
Abstract
Oral squamous cell carcinoma (OSCC), the most prevalent form of oral cancer, poses significant challenges to the medical community due to its high recurrence rate and low survival rate. Mitochondrial Damage-Related Genes (MDGs) have been closely associated with the occurrence, metastasis, and progression of OSCC. Consequently, we constructed a prognostic model for OSCC based on MDGs and identified potential mitochondrial damage-related biomarkers. Gene expression profiles and relevant clinical information were obtained from The Cancer Genome Atlas (TCGA) database. Differential analysis was conducted to identify MDGs associated with OSCC. COX analysis was employed to screen seven prognosis-related MDGs and build a prognostic prediction model for OSCC. Cases were categorized into low-risk or high-risk groups based on the optimal risk score threshold. Kaplan-Meier (KM) analysis revealed significant survival differences (P < 0.05). Additionally, the area under the ROC curve (AUC) for patient survival at 1 year, 3 years, and 5 years were 0.687, 0.704, and 0.70, respectively, indicating a high long-term predictive accuracy of the prognostic model. To enhance predictive accuracy, age, gender, risk score, and TN staging were incorporated into a nomogram and verified using calibration curves. Risk scoring based on MDGs was identified as a potential independent prognostic biomarker. Furthermore, BID and SLC25A20 were identified as two potential independent mitochondrial damage-related prognostic biomarkers, offering new therapeutic targets for OSCC.
Similar content being viewed by others
Introduction
Oral squamous cell carcinoma (OSCC) presents a global healthcare challenge, contributing to over 170,000 annual fatalities [1, 2]. Recent epidemiological investigations have underscored the correlation between OSCC occurrence and risk factors such as age, alcohol consumption, smoking, and betel nut chewing [3]. Although the oral cavity is relatively easy to examine, most patients with OSCC present with advanced-stage tumors at the time of diagnosis, resulting in poor treatment outcomes [4]. Based on tumor staging and pathological diagnosis, the primary treatment methods for OSCC include surgery, radiotherapy, and chemotherapy. Generally, early-stage OSCC is primarily treated with surgery alone, while locally advanced OSCC is managed with a combination of surgery and adjuvant immunochemotherapy (such as cisplatin, paclitaxel, pembrolizumab, and tislelizumab). For OSCC with distant metastasis, systemic therapy is the main approach [5]. Surgery is the most effective treatment for oral squamous cell carcinoma, but the surgical trauma significantly impacts the function and aesthetics of the oral and maxillofacial region [6]. Additionally, chemotherapy can cause hair loss, nausea, vomiting, and infection, while radiotherapy can result in temporary or permanent damage to healthy tissues, severely affecting the quality of life [7]. Moreover, approximately one-third of patients still face the risk of recurrence and the development of resistance to radiotherapy and chemotherapy after undergoing these traditional treatments, posing a serious threat to public health [8]. Currently, OSCC remains plagued by high local recurrence rates and frequent metastasis. Most diagnoses occur in advanced stages, leading to a disheartening five-year survival rate of less than 50% [9]. Consequently, unraveling the pathophysiological mechanisms underlying OSCC and identifying effective interventions or pharmaceuticals to impede or decelerate its progression, ultimately reducing mortality rates, stands as an imminent imperative.
The pathogenic mechanisms of OSCC are intricate, and despite extensive research, they remain incompletely elucidated [10]. Mitochondria, central hubs for cellular energy supply and metabolism, are pivotal in instigating metabolic perturbations. Mitochondrial damage contributes to the development of various cancers, including OSCC [11]. Impaired mitochondrial function during tumorigenesis results in significant cellular damage due to disrupted energy metabolism [12]. Ongoing research has unveiled that mitigating mitochondrial oxidative stress, promoting mitochondrial biogenesis, stimulating mitochondrial autophagy, and inhibiting mitochondrial apoptosis pathways can markedly attenuate tumorigenesis in cancer models [13]. These findings underscore the critical role of mitochