrial ROS-induced by Lon to promote inflammatory factors present in the TME and contribute to M2d macrophage (TAMs) polarization [185, 186]. For a long time, ROS have been considered as harmful metabolites of mitochondria [187]. More recently, evidence has shown that mtROS are signaling molecules necessary to prevent excessive immune responses, and this concept has also been extended to immunity, in particular to the function of macrophages in which cells predominate [188].
Another group of immune cells that initiate and control immune responses are DCs that can be differentiated from monocytes during inflammation [189]. DCs are crucial for eliciting anti-tumor immunity, due to their ability to present antigens and activate T cells. This antigen-identification process is done by pathogen-recognition receptors (PRRs), like toll-like receptor (TLRs). Starting from immature DCs (iDCs), acceptance of different stimuli guides iDCs to two different physiological types. Once accept proinflammatory cytokines or TLR ligands, LPS, IL-6, would lead to CD80, CD86, and IL-6 expressing mature DCs that drive effector T cell response [190]. While receiving regulatory factors, IL-10, TGF-β, vitamin D3, and corticosteroids, iDCs would become tolerogenic DCs, so-called regulatory DCs (regDCs) that express IL-10, indoleamine 2,3-dioxygenase (IDO), and PD-L1 then dampen effector T cell differentiation or activate Tregs [190]. The differentiation of regDCs and myeloid-derived suppressor cells (MDSC) by TAM promotes the immunosuppressive TME [191,192,193]. TGFβ and IL-10 secreted by cancer cells and TAMs also inhibit antigen presentation and adaptive immune response promoted by DCs [194,195,196]. Accumulating evidences have shown that ROS around the TME promote cytotoxicity or immunosuppressive effect of immune cells [197,198,199], and this issue is based on quantity of ROS in the TME [200, 201]. Moreover, prolong exposed under ROS microenvironment is thought to lead to a chronic inflammatory condition [198]. Diverse inflammatory environments decide antigens cross-presentation capability among DCs and T cells [202]. As a crucial signaling factor in the TME, different levels of ROS may provide some perspectives to elucidate the DC activation state. Various tension of ROS stress levels may result in different terminal outcomes of DC maturation status [202, 203]. Both DCs and T cells showed elevation of intracellular ROS during antigen presentation. This DC-T cell communication was thus inhibited by ebselen, a selenium-containing antioxidant [204]. However, in the dark side of the moon, some cases found that elevated levels of ROS hamper DC cross-presentation and following pro-inflammatory functions. One study showed that mtROS elevation in aged murine DCs obstructed later step of antigen presentation to T cell, this disruption of DC was mitigated after treating with ROS scavenger [205].
Mitochondrial ROS-induced mtDNA leakage/EV contributes to inflammation and PD-L1-mediated immunoescape
Although our body has strategies to inhibit or kill cancer cells, cancer cells have developed several ways to escape the killing. First, low expression of major histocompatibility complex (MHC) molecule of cancer cells makes cancer cells escape recognition from the immune system. Second, cancer cells gain the stress phenotypes and try to survive under hypoxic and highly oxidative stress of the TME in which immune surveillance will be suppressed. Third, suppression of immune surveillance by releasing inhibitory cytokines (e.g. TGF-β) into the TME and by inhibiting metabolic energy supply. Fourth, activate the immune checkpoint to inhibit the activity of T cells. Immune checkpoints are regulators of the immune system, which are crucial for self-tolerance and prevent the immune system from attacking cells indiscriminately. However, cancers are able to protect themselves from attack by stimulating the immune system.
As signal mediators, ROS also serve a key role in the immune monitoring of regulatory (Tregs) and effector T cells, which rely on toll-like receptors, and perception of the metabolic microenvironment [206]. Prolonged ROS production is considered to lead to chronic inflammation, and inflammatory cytokines and signaling pathways, such as NF-κB and TGF-β, are induced to cause cancer formation and progression [207]. Numerous reports indicated that ROS stress enhances DAMPs production, and mtDNA is pivotal for mitochondrial DAMPs. Due to the bacterial origin of mtDNA which can stimulate innate immune systems including TLR9, NLR family pyrin domain containing 3 (NLRP3), and cGAS-STING signaling pathways in the mammalian cells [158, 208]. ROS promote the mtDNA leakage from mitochondria through more than one mechanism including (1) Bax/Bak pores, (2) VDAC oligomers, (3) mitochondria permeability transition, (4) altered mitophagy, (5) mitochondrial dynamics, and (6) extracellular vesicles [209]. These suggest the extracellular/intracellular release of damaged mtDNA has some physiological role in response to tumor-induced mitochondrial stress. Bao D et al. have established the significance of cytosolic mtDNA stress in cancer progression after DRP1-induced mitochondrial dysfunction leading to tumor-associated macrophage infiltration through HCC secretion of CCL2 by TLR9-mediated NFκB signaling [210]. Their following recent work reported the activation of cGAS-STING signaling contributing to cytosolic mtDNA stress-induced autophagy in esophageal squamous cell carcinoma (ESCC) [211]. Previous evidence also show that oxidative stress can promote the damaged mtDNA escape to the cytosol to upregulate the expression interferon-stimulated genes (ISGs) and activate the interferon (IFN) signal pathway [131, 212,213,214]. The type II IFNγ is a pleiotropic cytokine with numerous effects on the innate and adaptive immune systems due to the broad expression of its receptors on immune cells [215]. IFN-γ augments the cytotoxic function of NK cells and CTLs to inhibit carcinogenesis [216]. On the contrary, IFN-γ induces many genes involved in cancer cell immunoevasion, such as PD-L1 and CTLA-4, stimulating immune-suppressive mechanisms [217, 218]. Cheng et al. discovered that mitochondrial ROS can promote the immunosuppression gene such as PD-L1 and IDO expression through the STING-IFN axis in various cancers [131]. It has also been reported that ROS-induced PD-L1 expression in macrophages and PD-L1 blockade revert this effect and synergizes with paclitaxel to diminish breast cancer [219]. In addition, mtDNA is vulnerable to damage by ROS and the mtDNA mutations play a role in chemotherapy resistance [220, 221]. The cisplatin resistant cancer cells showed mtDNA mutations and elevated ROS thereby activating NF-κB mediated inhibitor of apoptotic proteins and Ca2+-dependent inflammation [84, 222].
Notably, ROS stress can induce the secretion of extracellular vehicles (EVs), which carry mtDNA and PD-L1 to remodel the environment around cancer tissues [131, 223]. ROS-induced EVs further enhance the production of IFN and IL-6 from macrophages, which attenuates T-cell immunity in the TME (Fig. 1). Recent reports indicate that patients with various cancers have an increased level of exosomal PD-L1 which positively correlates with mtDNA and IFN-γ production [131, 224]. The constitute secretion of mtDNA and proteins into EVs is the important phenomenon mutually developed between the cells. The transported materials triggered many cellular events including inflammatory responses favoring or against the pathophysiological process. In the last decade, the extrusion of mitochondrial components to the EVs was reported through the newly included mitochondrial quality control (MQC) pathway called mitochondria-derived vesicles (MDV) which significantly contributes to the organelle homeostasis depending on the severity of dysfunction in mitochondria [225]. The MQC systems help to recover the vital functions of mitochondria. The mechanism of mitochondria-lysosome contact was recently included and considered as one among the MQC involved in cross-talk to deliver components into EVs [226]. MDVs are generated depending on the cargo molecules including proteins and nucleic acids which are limited to one or include cargos from many different compartments of mitochondria [227, 228]. It is more reasonable to consider the fact of MDVs role in eliciting an immune response by allowing oxidized mtDNA to enter the endo-lysosomal pathway and secreted to the extracellular space through exosomes and triggering many inflammatory and anti-inflammatory regulatory pathways [229]. In cancer, although many reported EVs with mtDNA are a critical component affecting the metabolic output and progress the tumor growth, the actual mechanism in MDVs governing the mtDNA transport to EVs is still uncertain. Overall, the MDV dependent MQC mechanisms are important for the cell in achieving both survival and inflammatory properties. More investigations on the biogenesis pathways of MDVs will open a new platform to understand the selection of cargo uptake and its contribution over mitochondrial homeostasis regulation.
The translational significance of mitochondrial ROS modulation in the prognosis and combination of cancer immunotherapy
In general, the level of ROS in cancer cells is typically higher than their normal surrounding cells. Redox homeostasis in cancer cells can be disrupted by enhancing ROS production or reducing ROS scavenging by inhibiting the antioxidant system. Here, we will focus on the translational and clinical significance of ROS modulation that combines chemo/radiotherapy and immunotherapy against the survival strategies of cancer cells (Table 2).
ROS-induced mutational mtDNA, one of the important cellular stresses, can directly regulate the delivery of signal components into the cytoplasm, resulting in mitochondrial retrograde signaling pathways that affect the nuclear gene expression and mitochondrial metabolites to cellular injury. Exosomes, one type of EVs and ranging in size from 30 to 150 nm, act as a medium of cell-to-cell communication to deliver the cargo, including RNA, DNA, proteins, lipids, mitochondria, and mtDNA, to the receptor cells. [229,230,231]. The secretion of exosomes or EVs indirectly changes the mitochondrial function through the uptake of cargo by the receptor cells such as tumor cells or immune cells [229,230,231]. The production and composition of EVs affect the oncologic settings, where their concentration is frequently higher in the blood of cancer patients when compared with healthy control [232]. For example, studies involving several cancers showed that tumor-derived exosomes can induce tumor cell proliferation [131, 233,234,235,236]. Due to the difference of carrying molecules from origin tumor cell to the peripheral circulation, increasing studies have been described that EVs are as sources of tumor biomarkers in liquid biopsies [237]. Therefore, it seems like EVs should be used to evaluate mtDNA as a biomarker candidate of mitochondrial DAMP. The purpose of liquid biopsy testing is to achieve personalized treatment by identifying the biomarkers of specific physiological and pathological conditions of patient blood. By analyzing EVs/exosomes, liquid biopsy can be used for early diagnosis and subsequent monitoring of disease through simple biosomal fluid testing [238, 239]. Today, the EVs are one of the most exciting and rapidly evolving areas of cancer research in biological fluids. It is recognized that EVs are involved in cell-to-cell communication and are involved in the development of cancer disease. The functionality of the EV makes it ideal for biomarkers based on liquid biopsy. Since EVs represent a mirror of the tissue-specific physiological and pathological condition [240,241,242], their cargos, RNA and DNA produced from nuclease degradation, can be used for early diagnosis and subsequent monitoring of disease through simple biosomal fluid testing. Therefore, EVs can help physicians choose the best treatment for each patient at all stages of the tumor disease. Taken together, exosomal PD-L1 and mtDNA can serve as a biomarker candidate for cancer diagnosis, prognosis, and cancer immunity therapeutic response [243].
The various antioxidants have been tested as chemo-preventive agents based on the rationale that ROS scavenging can reduce cancer incidence or delay cancer progression [244]. There are also other studies showing that overexpression or targeted catalase, and delivery of SOD or GSH can inhibit tumor growth [245,246,247]. An early study also demonstrates that administration of the antioxidant NAC suppresses tumor incidence in mice by inhibiting HIF1a-driven tumor growth [248]. But several large-scale clinical trials of dietary antioxidant supplements such as vitamin A, vitamin E, and beta-carotene failed to demonstrate significant antitumor benefits [249, 250]. In some cases, antioxidant supplements even increase the risk of certain cancers [251, 252]. Possible reasons for the unexpected failure of antioxidant approaches include insufficient tumor-promoting ROS scavenging efficiency in mitochondria, and/or interfering with the antitumor effects of ROS in cancer cells or stromal cells in the TME [253,254,255]. Treatment with paclitaxel can induce extracellular ROS which cause cytotoxic effects to the bystander cancer cells to lethal damage [256]. Paclitaxel also has cytostatic effects to inhibit angiogenesis [257]. A strain of Salmonella typhimurium (VNP20009) has been shown to target and replicate in hypoxia and necrotic areas within tumors with anti-tumor activity in different tumor models [258, 259]. Later, when VNP20009 synergic combined with an anti-angiogenesis inhibitor, endostatin which has no significant anti-tumor effect alone, this strategy significantly enhances therapeutic effects on tumor progressions and normalizes vessels [260].
Numerous studies have demonstrated that chemotherapeutic agents exert tumor-killing effects by generating free radicals that cause irreversible cell damage [261, 262]. Cisplatin, a widely used platinum-based chemotherapy, is known to induce tumor cell apoptosis involving the induction of superoxide but is largely DNA damage-independent, an effect that can be abolished by superoxide scavengers [263]. 5-Fluorouracil, an antimetabolite that interferes with DNA synthesis for the treatment of colon cancer, head and neck cancer, and other solid tumors, induces tumor cell apoptosis by inducing mtROS, and this effect can be inhibited by mitoQ, serves as a mitochondrial-selective antioxidant. [264]. Some chemotherapeutic agents such as taxanes (paclitaxel and docetaxel) and vinca alkaloids (vinblastine and vinblastine) induce the production of superoxide radicals and induce cell death [265, 266]. In addition to chemotherapeutic drugs, ionizing radiation can trigger tumor cell apoptosis through ROS induction and release of mitochondrial cytochrome c [267, 268]. Recently, some new prodrugs have been developed as DNA cross-linkers or ROS-activated alkylating agents. For example, leinamycin (LNM) is a potent antitumor antibiotic produced by Streptomyces atroolivaceus S-140. LNM E1 can be activated by cellular ROS oxidation to generate an intermediate with DNA alkylation activity, which exhibits strong cytotoxicity against prostate cancer cell lines with elevated ROS levels [269]. However, Wang et al. reported that fibroblasts facilitate platinum-resistance in ovarian cancer cells by modulating ROS in the TME [270].
In the chronic inflammation TME, tumor cells would balance the lethal level of ROS through regulating the several protective signaling pathways described above as survival strategies. When suffering from hypoxia and nutrient-deprived conditions, the tumor and surrounding stromal cells and endothelial cells begin to secrete pro-angiogenic factors, such as VEGF, angiopoietin, platelet-derived growth factor (PDGF), transforming growth factor beta (TGF-β), fibroblast growth factor (FGF), and some growth factors promotes angiogenesis [271]. Abnormal tumor vasculature is one of the major mechanisms of signaling imbalance induced by pro- and anti-angiogenic molecules [272]. The blood vessels in the TME are very chaotic, complex, irregular, and leaky, resulting in the inability of intratumor hypoxia to deliver antitumor drugs normally. As angiogenesis is the crucial process of tumor progression, targeting angiogenesis is a desirable anti-tumor therapy. Although anti-angiogenesis therapy of Bevacizumab (Avastin) has achieved great success in different cancer treatments, however, anti-angiogenesis is not efficacious as expected because a lot of patients showed resistance to anti-angiogenic therapy. Many papers have proved that antiangiogenic therapies destroy the tumor vasculature, causing intratumoral hypoxia that will promote tumor recurrence and metastasis [273,274,275,276,277,278]. This reflection indicated that complete inhibition of tumor angiogenesis may not a perfect therapeutic strategy. Abnormal tumor vasculature affects immune cell infiltration through the synthesis of pro-angiogenic factors VEGF and ANGPT2, and promotes TME-mediated immunosuppression [279]. Excessive VEGF in the TME can promote immunosuppression in several ways, such as: regulating T cells to inhibit CTL function [280], inhibiting DC antigen presentation and maturation hindering T cell activation [281], promoting immunosuppressive cells Treg cells, MDSCs and M2 TAM recruitment and proliferation [282]. In 2005, Jain first raised a postulate an emerging concept that “normalize” the abnormal structure and function of tumor vasculature as the anti-angiogenesis therapy [278]. Conceivably, this strategy may alleviate oxidative and hypoxic stresses in the TME and promote the regular vascular formation, immune cells infiltration, and drug delivery into the tumor. However, vascular normalization monotherapy met several challenges, such as the detailed functional mechanisms, the window of normalization monitoring, and time-consuming initiation with short-lived maintenance [278]. The combination idea of anti-angiogenesis and cytotoxic therapies was first postulated by Teicher with many clinical data supported afterward [283]. Vascular normalization has the potential to promote improved efficacy of immunotherapy, and restoring vascular normalization reduces interstitial fluid pressure and improves tumor perfusion, creating a positive feedback loop that not only increases immune cell infiltration within tumors, but also increases oxygen and supply of nutrients to achieve a good therapeutic effect [284, 285]. Therefore, normalization of tumor blood vessels is one of the approaches to solve cancer immunotherapy.
Since immune checkpoint inhibitors, e.g., anti-PD-1, have response rates of only 10–30% in solid tumors because of the immunosuppressive TME. Manipulating the TME therefore is more beneficial for controlling the progression of tumors and reverse the resistance of immunotherapy. Over the past decade, an increasing number of studies have revealed that regulation of the levels of ROS can exert anti-tumor effects by acting on the TME [81]. The combination of metformin with PD-1 blockade enhanced intratumor T cell activation and proliferation, leading to tumor clearance. This observation suggests that non-responders to PD-1 antibodies may have high mROS and more hypoxic microenvironment, which results in compromised T cell response. A study report that adoptive T-cell therapy (ACT) can significantly altered tumor metabolism, leading to GSH depletion and accompanying accumulation of ROS in tumor cells [286]. Some therapeutic molecules, including chemotherapeutics and anti-PD-L1 antibodies, can be delivered to and released within tumor cells or TME via ROS-responsive prodrugs or nanoparticles, thereby inhibiting tumor cell growth in vitro and in vivo [287,288,289].
Another burgeoning strategy is nanomaterial which can compose of different drugs and be target-specific delivery. To combine with other therapy, such as immunotherapy, many nanoparticles were designed to modulate the level of extracellular ROS to align the immunosuppressive microenvironment. Deng et al. created a nanoscavenger that can be delivered to the low pH microenvironment and anchor to ECM to release drugs to inhibit extrinsic ROS and enhance immunotherapy [168]. The nitric oxide (NO) releasing particles, NanoNO not only normalizes tumor vessels to reprogramme the immunosuppressive tumor microenvironment but also potentiates anti-cancer therapies [290]. A promising therapeutic strategy by dual-targeting particle targeting mTOR efficiently arrest tumor growth by reducing metabolic stresses, repolarized TAMs, inhibiting angiogenesis, reprogramming immune cells [291]. In short, the most important issue is how to deliver the nanoparticle to the target site. Therefore, the top strategy to design a nanoparticles based combination therapy is required to induce the normalized intratumoral vessels to improve immunosuppressive TME.
Conclusions and perspectives
Cancer is a disease caused by abnormal cell growth and uncontrolled cell death with an ability to spread to other distant tissues. The point of view of cancer research is evolving from a 'cancer cell-centric' perspective to consider tumor as a network of surrounding cells, called a TME. The TME not only includes tumor cells but also their neighbor cells, including CAFs, vascular cells, and immune cells. With extravagance growth, some of stress phenotypes detected in the TME are genome instability (replicative and mitotic stress), hypoxia (metabolic stress and sustained angiogenesis), and the increasing level of ROS (metabolic and mitochondrial stress). ROS act as a double-edged sword in carcinogenesis, which both support and inhibit malignant behavior and the evolution of cancer. ROS produced either by tumor cells or by the TME cells have very diverse effects depending on their level, location, and regulation.
Mitochondria are the major cellular source of ROS production, and mitochondrial ROS (mtROS) are produced during aerobic respiration or as a byproduct of metabolic enzymes. Mitochondria take important roles in cell survival as they contribute to various cellular functions, including ATP production, apoptosis, calcium signaling, mitophagy, and signaling through mtROS. Here, we focus on the impact of mitochondria and mtROS on cancer and immune cells in the TME with their relevance to cancer immunotherapy. Many studies have identified that mitochondrial Lon-ROS promote abnormal cell proliferation, migration, angiogenesis, resistance towards apoptosis, and inflammation. By mtROS-stimulated angiogenesis, migration, and the secretion of inflammatory cytokines and mtDNA/EVs, cancer cells interact with different components in the TME to escape from the immunosuppressive microenvironment.
Two opposing strategies have been attempted to modulate tumor redox as a way to prevent or treat cancer. One approach is to reduce the pro-tumor effects of ROS by reducing oxidative stress with antioxidants. Another approach is to increase cancer cell death by increasing ROS levels in cancer cells. Although many questions remain unanswered, but we know that the effects of ROS-modulating therapies will vary largely depending on ROS level, location, and stage of cancer progression. High levels of ROS induce cellular damage or even cell death. Low to moderate ROS levels promote cell proliferation, EMT, angiogenesis, and inflammation. The increased ROS from cancer cells and various types of myeloid cells in the TME is a characteristic of chronic inflammation, which is intimately involved in cancer development and progression. ROS in the TME are used by cancer cells, immunosuppressive macrophages, and DCs to create an immune tolerance environment for tumors, dampening the outcome of antitumor immunotherapy.
Although current mainstream of cancer therapies is still surgery, chemotherapy, or targeted therapy, the new concept of cancer therapy is trying to keep the tumor in the "hot" state for immunotherapy and to find the weakness of the non-oncogenic addiction for cancer cell survival, avoiding metastasis and recurrence. Combination therapies of the emerging ROS-modulating strategies and cancer immunotherapy enhanced the antitumor effects. Moreover, with help of vessel normalization will mitigate the excess ROS level and hypoxic resistance, which provides a route to drug delivery and immune cells. In summary, the equilibrium of ROS stress in the TME and the immunosurveillance function will optimize the window to enhance the therapeutic efficacy of immunotherapy for eradication of tumors. Rational combination of ROS-modulating agents and immunotherapy is emerging as a promising strategy of cancer treatment. Further research is needed to provide insights on the role of ROS modulators in an immunosuppressive TME to avoid the immunoescape and further recurrence and progression of cancer.
Availability of data and materials
Not applicable.
Abbreviations
- ACT:
-
Adoptive T-cell therapy
- ATP:
-
Adenosine triphosphate
- AMPK:
-
AMP-activated protein kinase
- ANGPT2:
-
Angiopoietin 2
- BAX:
-
Bcl-2-like protein 4
- Bcl-2:
-
B-cell lymphoma 2
- BNIP3:
-
BCL2/adenovirus E1B 19 kDa protein-interacting protein 3
- CAFs:
-
Cancer-associated fibroblasts
- CCCP:
-
Carbonyl cyanide m-chlorophenyl hydrazone
- CD73:
-
Cluster of Differentiation 73
- cGAS:
-
Cyclic GMP-AMP Synthase
- CTL:
-
Cytotoxic T lymphocytes
- CTLA-4:
-
Cytotoxic T-lymphocyte-associated antigen 4
- CuZnSOD:
-
Copper/Zinc superoxide dismutase
- DAMPs:
-
Damage-associated molecular patterns
- DC:
-
Dendritic cell
- DRP1:
-
Dynamin-related protein 1
- ECM:
-
Extracellular matrix
- EMT:
-
Endothelial mesenchymal transition
- ER:
-
Endoplasmic reticulum
- ERK:
-
Extracellular signal-regulated kinases
- ESCC:
-
Esophageal squamous cell carcinoma
- ETC:
-
Electron transport chain
- EVs:
-
Extracellular vesicles
- FGF:
-
Fibroblast growth factor
- FUNC1:
-
FUN14 domain-containing 1
- GPx:
-
Glutathione peroxidase
- GRP-75:
-
75-KDa Glucose-regulated protein
- GSH:
-
Glutathione
- HIF-1:
-
Hypoxia-inducible factor-1
- HMGB1:
-
High mobility group box 1
- HEY1:
-
Hairy/enhancer-of-split related with YRPW motif protein 1
- HMGB1:
-
High mobility group box 1
- ICD:
-
Immunogenic cell death
- iDCs:
-
Immature dendritic cells
- IFN:
-
Interferon
- IL-6:
-
Interleukin-6
- ISGs:
-
Interferon-stimulated genes
- IRF:
-
Interferon regulatory factors
- JNK:
-
C-Jun N-terminal kinase
- KLF4:
-
Kruppel-like factor 4
- H2O2 :
-
Hydrogen peroxide
- HIF:
-
Hypoxia-inducible factor
- LNM:
-
Leinamycin
- MAM:
-
Mitochondria-associated membranes
- MAPK:
-
Mitogen-activated protein kinase
- MCU:
-
Mitochondrial calcium uniporter
- MCL-1:
-
Myeloid cell leukemia-1
- MCP-1:
-
Monocyte chemoattractant protein-1
- MDSCs:
-
Myeloid-derived suppressor cells
- MDV:
-
Mitochondria-derived vesicles
- MHC:
-
Major histocompatibility complex
- mtDNA:
-
Mitochondrial DNA
- mtHSP70:
-
Mitochondrial heat shock protein 70
- mtPQC:
-
Mitochondrial protein quality control
- mtROS:
-
Mitochondrial ROS
- MnSOD:
-
Manganese superoxide dismutase
- MQC:
-
Mitochondrial quality control
- NAC:
-
N-acetyl-L-cysteine
- NAD + :
-
Nicotinamide adenine dinucleotide
- NADPH:
-
Nicotinamide-adenine dinucleotide phosphate
- NCLX:
-
Mitochondrial sodium/calcium exchanger
- NDFUS8:
-
NADH Ubiquinone Oxidoreductase Core Subunit S8
- NOX:
-
NADPH oxidase
- NF-κB:
-
Nuclear factor kappa-light-chain-enhancer of activated B cells
- O2•–:
-
Superoxide
- •OH:
-
Hydroxyl radical
- 8-OHdG:
-
8-Hydroxydeoxyguanosine
- OTC∆:
-
Mitochondrial misfolded ornithine transcarbamylase
- OXPHOS:
-
Oxidative phosphorylation
- PD-L1:
-
Programmed death-ligand 1
- PDGF:
-
Platelet-derived growth factor
- PDPN:
-
Podoplanin
- PI3K:
-
Phosphoinositide 3-kinase
- PINK1:
-
PTEN-induced kinase 1
- PRRs:
-
Pathogen-recognition receptors
- Prx:
-
Peroxiredoxins
- PYCR1:
-
Pyrroline-5-carboxylate reductase 1
- PYK2:
-
Proline-rich tyrosine kinase 2
- regDCs:
-
Regulatory dendritic cells
- ROS:
-
Reactive oxygen species
- RT:
-
Radiotherapy
- RYR:
-
Ryanodine receptor
- SERCA:
-
Sarco/endoplasmic reticulum Ca+ 2–ATPase
- SIRT3:
-
Sirtuin 3
- SOCE:
-
Store operated calcium entry
- SOD1/2:
-
Super oxide dismutase 1/2
- STAT3:
-
Signal transducer and activator of transcription 3
- STIM1:
-
Stromal interaction molecule 1
- STING:
-
Stimulator of Interferon Genes
- TAMs:
-
Tumor-associated macrophages
- TGF-β:
-
Transforming growth factor beta
- TLR9:
-
Toll-like receptor 9
- TME:
-
Tumor microenvironment
- Tregs:
-
Regulatory T cells
- TRP:
-
Transient receptor potential channel
- TRPA1:
-
Transient receptor potential cation channel subfamily A member 1
- UPRmt:
-
Mitochondrial unfolded protein response
- VDAC:
-
Voltage dependent anion channel
- VDCC:
-
Voltage-gated calcium channel
- VEGF:
-
Vascular endothelial growth factor
- YME1L1:
-
ATP-dependent zinc metalloprotease
References
Holmström KM, Finkel T. Cellular mechanisms and physiological consequences of redox-dependent signalling. Nat Rev Mol Cell Biol. 2014;15(6):411–21.
D’Autréaux B, Toledano MB. ROS as signalling molecules: mechanisms that generate specificity in ROS homeostasis. Nat Rev Mol Cell Biol. 2007;8(10):813–24.
Murphy MP. How mitochondria produce reactive oxygen species. Biochem J. 2009;417(1):1–13.
Stowe DF, Camara AK. Mitochondrial reactive oxygen species production in excitable cells: modulators of mitochondrial and cell function. Antioxid Redox Signal. 2009;11(6):1373–414.
Cheung EC, Vousden KH. The role of ROS in tumour development and progression. Nat Rev Cancer. 2022. Epub ahead of print.
Sanchez-Madrid F, Serrador JM. Bringing up the rear: defining the roles of the uropod. Nat Rev Mol Cell Biol. 2009;10(5):353–9.
Shu L, Hu C, Xu M, Yu J, He H, Lin J, et al. ATAD3B is a mitophagy receptor mediating clearance of oxidative stress-induced damaged mitochondrial DNA. EMBO J. 2021;40(8): e106283.
Brand MD. Uncoupling to survive? The role of mitochondrial inefficiency in ageing. Exp Gerontol. 2000;35(6–7):811–20.
Cadenas S. Mitochondrial uncoupling, ROS generation and cardioprotection. Biochim Biophys Acta. 2018;1859(9):940–50.
Zakharova VV, Pletjushkina OY, Zinovkin RA, Popova EN, Chernyak BV. Mitochondria-targeted antioxidants and uncouplers of oxidative phosphorylation in treatment of the systemic inflammatory response syndrome (SIRS). J Cell Physiol. 2017;232(5):904–12.
Shrestha R, Johnson E, Byrne FL. Exploring the therapeutic potential of mitochondrial uncouplers in cancer. Mol Metab. 2021;51: 101222.
Brookes PS, Yoon Y, Robotham JL, Anders MW, Sheu SS. Calcium, ATP, and ROS: a mitochondrial love-hate triangle. Am J Physiol Cell Physiol. 2004;287(4):C817–33.
Peng TI, Jou MJ. Oxidative stress caused by mitochondrial calcium overload. Ann N Y Acad Sci. 2010;1201:183–8.
Ma S, Zhang X, Zheng L, Li Z, Zhao X, Lai W, et al. Peroxiredoxin 6 is a crucial factor in the initial step of mitochondrial clearance and is upstream of the PINK1-parkin pathway. Antioxid Redox Signal. 2016;24(9):486–501.
Church SL, Grant JW, Ridnour LA, Oberley LW, Swanson PE, Meltzer PS, et al. Increased manganese superoxide dismutase expression suppresses the malignant phenotype of human melanoma cells. Proc Natl Acad Sci U S A. 1993;90(7):3113–7.
Hybertson BM, Gao B, Bose SK, McCord JM. Oxidative stress in health and disease: the therapeutic potential of Nrf2 activation. Mol Aspects Med. 2011;32(4–6):234–46.
Runchel C, Matsuzawa A, Ichijo H. Mitogen-activated protein kinases in mammalian oxidative stress responses. Antioxid Redox Signal. 2011;15(1):205–18.
Wada T, Penninger JM. Mitogen-activated protein kinases in apoptosis regulation. Oncogene. 2004;23(16):2838–49.
Wagner EF, Nebreda AR. Signal integration by JNK and p38 MAPK pathways in cancer development. Nat Rev Cancer. 2009;9(8):537–49.
Schumacker PT. Reactive oxygen species in cancer cells: live by the sword, die by the sword. Cancer Cell. 2006;10(3):175–6.
Hamanaka RB, Chandel NS. Mitochondrial reactive oxygen species regulate cellular signaling and dictate biological outcomes. Trends Biochem Sci. 2010;35(9):505–13.
Wu WS. The signaling mechanism of ROS in tumor progression. Cancer Metastasis Rev. 2006;25(4):695–705.
Sullivan LB, Chandel NS. Mitochondrial reactive oxygen species and cancer. Cancer Metab. 2014;2:17.
Wang Y, Qi H, Liu Y, Duan C, Liu X, Xia T, et al. The double-edged roles of ROS in cancer prevention and therapy. Theranostics. 2021;11(10):4839–57.
Garg M, Moloney JN, Cotter TG. ROS signalling in the biology of cancer. Cell Mol Life Sci CMLS. 2018;80:50–64.
Fang J, Seki T, Maeda H. Therapeutic strategies by modulating oxygen stress in cancer and inflammation. Adv Drug Deliv Rev. 2009;61(4):290–302.
Rai P. Oxidation in the nucleotide pool, the DNA damage response and cellular senescence: defective bricks build a defective house. Mutat Res. 2010;703(1):71–81.
Lee HC, Wei YH. Mitochondrial DNA instability and metabolic shift in human cancers. Int J Mol Sci. 2009;10(2):674–701.
Karihtala P, Soini Y. Reactive oxygen species and antioxidant mechanisms in human tissues and their relation to malignancies. APMIS. 2007;115(2):81–103.
McCord JM, Fridovich I. Superoxide dismutase. An enzymic function for erythrocuprein (hemocuprein). J Biol Chem. 1969;244(22):6049–55.
Valko M, Leibfritz D, Moncol J, Cronin MT, Mazur M, Telser J. Free radicals and antioxidants in normal physiological functions and human disease. Int J Biochem Cell Biol. 2007;39(1):44–84.
Lubos E, Loscalzo J, Handy DE. Glutathione peroxidase-1 in health and disease: from molecular mechanisms to therapeutic opportunities. Antioxid Redox Signal. 2011;15(7):1957–97.
Zhang Y, Qi H, Taylor R, Xu W, Liu LF, Jin S. The role of autophagy in mitochondria maintenance: characterization of mitochondrial functions in autophagy-deficient S. cerevisiae strains. Autophagy. 2007;3(4):337–46.
Mathew R, Karp CM, Beaudoin B, Vuong N, Chen G, Chen HY, et al. Autophagy suppresses tumorigenesis through elimination of p62. Cell. 2009;137(6):1062–75.
Xu Y, Shen J, Ran Z. Emerging views of mitophagy in immunity and autoimmune diseases. Autophagy. 2020;16(1):3–17.
Greten FR, Grivennikov SI. Inflammation and cancer: triggers, mechanisms, and consequences. Immunity. 2019;51(1):27–41.
Luo J, Solimini NL, Elledge SJ. Principles of cancer therapy: oncogene and non-oncogene addiction. Cell. 2009;136(5):823–37.
Solimini NL, Luo J, Elledge SJ. Non-oncogene addiction and the stress phenotype of cancer cells. Cell. 2007;130(6):986–8.
Policastro LL, Ibanez IL, Notcovich C, Duran HA, Podhajcer OL. The tumor microenvironment: characterization, redox considerations, and novel approaches for reactive oxygen species-targeted gene therapy. Antioxid Redox Signal. 2013;19(8):854–95.
Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–74.
Paget S. The distribution of secondary growths in cancer of the breast. 1889. Cancer Metastasis Rev. 1989;8(2):98–101.
Balkwill F, Mantovani A. Inflammation and cancer: back to Virchow? Lancet. 2001;357(9255):539–45.
Federico A, Morgillo F, Tuccillo C, Ciardiello F, Loguercio C. Chronic inflammation and oxidative stress in human carcinogenesis. Int J Cancer. 2007;121(11):2381–6.
Joyce JA, Pollard JW. Microenvironmental regulation of metastasis. Nat Rev Cancer. 2009;9(4):239–52.
Stewart T, Tsai SC, Grayson H, Henderson R, Opelz G. Incidence of de-novo breast cancer in women chronically immunosuppressed after organ transplantation. Lancet. 1995;346(8978):796–8.
Seledtsov VI, Goncharov AG, Seledtsova GV. Clinically feasible approaches to potentiating cancer cell-based immunotherapies. Hum Vaccin Immunother. 2015;11(4):851–69.
Marchi S, Guilbaud E, Tait SWG, Yamazaki T, Galluzzi L. Mitochondrial control of inflammation. Nat Rev Immunol. 2022:1–15.
Cheng CW, Kuo CY, Fan CC, Fang WC, Jiang SS, Lo YK, et al. Overexpression of Lon contributes to survival and aggressive phenotype of cancer cells through mitochondrial complex I-mediated generation of reactive oxygen species. Cell Death Dis. 2013;4: e681.
Sabharwal SS, Schumacker PT. Mitochondrial ROS in cancer: initiators, amplifiers or an Achilles’ heel? Nat Rev Cancer. 2014;14(11):709–21.
Pan JS, Hong MZ, Ren JL. Reactive oxygen species: a double-edged sword in oncogenesis. World J Gastroenterol. 2009;15(14):1702–7.
Hanahan D. Hallmarks of cancer: new dimensions. Cancer Discov. 2022;12(1):31–46.
Zhao Q, Wang J, Levichkin IV, Stasinopoulos S, Ryan MT, Hoogenraad NJ. A mitochondrial specific stress response in mammalian cells. EMBO J. 2002;21(17):4411–9.
Voos W, Pollecker K. The mitochondrial lon protease: novel functions off the beaten track? Biomolecules. 2020;10(2).
Gibellini L, De Gaetano A, Mandrioli M, Van Tongeren E, Bortolotti CA, Cossarizza A, et al. The biology of Lonp1: more than a mitochondrial protease. Int Rev Cell Mol Biol. 2020;354:1–61.
Jovaisaite V, Mouchiroud L, Auwerx J. The mitochondrial unfolded protein response, a conserved stress response pathway with implications in health and disease. J Exp Biol. 2014;217(Pt 1):137–43.
Naresh NU, Haynes CM. Signaling and regulation of the mitochondrial unfolded protein response. Cold Spring Harb Perspect Biol. 2019;11(6).
Czarnecka AM, Campanella C, Zummo G, Cappello F. Mitochondrial chaperones in cancer: from molecular biology to clinical diagnostics. Cancer Biol Ther. 2006;5(7):714–20.
Karpel-Massler G, Horst BA, Shu C, Chau L, Tsujiuchi T, Bruce JN, et al. A synthetic cell-penetrating dominant-negative ATF5 peptide exerts anticancer activity against a broad spectrum of treatment-resistant cancers. Clin Cancer Res. 2016;22(18):4698–711.
Deng P, Haynes CM. Mitochondrial dysfunction in cancer: potential roles of ATF5 and the mitochondrial UPR. Semin Cancer Biol. 2017;47:43–9.
Ghosh JC, Dohi T, Kang BH, Altieri DC. Hsp60 regulation of tumor cell apoptosis. J Biol Chem. 2008;283(8):5188–94.
Kim W, Ryu J, Kim JE. CCAR2/DBC1 and Hsp60 positively regulate expression of survivin in neuroblastoma cells. Int J Mol Sci. 2019;20(1).
Mylonis I, Kourti M, Samiotaki M, Panayotou G, Simos G. Mortalin-mediated and ERK-controlled targeting of HIF-1alpha to mitochondria confers resistance to apoptosis under hypoxia. J Cell Sci. 2017;130(2):466–79.
Tsuneki M, Maruyama S, Yamazaki M, Xu B, Essa A, Abe T, et al. Extracellular heat shock protein A9 is a novel interaction partner of podoplanin in oral squamous cell carcinoma cells. Biochem Biophys Res Commun. 2013;434(1):124–30.
Goard CA, Schimmer AD. Mitochondrial matrix proteases as novel therapeutic targets in malignancy. Oncogene. 2014;33(21):2690–9.
Ngo JK, Davies KJ. Mitochondrial Lon protease is a human stress protein. Free Radical Biol Med. 2009;46(8):1042–8.
Pryde KR, Taanman JW, Schapira AH. A LON-ClpP proteolytic axis degrades complex I to extinguish ROS production in depolarized mitochondria. Cell Rep. 2016;17(10):2522–31.
Ghosh JC, Seo JH, Agarwal E, Wang Y, Kossenkov AV, Tang HY, et al. Akt phosphorylation of mitochondrial Lonp1 protease enables oxidative metabolism and advanced tumor traits. Oncogene. 2019;38(43):6926–39.
Li J, Agarwal E, Bertolini I, Seo JH, Caino MC, Ghosh JC, et al. The mitophagy effector FUNDC1 controls mitochondrial reprogramming and cellular plasticity in cancer cells. Sci Signal. 2020;13(642).
Li Y, Xue Y, Xu X, Wang G, Liu Y, Wu H, et al. A mitochondrial FUNDC1/HSC70 interaction organizes the proteostatic stress response at the risk of cell morbidity. EMBO J. 2019;38(3).
Lee YG, Kim HW, Nam Y, Shin KJ, Lee YJ, Park DH, et al. LONP1 and ClpP cooperatively regulate mitochondrial proteostasis for cancer cell survival. Oncogenesis. 2021;10(2):18.
Wang HM, Cheng KC, Lin CJ, Hsu SW, Fang WC, Hsu TF, et al. Obtusilactone A and (-)-sesamin induce apoptosis in human lung cancer cells by inhibiting mitochondrial Lon protease and activating DNA damage checkpoints. Cancer Sci. 2010;101(12):2612–20.
Ganta KK, Mandal A, Chaubey B. Depolarization of mitochondrial membrane potential is the initial event in non-nucleoside reverse transcriptase inhibitor efavirenz induced cytotoxicity. Cell Biol Toxicol. 2017;33(1):69–82.
Bernstein SH, Venkatesh S, Li M, Lee J, Lu B, Hilchey SP, et al. The mitochondrial ATP-dependent Lon protease: a novel target in lymphoma death mediated by the synthetic triterpenoid CDDO and its derivatives. Blood. 2012;119(14):3321–9.
Nie X, Li M, Lu B, Zhang Y, Lan L, Chen L, et al. Down-regulating overexpressed human Lon in cervical cancer suppresses cell proliferation and bioenergetics. PLoS ONE. 2013;8(11): e81084.
Liu Y, Lan L, Huang K, Wang R, Xu C, Shi Y, et al. Inhibition of Lon blocks cell proliferation, enhances chemosensitivity by promoting apoptosis and decreases cellular bioenergetics of bladder cancer: potential roles of Lon as a prognostic marker and therapeutic target in baldder cancer. Oncotarget. 2014;5(22):11209–24.
Varambally S, Yu J, Laxman B, Rhodes DR, Mehra R, Tomlins SA, et al. Integrative genomic and proteomic analysis of prostate cancer reveals signatures of metastatic progression. Cancer Cell. 2005;8(5):393–406.
Quiros PM, Espanol Y, Acin-Perez R, Rodriguez F, Barcena C, Watanabe K, et al. ATP-dependent Lon protease controls tumor bioenergetics by reprogramming mitochondrial activity. Cell Rep. 2014;8(2):542–56.
Gibellini L, Losi L, De Biasi S, Nasi M, Lo Tartaro D, Pecorini S, et al. LonP1 differently modulates mitochondrial function and bioenergetics of primary versus metastatic colon cancer cells. Front Oncol. 2018;8:254.
Bota DA, Ngo JK, Davies KJ. Downregulation of the human Lon protease impairs mitochondrial structure and function and causes cell death. Free Radical Biol Med. 2005;38(5):665–77.
Gibellini L, Pinti M, Bartolomeo R, De Biasi S, Cormio A, Musicco C, et al. Inhibition of Lon protease by triterpenoids alters mitochondria and is associated to cell death in human cancer cells. Oncotarget. 2015;6(28):25466–83.
Kuo CL, Chou HY, Chiu YC, Cheng AN, Fan CC, Chang YN, et al. Mitochondrial oxidative stress by Lon-PYCR1 maintains an immunosuppressive tumor microenvironment that promotes cancer progression and metastasis. Cancer Lett. 2020;474:138–50.
Kao TY, Chiu YC, Fang WC, Cheng CW, Kuo CY, Juan HF, et al. Mitochondrial Lon regulates apoptosis through the association with Hsp60-mtHsp70 complex. Cell Death Dis. 2015;6: e1642.
Sung YJ, Kao TY, Kuo CL, Fan CC, Cheng AN, Fang WC, et al. Mitochondrial Lon sequesters and stabilizes p53 in the matrix to restrain apoptosis under oxidative stress via its chaperone activity. Cell Death Dis. 2018;9(6):697.
Tangeda V, Lo YK, Babuharisankar AP, Chou HY, Kuo CL, Kao YH, et al. Lon upregulation contributes to cisplatin resistance by triggering NCLX-mediated mitochondrial Ca(2+) release in cancer cells. Cell Death Dis. 2022;13(3):241.
Wang H, Liu C, Zhao Y, Zhang W, Xu K, Li D, et al. Inhibition of LONP1 protects against erastin-induced ferroptosis in Pancreatic ductal adenocarcinoma PANC1 cells. Biochem Biophys Res Commun. 2020;522(4):1063–8.
Cheng X, Kanki T, Fukuoh A, Ohgaki K, Takeya R, Aoki Y, et al. PDIP38 associates with proteins constituting the mitochondrial DNA nucleoid. J Biochem. 2005;138(6):673–8.
Chang TP, Poltoratsky V, Vancurova I. Bortezomib inhibits expression of TGF-beta1, IL-10, and CXCR4, resulting in decreased survival and migration of cutaneous T cell lymphoma cells. J Immunol. 2015;194(6):2942–53.
Wu J, Niu J, Li X, Wang X, Guo Z, Zhang F. TGF-beta1 induces senescence of bone marrow mesenchymal stem cells via increase of mitochondrial ROS production. BMC Dev Biol. 2014;14:21.
Ishikawa F, Kaneko E, Sugimoto T, Ishijima T, Wakamatsu M, Yuasa A, et al. A mitochondrial thioredoxin-sensitive mechanism regulates TGF-beta-mediated gene expression associated with epithelial-mesenchymal transition. Biochem Biophys Res Commun. 2014;443(3):821–7.
Jain M, Rivera S, Monclus EA, Synenki L, Zirk A, Eisenbart J, et al. Mitochondrial reactive oxygen species regulate transforming growth factor-beta signaling. J Biol Chem. 2013;288(2):770–7.
Byun HO, Jung HJ, Seo YH, Lee YK, Hwang SC, Hwang ES, et al. GSK3 inactivation is involved in mitochondrial complex IV defect in transforming growth factor (TGF) beta1-induced senescence. Exp Cell Res. 2012;318(15):1808–19.
Yoon YS, Lee JH, Hwang SC, Choi KS, Yoon G. TGF beta1 induces prolonged mitochondrial ROS generation through decreased complex IV activity with senescent arrest in Mv1Lu cells. Oncogene. 2005;24(11):1895–903.
Lamouille S, Xu J, Derynck R. Molecular mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell Biol. 2014;15(3):178–96.
Rhyu DY, Yang Y, Ha H, Lee GT, Song JS, Uh ST, et al. Role of reactive oxygen species in TGF-beta1-induced mitogen-activated protein kinase activation and epithelial-mesenchymal transition in renal tubular epithelial cells. J Am Soc Nephrol. 2005;16(3):667–75.
Gorlach A, Bertram K, Hudecova S, Krizanova O. Calcium and ROS: a mutual interplay. Redox Biol. 2015;6:260–71.
Ma L, Wang H, Wang C, Su J, Xie Q, Xu L, et al. Failure of elevating calcium induces oxidative stress tolerance and imparts cisplatin resistance in ovarian cancer cells. Aging Dis. 2016;7(3):254–66.
Bogeski I, Kummerow C, Al-Ansary D, Schwarz EC, Koehler R, Kozai D, et al. Differential redox regulation of ORAI ion channels: a mechanism to tune cellular calcium signaling. Sci Signal. 2010;3(115):ra24.
Bansaghi S, Golenar T, Madesh M, Csordas G, RamachandraRao S, Sharma K, et al. Isoform- and species-specific control of inositol 1,4,5-trisphosphate (IP3) receptors by reactive oxygen species. J Biol Chem. 2014;289(12):8170–81.
Bird GS, Burgess GM, Putney JW Jr. Sulfhydryl reagents and cAMP-dependent kinase increase the sensitivity of the inositol 1,4,5-trisphosphate receptor in hepatocytes. J Biol Chem. 1993;268(24):17917–23.
Giorgi C, Ito K, Lin HK, Santangelo C, Wieckowski MR, Lebiedzinska M, et al. PML regulates apoptosis at endoplasmic reticulum by modulating calcium release. Science. 2010;330(6008):1247–51.
Takahashi N, Chen HY, Harris IS, Stover DG, Selfors LM, Bronson RT, et al. Cancer cells co-opt the neuronal redox-sensing channel TRPA1 to promote oxidative-stress tolerance. Cancer Cell. 2018;33(6):985-1003 e7.
Reczek CR, Chandel NS. ROS promotes cancer cell survival through calcium signaling. Cancer Cell. 2018;33(6):949–51.
Chandel NS, Maltepe E, Goldwasser E, Mathieu CE, Simon MC, Schumacker PT. Mitochondrial reactive oxygen species trigger hypoxia-induced transcription. Proc Natl Acad Sci U S A. 1998;95(20):11715–20.
Tosatto A, Sommaggio R, Kummerow C, Bentham RB, Blacker TS, Berecz T, et al. The mitochondrial calcium uniporter regulates breast cancer progression via HIF-1alpha. EMBO Mol Med. 2016;8(5):569–85.
Ren T, Zhang H, Wang J, Zhu J, Jin M, Wu Y, et al. MCU-dependent mitochondrial Ca(2+) inhibits NAD(+)/SIRT3/SOD2 pathway to promote ROS production and metastasis of HCC cells. Oncogene. 2017;36(42):5897–909.
Dong Z, Shanmughapriya S, Tomar D, Siddiqui N, Lynch S, Nemani N, et al. Mitochondrial Ca(2+) uniporter is a mitochondrial luminal redox sensor that augments MCU channel activity. Mol Cell. 2017;65(6):1014-28 e7.
Hernansanz-Agustin P, Choya-Foces C, Carregal-Romero S, Ramos E, Oliva T, Villa-Pina T, et al. Na(+) controls hypoxic signalling by the mitochondrial respiratory chain. Nature. 2020;586(7828):287–91.
Li Y, Guo B, Xie Q, Ye D, Zhang D, Zhu Y, et al. STIM1 mediates hypoxia-driven hepatocarcinogenesis via interaction with HIF-1. Cell Rep. 2015;12(3):388–95.
Szabadkai G, Bianchi K, Varnai P, De Stefani D, Wieckowski MR, Cavagna D, et al. Chaperone-mediated coupling of endoplasmic reticulum and mitochondrial Ca2+ channels. J Cell Biol. 2006;175(6):901–11.
Booth DM, Enyedi B, Geiszt M, Varnai P, Hajnoczky G. Redox nanodomains are induced by and control calcium signaling at the ER-mitochondrial interface. Mol Cell. 2016;63(2):240–8.
Ma K, Chen G, Li W, Kepp O, Zhu Y, Chen Q. Mitophagy, mitochondrial homeostasis, and cell fate. Front Cell Dev Biol. 2020;8:467.
Okado-Matsumoto A, Fridovich I. Subcellular distribution of superoxide dismutases (SOD) in rat liver: Cu, Zn-SOD in mitochondria. J Biol Chem. 2001;276(42):38388–93.
Wang Y, Branicky R, Noe A, Hekimi S. Superoxide dismutases: dual roles in controlling ROS damage and regulating ROS signaling. J Cell Biol. 2018;217(6):1915–28.
Fang Y, Tan J, Zhang Q. Signaling pathways and mechanisms of hypoxia-induced autophagy in the animal cells. Cell Biol Int. 2015;39(8):891–8.
Bellot G, Garcia-Medina R, Gounon P, Chiche J, Roux D, Pouyssegur J, et al. Hypoxia-induced autophagy is mediated through hypoxia-inducible factor induction of BNIP3 and BNIP3L via their BH3 domains. Mol Cell Biol. 2009;29(10):2570–81.
Liu L, Feng D, Chen G, Chen M, Zheng Q, Song P, et al. Mitochondrial outer-membrane protein FUNDC1 mediates hypoxia-induced mitophagy in mammalian cells. Nat Cell Biol. 2012;14(2):177–85.
Sowter HM, Ratcliffe PJ, Watson P, Greenberg AH, Harris AL. HIF-1-dependent regulation of hypoxic induction of the cell death factors BNIP3 and NIX in human tumors. Can Res. 2001;61(18):6669–73.
Jung J, Zhang Y, Celiku O, Zhang W, Song H, Williams BJ, et al. Mitochondrial NIX promotes tumor survival in the hypoxic niche of glioblastoma. Can Res. 2019;79(20):5218–32.
Melser S, Chatelain EH, Lavie J, Mahfouf W, Jose C, Obre E, et al. Rheb regulates mitophagy induced by mitochondrial energetic status. Cell Metab. 2013;17(5):719–30.
Abdrakhmanov A, Yapryntseva MA, Kaminskyy VO, Zhivotovsky B, Gogvadze V. Receptor-mediated mitophagy rescues cancer cells under hypoxic conditions. Cancers (Basel). 2021;13(16).
Kung-Chun Chiu D, Pui-Wah Tse A, Law CT, Ming-Jing XuI, Lee D, Chen M, et al. Hypoxia regulates the mitochondrial activity of hepatocellular carcinoma cells through HIF/HEY1/PINK1 pathway. Cell Death Dis. 2019;10(12):934.
Chen M, Chen Z, Wang Y, Tan Z, Zhu C, Li Y, et al. Mitophagy receptor FUNDC1 regulates mitochondrial dynamics and mitophagy. Autophagy. 2016;12(4):689–702.
Wang C, Dai X, Wu S, Xu W, Song P, Huang K. FUNDC1-dependent mitochondria-associated endoplasmic reticulum membranes are involved in angiogenesis and neoangiogenesis. Nat Commun. 2021;12(1):2616.
O’Donnell JS, Teng MWL, Smyth MJ. Cancer immunoediting and resistance to T cell-based immunotherapy. Nat Rev Clin Oncol. 2019;16(3):151–67.
Kunimasa K, Goto T. Immunosurveillance and immunoediting of lung cancer: current perspectives and challenges. Int J Mol Sci. 2020;21(2).
Tang S, Ning Q, Yang L, Mo Z, Tang S. Mechanisms of immune escape in the cancer immune cycle. Int Immunopharmacol. 2020;86: 106700.
Valacchi G, Virgili F, Cervellati C, Pecorelli A. OxInflammation: from subclinical condition to pathological biomarker. Front Physiol. 2018;9:858.
Weinberg SE, Singer BD, Steinert EM, Martinez CA, Mehta MM, Martinez-Reyes I, et al. Mitochondrial complex III is essential for suppressive function of regulatory T cells. Nature. 2019;565(7740):495–9.
Weinberg SE, Sena LA, Chandel NS. Mitochondria in the regulation of innate and adaptive immunity. Immunity. 2015;42(3):406–17.
Aboelella NS, Brandle C, Kim T, Ding ZC, Zhou G. Oxidative stress in the tumor microenvironment and its relevance to cancer immunotherapy. Cancers (Basel). 2021;13(5).
Cheng AN, Cheng LC, Kuo CL, Lo YK, Chou HY, Chen CH, et al. Mitochondrial Lon-induced mtDNA leakage contributes to PD-L1-mediated immunoescape via STING-IFN signaling and extracellular vesicles. J Immunother Cancer. 2020;8(2).
Pinti M, Gibellini L, Nasi M, De Biasi S, Bortolotti CA, Iannone A, et al. Emerging role of Lon protease as a master regulator of mitochondrial functions. Biochem Biophys Acta. 2016;1857(8):1300–6.
Hoesel B, Schmid JA. The complexity of NF-kappaB signaling in inflammation and cancer. Mol Cancer. 2013;12:86.
Yao H, de Boer WI, Rahman I. Targeting lung inflammation: novel therapies for the treatment of COPD. Curr Respir Med Rev. 2008;4(1):57–68.
Fang JY, Richardson BC. The MAPK signalling pathways and colorectal cancer. Lancet Oncol. 2005;6(5):322–7.
Shuto T, Xu H, Wang B, Han J, Kai H, Gu XX, et al. Activation of NF-kappa B by nontypeable Hemophilus influenzae is mediated by toll-like receptor 2-TAK1-dependent NIK-IKK alpha /beta-I kappa B alpha and MKK3/6-p38 MAP kinase signaling pathways in epithelial cells. Proc Natl Acad Sci U S A. 2001;98(15):8774–9.
Wang J, Huang J, Wang L, Chen C, Yang D, Jin M, et al. Urban particulate matter triggers lung inflammation via the ROS-MAPK-NF-kappaB signaling pathway. J Thorac Dis. 2017;9(11):4398–412.
Lu H, Chen I, Shimoda LA, Park Y, Zhang C, Tran L, et al. Chemotherapy-induced Ca(2+) release stimulates breast cancer stem cell enrichment. Cell Rep. 2017;18(8):1946–57.
Curiel TJ, Wei S, Dong H, Alvarez X, Cheng P, Mottram P, et al. Blockade of B7–H1 improves myeloid dendritic cell-mediated antitumor immunity. Nat Med. 2003;9(5):562–7.
Kryczek I, Zou L, Rodriguez P, Zhu G, Wei S, Mottram P, et al. B7–H4 expression identifies a novel suppressive macrophage population in human ovarian carcinoma. J Exp Med. 2006;203(4):871–81.
Bailly C. Regulation of PD-L1 expression on cancer cells with ROS-modulating drugs. Life Sci. 2020;246: 117403.
Shima T, Shimoda M, Shigenobu T, Ohtsuka T, Nishimura T, Emoto K, et al. Infiltration of tumor-associated macrophages is involved in tumor programmed death-ligand 1 expression in early lung adenocarcinoma. Cancer Sci. 2020;111(2):727–38.
Griess B, Mir S, Datta K, Teoh-Fitzgerald M. Scavenging reactive oxygen species selectively inhibits M2 macrophage polarization and their pro-tumorigenic function in part, via Stat3 suppression. Free Radic Biol Med. 2020;147:48–60.
Zou W. Regulatory T cells, tumour immunity and immunotherapy. Nat Rev Immunol. 2006;6(4):295–307.
Maj T, Wang W, Crespo J, Zhang H, Wang W, Wei S, et al. Oxidative stress controls regulatory T cell apoptosis and suppressor activity and PD-L1-blockade resistance in tumor. Nat Immunol. 2017;18(12):1332–41.
Nagaraj S, Gupta K, Pisarev V, Kinarsky L, Sherman S, Kang L, et al. Altered recognition of antigen is a mechanism of CD8+ T cell tolerance in cancer. Nat Med. 2007;13(7):828–35.
Rodriguez PC, Ernstoff MS, Hernandez C, Atkins M, Zabaleta J, Sierra R, et al. Arginase I-producing myeloid-derived suppressor cells in renal cell carcinoma are a subpopulation of activated granulocytes. Cancer Res. 2009;69(4):1553–60.
Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol. 2009;9(3):162–74.
Huang B, Pan PY, Li Q, Sato AI, Levy DE, Bromberg J, et al. Gr-1+CD115+ immature myeloid suppressor cells mediate the development of tumor-induced T regulatory cells and T-cell anergy in tumor-bearing host. Cancer Res. 2006;66(2):1123–31.
Semenza GL. Hypoxia-inducible factors in physiology and medicine. Cell. 2012;148(3):399–408.
Liberti MV, Locasale JW. The Warburg Effect: how does it benefit cancer cells? Trends Biochem Sci. 2016;41(3):211–8.
van Gisbergen MW, Offermans K, Voets AM, Lieuwes NG, Biemans R, Hoffmann RF, et al. Mitochondrial dysfunction inhibits hypoxia-induced HIF-1alpha stabilization and expression of its downstream targets. Front Oncol. 2020;10:770.
Dan Dunn J, Alvarez LAJ, Zhang X, Soldati T. Reactive oxygen species and mitochondria: a nexus of cellular homeostasis. Redox Biol. 2015;6:472–85.
Hernansanz-Agustín P, Choya-Foces C, Carregal-Romero S. Na(+) controls hypoxic signalling by the mitochondrial respiratory chain. Nature. 2020;586(7828):287–91.
Kuo CW, Tsai MH, Lin TK, Tiao MM, Wang PW, Chuang JH, et al. mtDNA as a mediator for expression of hypoxia-inducible factor 1alpha and ros in hypoxic neuroblastoma cells. Int J Mol Sci. 2017;18(6).
Kim MC, Hwang SH, Yang Y, Kim NY, Kim Y. Reduction in mitochondrial oxidative stress mediates hypoxia-induced resistance to cisplatin in human transitional cell carcinoma cells. Neoplasia. 2021;23(7):653–62.
Bousquet PA, Meltzer S, Sonstevold L, Esbensen Y, Dueland S, Flatmark K, et al. Markers of mitochondrial metabolism in tumor hypoxia, systemic inflammation, and adverse outcome of rectal cancer. Transl Oncol. 2019;12(1):76–83.
West AP, Shadel GS. Mitochondrial DNA in innate immune responses and inflammatory pathology. Nat Rev Immunol. 2017;17(6):363–75.
Zhang Q, Raoof M, Chen Y, Sumi Y, Sursal T, Junger W, et al. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature. 2010;464(7285):104–7.
Park JE, Dutta B, Tse SW, Gupta N, Tan CF, Low JK, et al. Hypoxia-induced tumor exosomes promote M2-like macrophage polarization of infiltrating myeloid cells and microRNA-mediated metabolic shift. Oncogene. 2019;38(26):5158–73.
Kennel KB, Greten FR. Immune cell - produced ROS and their impact on tumor growth and metastasis. Redox Biol. 2021;42: 101891.
Checa J, Aran JM. Reactive oxygen species: drivers of physiological and pathological processes. J Inflamm Res. 2020;13:1057–73.
Yang Y, Neo SY, Chen Z, Cui W, Chen Y, Guo M, et al. Thioredoxin activity confers resistance against oxidative stress in tumor-infiltrating NK cells. J Clin Invest. 2020;130(10):5508–22.
Hildeman DA, Mitchell T, Teague TK, Henson P, Day BJ, Kappler J, et al. Reactive oxygen species regulate activation-induced T cell apoptosis. Immunity. 1999;10(6):735–44.
Weiskopf D, Schwanninger A, Weinberger B, Almanzar G, Parson W, Buus S, et al. Oxidative stress can alter the antigenicity of immunodominant peptides. J Leukoc Biol. 2010;87(1):165–72.
Bianchi ME. DAMPs, PAMPs and alarmins: all we need to know about danger. J Leukoc Biol. 2007;81(1):1–5.
Panaretakis T, Kepp O, Brockmeier U, Tesniere A, Bjorklund AC, Chapman DC, et al. Mechanisms of pre-apoptotic calreticulin exposure in immunogenic cell death. EMBO J. 2009;28(5):578–90.
Deng H, Yang W, Zhou Z, Tian R, Lin L, Ma Y, et al. Targeted scavenging of extracellular ROS relieves suppressive immunogenic cell death. Nat Commun. 2020;11(1):4951.
Iida N, Dzutsev A, Stewart CA, Smith L, Bouladoux N, Weingarten RA, et al. Commensal bacteria control cancer response to therapy by modulating the tumor microenvironment. Science. 2013;342(6161):967–70.
Shrihari TG. Dual role of inflammatory mediators in cancer. Ecancermedicalscience. 2017;11:721.
Mougiakakos D, Johansson CC, Jitschin R, Bottcher M, Kiessling R. Increased thioredoxin-1 production in human naturally occurring regulatory T cells confers enhanced tolerance to oxidative stress. Blood. 2011;117(3):857–61.
Mougiakakos D, Johansson CC, Kiessling R. Naturally occurring regulatory T cells show reduced sensitivity toward oxidative stress-induced cell death. Blood. 2009;113(15):3542–5.
Kurniawan H, Franchina DG, Guerra L, Bonetti L, Baguet LS, Grusdat M, et al. Glutathione restricts serine metabolism to preserve regulatory T cell function. Cell Metab. 2020;31(5):920-36 e7.
Mantovani A, Sozzani S, Locati M, Schioppa T, Saccani A, Allavena P, et al. Infiltration of tumours by macrophages and dendritic cells: tumour-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Novartis Found Symp. 2004;256:137–45; discussion 46-8, 259–69.
Biswas SK, Mantovani A. Orchestration of metabolism by macrophages. Cell Metab. 2012;15(4):432–7.
Dehne N, Mora J, Namgaladze D, Weigert A, Brune B. Cancer cell and macrophage cross-talk in the tumor microenvironment. Curr Opin Pharmacol. 2017;35:12–9.
Mantovani A, Biswas SK, Galdiero MR, Sica A, Locati M. Macrophage plasticity and polarization in tissue repair and remodelling. J Pathol. 2013;229(2):176–85.
Zhou J, Tang Z, Gao S, Li C, Feng Y, Zhou X. Tumor-associated macrophages: recent insights and therapies. Front Oncol. 2020;10(188).
Rendra E, Riabov V, Mossel DM, Sevastyanova T, Harmsen MC, Kzhyshkowska J. Reactive oxygen species (ROS) in macrophage activation and function in diabetes. Immunobiology. 2019;224(2):242–53.
Tan HY, Wang N, Li S, Hong M, Wang X, Feng Y. The reactive oxygen species in macrophage polarization: reflecting its dual role in progression and treatment of human diseases. Oxid Med Cell Longev. 2016;2016:2795090.
Dey N, Sinha M, Gupta S, Gonzalez MN, Fang R, Endsley JJ, et al. Caspase-1/ASC inflammasome-mediated activation of IL-1β-ROS-NF-κB pathway for control of Trypanosoma cruzi replication and survival is dispensable in NLRP3-/- macrophages. PLoS ONE. 2014;9(11): e111539.
Coffelt SB, Tal AO, Scholz A, De Palma M, Patel S, Urbich C, et al. Angiopoietin-2 regulates gene expression in TIE2-expressing monocytes and augments their inherent proangiogenic functions. Cancer Res. 2010;70(13):5270–80.
Pollard JW. Tumour-educated macrophages promote tumour progression and metastasis. Nat Rev Cancer. 2004;4(1):71–8.
Van Ginderachter JA, Movahedi K, HassanzadehGhassabeh G, Meerschaut S, Beschin A, Raes G, et al. Classical and alternative activation of mononuclear phagocytes: picking the best of both worlds for tumor promotion. Immunobiology. 2006;211(6–8):487–501.
Duluc D, Delneste Y, Tan F, Moles MP, Grimaud L, Lenoir J, et al. Tumor-associated leukemia inhibitory factor and IL-6 skew monocyte differentiation into tumor-associated macrophage-like cells. Blood. 2007;110(13):4319–30.
Wang Q, Ni H, Lan L, Wei X, Xiang R, Wang Y. Fra-1 protooncogene regulates IL-6 expression in macrophages and promotes the generation of M2d macrophages. Cell Res. 2010;20(6):701–12.
Zorov DB, Juhaszova M, Sollott SJ. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol Rev. 2014;94(3):909–50.
Silwal P, Kim JK, Kim YJ, Jo EK. Mitochondrial reactive oxygen species: double-edged weapon in host defense and pathological inflammation during infection. Front Immunol. 2020;11:1649.
Collin M, Bigley V. Human dendritic cell subsets: an update. Immunology. 2018;154(1):3–20.
Fucikova J, Palova-Jelinkova L, Bartunkova J, Spisek R. Induction of tolerance and immunity by dendritic cells: mechanisms and clinical applications. Front Immunol. 2019;10:2393.
Michielsen AJ, Noonan S, Martin P, Tosetto M, Marry J, Biniecka M, et al. Inhibition of dendritic cell maturation by the tumor microenvironment correlates with the survival of colorectal cancer patients following bevacizumab treatment. Mol Cancer Ther. 2012;11(8):1829–37.
Schmidt SV, Nino-Castro AC, Schultze JL. Regulatory dendritic cells: there is more than just immune activation. Front Immunol. 2012;3:274.
Zong J, Keskinov AA, Shurin GV, Shurin MR. Tumor-derived factors modulating dendritic cell function. Cancer Immunol Immunother. 2016;65(7):821–33.
Ito M, Minamiya Y, Kawai H, Saito S, Saito H, Nakagawa T, et al. Tumor-derived TGFbeta-1 induces dendritic cell apoptosis in the sentinel lymph node. J Immunol. 2006;176(9):5637–43.
Ruffell B, Chang-Strachan D, Chan V, Rosenbusch A, Ho CM, Pryer N, et al. Macrophage IL-10 blocks CD8+ T cell-dependent responses to chemotherapy by suppressing IL-12 expression in intratumoral dendritic cells. Cancer Cell. 2014;26(5):623–37.
Weber F, Byrne SN, Le S, Brown DA, Breit SN, Scolyer RA, et al. Transforming growth factor-beta1 immobilises dendritic cells within skin tumours and facilitates tumour escape from the immune system. Cancer Immunol Immunother. 2005;54(9):898–906.
Yang Y, Bazhin AV, Werner J, Karakhanova S. Reactive oxygen species in the immune system. Int Rev Immunol. 2013;32(3):249–70.
Multhoff G, Molls M, Radons J. Chronic inflammation in cancer development. Front Immunol. 2011;2:98.
Nahrendorf M, Swirski FK. Abandoning M1/M2 for a network model of macrophage function. Circ Res. 2016;119(3):414–7.
Wang J, Yi J. Cancer cell killing via ROS: to increase or decrease, that is the question. Cancer Biol Ther. 2008;7(12):1875–84.
Yang Y, Karakhanova S, Werner J, Bazhin AV. Reactive oxygen species in cancer biology and anticancer therapy. Curr Med Chem. 2013;20(30):3677–92.
Joffre OP, Segura E, Savina A, Amigorena S. Cross-presentation by dendritic cells. Nat Rev Immunol. 2012;12(8):557–69.
Paardekooper LM, Vos W, van den Bogaart G. Oxygen in the tumor microenvironment: effects on dendritic cell function. Oncotarget. 2019;10(8):883–96.
Matsue H, Edelbaum D, Shalhevet D, Mizumoto N, Yang C, Mummert ME, et al. Generation and function of reactive oxygen species in dendritic cells during antigen presentation. J Immunol. 2003;171(6):3010–8.
Chougnet CA, Thacker RI, Shehata HM, Hennies CM, Lehn MA, Lages CS, et al. Loss of phagocytic and antigen cross-presenting capacity in aging dendritic cells is associated with mitochondrial dysfunction. J Immunol. 2015;195(6):2624–32.
Augustin RC, Delgoffe GM, Najjar YG. Characteristics of the tumor microenvironment that influence immune cell functions: hypoxia, oxidative stress, metabolic alterations. Cancers (Basel). 2020;12(12).
Massague J. TGFbeta signalling in context. Nat Rev Mol Cell Biol. 2012;13(10):616–30.
Ou L, Zhang A, Cheng Y, Chen Y. The cGAS-STING Pathway: A Promising Immunotherapy Target. Frontiers in Immunology. 2021;12.
Pérez-Treviño P, Velásquez M, García N. Mechanisms of mitochondrial DNA escape and its relationship with different metabolic diseases. Biochim Biophys Acta (BBA) Mol Basis Dis. 2020;1866(6): 165761.
Bao D, Zhao J, Zhou X, Yang Q, Chen Y, Zhu J, et al. Mitochondrial fission-induced mtDNA stress promotes tumor-associated macrophage infiltration and HCC progression. Oncogene. 2019;38(25):5007–20.
Li Y, Chen H, Yang Q, Wan L, Zhao J, Wu Y, et al. Increased Drp1 promotes autophagy and ESCC progression by mtDNA stress mediated cGAS-STING pathway. J Exp Clin Cancer Res. 2022;41(1):76.
Hu M, Zhou M, Bao X, Pan D, Jiao M, Liu X, et al. ATM inhibition enhances cancer immunotherapy by promoting mtDNA leakage and cGAS/STING activation. J Clin Investig. 2021;131(3).
Kanneganti T-D, Kundu M, Green DR. Innate immune recognition of mtDNA—an undercover signal? Cell Metab. 2015;21(6):793–4.
Zhou L, Zhang Y-F, Yang F-H, Mao H-Q, Chen Z, Zhang L. Mitochondrial DNA leakage induces odontoblast inflammation via the cGAS-STING pathway. Cell Commun Signal. 2021;19(1):58.
Mendoza JL, Escalante NK, Jude KM, SotolongoBellon J, Su L, Horton TM, et al. Structure of the IFNγ receptor complex guides design of biased agonists. Nature. 2019;567(7746):56–60.
Maimela NR, Liu S, Zhang Y. Fates of CD8+ T cells in tumor microenvironment. Comput Struct Biotechnol J. 2018;17:1–13.
Mojic M, Takeda K, Hayakawa Y. The dark side of IFN-γ: its role in promoting cancer immunoevasion. Int J Mol Sci. 2018;19(1):89.
Zaidi MR, Davis S, Noonan FP, Graff-Cherry C, Hawley TS, Walker RL, et al. Interferon-γ links ultraviolet radiation to melanomagenesis in mice. Nature. 2011;469(7331):548–53.
Roux C, Jafari SM, Shinde R, Duncan G, Cescon DW, Silvester J, et al. Reactive oxygen species modulate macrophage immunosuppressive phenotype through the up-regulation of PD-L1. Proc Natl Acad Sci. 2019;116(10):4326–35.
Cocetta V, Ragazzi E, Montopoli M. Mitochondrial involvement in cisplatin resistance. Int J Mol Sci. 2019;20(14).
Guerra F, Perrone AM, Kurelac I, Santini D, Ceccarelli C, Cricca M, et al. Mitochondrial DNA mutation in serous ovarian cancer: implications for mitochondria-coded genes in chemoresistance. J Clin Oncol. 2012;30(36):e373–8.
Horibe S, Ishikawa K, Nakada K, Wake M, Takeda N, Tanaka T, et al. Mitochondrial DNA mutations are involved in the acquisition of cisplatin resistance in human lung cancer A549 cells. Oncol Rep. 2022;47(2).
Chen G, Huang AC, Zhang W, Zhang G, Wu M, Xu W, et al. Exosomal PD-L1 contributes to immunosuppression and is associated with anti-PD-1 response. Nature. 2018;560(7718):382–6.
Theodoraki MN, Yerneni SS, Hoffmann TK, Gooding WE, Whiteside TL. Clinical significance of PD-L1(+) exosomes in plasma of head and neck cancer patients. Clin Cancer Res. 2018;24(4):896–905.
Sugiura A, McLelland GL, Fon EA, McBride HM. A new pathway for mitochondrial quality control: mitochondrial-derived vesicles. EMBO J. 2014;33(19):2142–56.
Miyamoto Y, Kitamura N, Nakamura Y, Futamura M, Miyamoto T, Yoshida M, et al. Possible existence of lysosome-like organella within mitochondria and its role in mitochondrial quality control. PLoS ONE. 2011;6(1): e16054.
Neuspiel M, Schauss AC, Braschi E, Zunino R, Rippstein P, Rachubinski RA, et al. Cargo-selected transport from the mitochondria to peroxisomes is mediated by vesicular carriers. Curr Biol. 2008;18(2):102–8.
Soubannier V, McLelland GL, Zunino R, Braschi E, Rippstein P, Fon EA, et al. A vesicular transport pathway shuttles cargo from mitochondria to lysosomes. Curr Biol. 2012;22(2):135–41.
Zhang Y, Tan J, Miao Y, Zhang Q. The effect of extracellular vesicles on the regulation of mitochondria under hypoxia. Cell Death Dis. 2021;12(4):358.
Asare-Werehene M, Nakka K, Reunov A, Chiu CT, Lee WT, Abedini MR, et al. The exosome-mediated autocrine and paracrine actions of plasma gelsolin in ovarian cancer chemoresistance. Oncogene. 2020;39(7):1600–16.
Luga V, Zhang L, Viloria-Petit Alicia M, Ogunjimi Abiodun A, Inanlou Mohammad R, Chiu E, et al. Exosomes mediate stromal mobilization of autocrine Wnt-PCP signaling in breast cancer cell migration. Cell. 2012;151(7):1542–56.
Kalluri R. The biology and function of exosomes in cancer. J Clin Invest. 2016;126(4):1208–15.
Liu C, Wu H, Mao Y, Chen W, Chen S. Exosomal microRNAs in hepatocellular carcinoma. Cancer Cell Int. 2021;21(1):254.
Matsumoto A, Takahashi Y, Nishikawa M, Sano K, Morishita M, Charoenviriyakul C, et al. Accelerated growth of B16BL6 tumor in mice through efficient uptake of their own exosomes by B16BL6 cells. Cancer Sci. 2017;108(9):1803–10.
Qu JL, Qu XJ, Zhao MF, Teng YE, Zhang Y, Hou KZ, et al. Gastric cancer exosomes promote tumour cell proliferation through PI3K/Akt and MAPK/ERK activation. Digest Liver Dis. 2009;41(12):875–80.
Raimondo S, Saieva L, Corrado C, Fontana S, Flugy A, Rizzo A, et al. Chronic myeloid leukemia-derived exosomes promote tumor growth through an autocrine mechanism. Cell Commun Signal. 2015;13(1):8.
Maia J, Caja S, Strano Moraes MC, Couto N, Costa-Silva B. Exosome-based cell-cell communication in the tumor microenvironment. Front Cell Dev Biol. 2018;6.
Matheoud D, Sugiura A, Bellemare-Pelletier A, Laplante A, Rondeau C, Chemali M, et al. Parkinson disease-related proteins PINK1 and Parkin repress mitochondrial antigen presentation. Cell. 2016;166(2):314–27.
Chao T, Shih HT, Hsu SC, Chen PJ, Fan YS, Jeng YM, et al. Autophagy restricts mitochondrial DNA damage-induced release of ENDOG (endonuclease G) to regulate genome stability. Autophagy. 2021;17(11):3444–60.
de la Torre Gomez C, Goreham RV, Bech Serra JJ, Nann T, Kussmann M. “Exosomics”—a review of biophysics, biology and biochemistry of exosomes with a focus on human breast milk. Front Genet. 2018;9.
Wu D, Yan J, Shen X, Sun Y, Thulin M, Cai Y, et al. Profiling surface proteins on individual exosomes using a proximity barcoding assay. Nat Commun. 2019;10(1):3854.
Vallabhajosyula P, Korutla L, Habertheuer A, Yu M, Rostami S, Yuan C-X, et al. Tissue-specific exosome biomarkers for noninvasively monitoring immunologic rejection of transplanted tissue. J Clin Investig. 2017;127(4):1375–91.
Ayala-Mar S, Donoso-Quezada J, González-Valdez J. Clinical implications of exosomal PD-L1 in cancer immunotherapy. J Immunol Res. 2021;2021:8839978.
Khan N, Afaq F, Mukhtar H. Cancer chemoprevention through dietary antioxidants: progress and promise. Antioxid Redox Signal. 2008;10(3):475–510.
Nelson SK, Bose SK, Grunwald GK, Myhill P, McCord JM. The induction of human superoxide dismutase and catalase in vivo: a fundamentally new approach to antioxidant therapy. Free Radic Biol Med. 2006;40(2):341–7.
Liu J, Du J, Zhang Y, Sun W, Smith BJ, Oberley LW, et al. Suppression of the malignant phenotype in pancreatic cancer by overexpression of phospholipid hydroperoxide glutathione peroxidase. Hum Gene Ther. 2006;17(1):105–16.
Teoh-Fitzgerald ML, Fitzgerald MP, Zhong W, Askeland RW, Domann FE. Epigenetic reprogramming governs EcSOD expression during human mammary epithelial cell differentiation, tumorigenesis and metastasis. Oncogene. 2014;33(3):358–68.
Gao P, Zhang H, Dinavahi R, Li F, Xiang Y, Raman V, et al. HIF-dependent antitumorigenic effect of antioxidants in vivo. Cancer Cell. 2007;12(3):230–8.
Alpha-Tocopherol BCCPSG. The effect of vitamin E and beta carotene on the incidence of lung cancer and other cancers in male smokers. N Engl J Med. 1994;330(15):1029–35.
Omenn GS, Goodman GE, Thornquist MD, Balmes J, Cullen MR, Glass A, et al. Effects of a combination of beta carotene and vitamin A on lung cancer and cardiovascular disease. N Engl J Med. 1996;334(18):1150–5.
Seifried HE, Anderson DE, Fisher EI, Milner JA. A review of the interaction among dietary antioxidants and reactive oxygen species. J Nutr Biochem. 2007;18(9):567–79.
Klein EA, Thompson IM Jr, Tangen CM, Crowley JJ, Lucia MS, Goodman PJ, et al. Vitamin E and the risk of prostate cancer: the Selenium and Vitamin E Cancer Prevention Trial (SELECT). JAMA. 2011;306(14):1549–56.
Peiris-Pages M, Martinez-Outschoorn UE, Sotgia F, Lisanti MP. Metastasis and oxidative stress: are antioxidants a metabolic driver of progression? Cell Metab. 2015;22(6):956–8.
Piskounova E, Agathocleous M, Murphy MM, Hu Z, Huddlestun SE, Zhao Z, et al. Oxidative stress inhibits distant metastasis by human melanoma cells. Nature. 2015;527(7577):186–91.
Wiel C, Le Gal K, Ibrahim MX, Jahangir CA, Kashif M, Yao H, et al. BACH1 stabilization by antioxidants stimulates lung cancer metastasis. Cell. 2019;178(2):330-45 e22.
Alexandre J, Hu Y, Lu W, Pelicano H, Huang P. Novel action of paclitaxel against cancer cells: bystander effect mediated by reactive oxygen species. Can Res. 2007;67(8):3512–7.
Pasquier E, Carre M, Pourroy B, Camoin L, Rebai O, Briand C, et al. Antiangiogenic activity of paclitaxel is associated with its cytostatic effect, mediated by the initiation but not completion of a mitochondrial apoptotic signaling pathway. Mol Cancer Ther. 2004;3(10):1301–10.
Luo X, Li Z, Lin S, Le T, Ittensohn M, Bermudes D, et al. Antitumor effect of VNP20009, an attenuated Salmonella, in murine tumor models. Oncol Res. 2001;12(11–12):501–8.
Zheng LM, Luo X, Feng M, Li Z, Le T, Ittensohn M, et al. Tumor amplified protein expression therapy: Salmonella as a tumor-selective protein delivery vector. Oncol Res. 2000;12(3):127–35.
Jia LJ, Xu HM, Ma DY, Hu QG, Huang XF, Jiang WH, et al. Enhanced therapeutic effect by combination of tumor-targeting Salmonella and endostatin in murine melanoma model. Cancer Biol Ther. 2005;4(8):840–5.
Chang HW, Kim MR, Lee HJ, Lee HM, Kim GC, Lee YS, et al. p53/BNIP3-dependent mitophagy limits glycolytic shift in radioresistant cancer. Oncogene. 2019;38(19):3729–42.
Firczuk M, Bajor M, Graczyk-Jarzynka A, Fidyt K, Goral A, Zagozdzon R. Harnessing altered oxidative metabolism in cancer by augmented prooxidant therapy. Cancer Lett. 2020;471:1–11.
Berndtsson M, Hagg M, Panaretakis T, Havelka AM, Shoshan MC, Linder S. Acute apoptosis by cisplatin requires induction of reactive oxygen species but is not associated with damage to nuclear DNA. Int J Cancer. 2007;120(1):175–80.
Hwang PM, Bunz F, Yu J, Rago C, Chan TA, Murphy MP, et al. Ferredoxin reductase affects p53-dependent, 5-fluorouracil-induced apoptosis in colorectal cancer cells. Nat Med. 2001;7(10):1111–7.
Varbiro G, Veres B, Gallyas F Jr, Sumegi B. Direct effect of Taxol on free radical formation and mitochondrial permeability transition. Free Radic Biol Med. 2001;31(4):548–58.
Chiu WH, Luo SJ, Chen CL, Cheng JH, Hsieh CY, Wang CY, et al. Vinca alkaloids cause aberrant ROS-mediated JNK activation, Mcl-1 downregulation, DNA damage, mitochondrial dysfunction, and apoptosis in lung adenocarcinoma cells. Biochem Pharmacol. 2012;83(9):1159–71.
Ogura A, Oowada S, Kon Y, Hirayama A, Yasui H, Meike S, et al. Redox regulation in radiation-induced cytochrome c release from mitochondria of human lung carcinoma A549 cells. Cancer Lett. 2009;277(1):64–71.
Maier P, Hartmann L, Wenz F, Herskind C. Cellular pathways in response to ionizing radiation and their targetability for tumor radiosensitization. Int J Mol Sci. 2016;17(1).
Huang SX, Yun BS, Ma M, Basu HS, Church DR, Ingenhorst G, et al. Leinamycin E1 acting as an anticancer prodrug activated by reactive oxygen species. Proc Natl Acad Sci U S A. 2015;112(27):8278–83.
Wang W, Kryczek I, Dostal L, Lin H, Tan L, Zhao L, et al. Effector T cells abrogate stroma-mediated chemoresistance in ovarian cancer. Cell. 2016;165(5):1092–105.
Rust R, Gantner C, Schwab ME. Pro- and antiangiogenic therapies: current status and clinical implications. Faseb J. 2019;33(1):34–48.
Fukumura D, Kloepper J, Amoozgar Z, Duda DG, Jain RK. Enhancing cancer immunotherapy using antiangiogenics: opportunities and challenges. Nat Rev Clin Oncol. 2018;15(5):325–40.
Ebos JM, Lee CR, Cruz-Munoz W, Bjarnason GA, Christensen JG, Kerbel RS. Accelerated metastasis after short-term treatment with a potent inhibitor of tumor angiogenesis. Cancer Cell. 2009;15(3):232–9.
Kudo-Saito C, Shirako H, Takeuchi T, Kawakami Y. Cancer metastasis is accelerated through immunosuppression during Snail-induced EMT of cancer cells. Cancer Cell. 2009;15(3):195–206.
Paez-Ribes M, Allen E, Hudock J, Takeda T, Okuyama H, Vinals F, et al. Antiangiogenic therapy elicits malignant progression of tumors to increased local invasion and distant metastasis. Cancer Cell. 2009;15(3):220–31.
Rapisarda A, Melillo G. Overcoming disappointing results with antiangiogenic therapy by targeting hypoxia. Nat Rev Clin Oncol. 2012;9(7):378–90.
Tseng FJ, Chen YC, Lin YL, Tsai NM, Lee RP, Chung YS, et al. A fusion protein with the receptor-binding domain of vascular endothelial growth factor-A (VEGF-A) is an antagonist of angiogenesis in cancer treatment: Simultaneous blocking of VEGF receptor-1 and 2. Cancer Biol Ther. 2010;10(9):865–73.
Jain RK. Normalization of tumor vasculature: an emerging concept in antiangiogenic therapy. Science. 2005;307(5706):58–62.
Guo F, Cui J. Anti-angiogenesis: opening a new window for immunotherapy. Life Sci. 2020;258: 118163.
Voron T, Colussi O, Marcheteau E, Pernot S, Nizard M, Pointet AL, et al. VEGF-A modulates expression of inhibitory checkpoints on CD8+ T cells in tumors. J Exp Med. 2015;212(2):139–48.
Gabrilovich D, Ishida T, Oyama T, Ran S, Kravtsov V, Nadaf S, et al. Vascular endothelial growth factor inhibits the development of dendritic cells and dramatically affects the differentiation of multiple hematopoietic lineages in vivo. Blood. 1998;92(11):4150–66.
Chaudhary B, Khaled YS, Ammori BJ, Elkord E. Neuropilin 1: function and therapeutic potential in cancer. Cancer Immunol Immunother. 2014;63(2):81–99.
Teicher BA. A systems approach to cancer therapy (Antioncogenics + standard cytotoxics–>mechanism(s) of interaction). Cancer Metastasis Rev. 1996;15(2):247–72.
Yang T, Xiao H, Liu X, Wang Z, Zhang Q, Wei N, et al. Vascular normalization: a new window opened for cancer therapies. Front Oncol. 2021;11: 719836.
Kuo CL, Chou HY, Lien HW, Yeh CA, Wang JR, Chen CH, et al. A Fc-VEGF chimeric fusion enhances PD-L1 immunotherapy via inducing immune reprogramming and infiltration in the immunosuppressive tumor microenvironment. Cancer Immunol Immunotherapy. 2022.
Habtetsion T, Ding ZC, Pi W, Li T, Lu C, Chen T, et al. Alteration of tumor metabolism by CD4+ T cells leads to TNF-alpha-dependent intensification of oxidative stress and tumor cell death. Cell Metab. 2018;28(2):228–426.
Xu X, Saw PE, Tao W, Li Y, Ji X, Bhasin S, et al. ROS-responsive polyprodrug nanoparticles for triggered drug delivery and effective cancer therapy. Adv Mater. 2017;29(33).
Tao W, He Z. ROS-responsive drug delivery systems for biomedical applications. Asian J Pharm Sci. 2018;13(2):101–12.
Yang B, Gao J, Pei Q, Xu H, Yu H. Engineering prodrug nanomedicine for cancer immunotherapy. Adv Sci (Weinh). 2020;7(23):2002365.
Sung YC, Jin PR, Chu LA, Hsu FF, Wang MR, Chang CC, et al. Delivery of nitric oxide with a nanocarrier promotes tumour vessel normalization and potentiates anti-cancer therapies. Nat Nanotechnol. 2019;14(12):1160–9.
Chen B, Gao A, Tu B, Wang Y, Yu X, Wang Y, et al. Metabolic modulation via mTOR pathway and anti-angiogenesis remodels tumor microenvironment using PD-L1-targeting codelivery. Biomaterials. 2020;255: 120187.
Chung LY, Tang SJ, Wu YC, Yang KC, Huang HJ, Sun GH, et al. Platinum-based combination chemotherapy triggers cancer cell death through induction of BNIP3 and ROS, but not autophagy. J Cell Mol Med. 2020;24(2):1993–2003.
Yan J, Yun H, Yang Y, Jing B, Feng C, Song-bin F. Upregulation of BNIP3 promotes apoptosis of lung cancer cells that were induced by p53. Biochem Biophys Res Commun. 2006;346(2):501–7.
Rikka S, Quinsay MN, Thomas RL, Kubli DA, Zhang X, Murphy AN, et al. Bnip3 impairs mitochondrial bioenergetics and stimulates mitochondrial turnover. Cell Death Differ. 2011;18(4):721–31.
Glick D, Zhang W, Beaton M, Marsboom G, Gruber M, Simon MC, et al. BNip3 regulates mitochondrial function and lipid metabolism in the liver. Mol Cell Biol. 2012;32(13):2570–84.
Mercy L, Pauw A, Payen L, Tejerina S, Houbion A, Demazy C, et al. Mitochondrial biogenesis in mtDNA-depleted cells involves a Ca2+-dependent pathway and a reduced mitochondrial protein import. FEBS J. 2005;272(19):5031–55.
Sun Y, Lu D, Yin Y, Song J, Liu Y, Hao W, et al. PTENalpha functions as an immune suppressor and promotes immune resistance in PTEN-mutant cancer. Nat Commun. 2021;12(1):5147.
Liu Y, Yan W, Tohme S, Chen M, Fu Y, Tian D, et al. Hypoxia induced HMGB1 and mitochondrial DNA interactions mediate tumor growth in hepatocellular carcinoma through Toll-like receptor 9. J Hepatol. 2015;63(1):114–21.
Yang L, Ye F, Zeng L, Li Y, Chai W. Knockdown of HMGB1 suppresses hypoxia-induced mitochondrial biogenesis in pancreatic cancer cells. Onco Targets Ther. 2020;13:1187–98.
Chanmee T, Ontong P, Konno K, Itano N. Tumor-associated macrophages as major players in the tumor microenvironment. Cancers (Basel). 2014;6(3):1670–90.
Veglia F, Tyurin VA, Mohammadyani D, Blasi M, Duperret EK, Donthireddy L, et al. Lipid bodies containing oxidatively truncated lipids block antigen cross-presentation by dendritic cells in cancer. Nat Commun. 2017;8(1):2122.
Luís A, Martins JD, Silva A, Ferreira I, Cruz MT, Neves BM. Oxidative stress-dependent activation of the eIF2α–ATF4 unfolded protein response branch by skin sensitizer 1-fluoro-2,4-dinitrobenzene modulates dendritic-like cell maturation and inflammatory status in a biphasic manner [corrected]. Free Radic Biol Med. 2014;77:217–29.
Fundora Ramos MI, Maden LB, Casanova FO, Cruz FH, Reyes CS, Gato AH, et al. Oncoxin-Viusid(®) may improve quality of life and survival in patients with hormone-refractory prostate cancer undergoing onco-specific treatments. Mol Clin Oncol. 2021;14(1):5.
Motzer RJ, Powles T, Atkins MB, Escudier B, McDermott DF, Alekseev BY, et al. Final overall survival and molecular analysis in IMmotion151, a Phase 3 trial comparing atezolizumab plus bevacizumab vs sunitinib in patients with previously untreated metastatic renal cell carcinoma. JAMA Oncol. 2022;8(2):275–80.
Kojima T, Shah MA. Randomized phase III KEYNOTE-181 study of pembrolizumab versus chemotherapy in advanced esophageal cancer. J Clin Oncol. 2020;38(35):4138–48.
Adenis A, Kulkarni AS, Girotto GC, Al-Rajabi R, Norquist J, Amonkar M, et al. Impact of pembrolizumab versus chemotherapy as second-line therapy for advanced esophageal cancer on health-related quality of life in KEYNOTE-181. J Clin Oncol. 2022;40(4):382–91.
Uppaluri R, Campbell KM, Mudianto T, Riley R, Zhou L, Jo VY, et al. Neoadjuvant and adjuvant pembrolizumab in resectable locally advanced, human papillomavirus-unrelated head and neck cancer: a multicenter Phase II Trial. Clin Cancer Res. 2020;26(19):5140–52.
Acknowledgements
Not applicable.
Funding
This work was supported by grants from the Ministry of Science and Technology (MOST107-2314-B-006-051, MOST108-2320-B-400-008-MY3, MOST108-2314-B-006-005, MOST109-2314-B-400-045) and the National Health Research Institutes (109/110/111A1-CA-PP-07), Taiwan to A. Y.-L. Lee.
Author information
Authors and Affiliations
Contributions
C.-L.K., A.P.B., and A.Y.-L.L. conceived the manuscript with input from all authors. The background was written by C.-L.K., A.P.B., and Y. K.L. The role of oxidative stress in the tumor progression and cancer therapy was written by A.P.B., Y.-C.L., Y. K.L., and H.-Y.C. The survival signalings of mitochondrial oxidative stress by chaperones in cancer cells was written by C.-L.K., A.P.B., and V.T. The impact of mitochondrial ROS-induced inflammation on the TME was written by C.-L.K., Y.-C.L., H.-Y.C., and H.-W.L. Mitochondrial ROS-induced mtDNA/EV contributes to inflammation and PD-L1-mediated immunoescape was written by A.P.B., L.-C.C., and A.N.C.. ROS-modulating strategy in the combinatory cancer immunotherapy for reprogramming immunosuppressive TME was written by C.-L.K., Y. K.L., V.T., and H.-Y.C. Conclusions and Perspectives was written by C.-L.K. and A.Y.-L.L. And C.-L.K., A.P.B., and AY-LL was responsible for the final revision of the manuscript. XA.P.B. and C.-L.K. provided Graphic abstract of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors have no conflicts of interest to declare.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Kuo, CL., Ponneri Babuharisankar, A., Lin, YC. et al. Mitochondrial oxidative stress in the tumor microenvironment and cancer immunoescape: foe or friend?. J Biomed Sci 29, 74 (2022). https://doi.org/10.1186/s12929-022-00859-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12929-022-00859-2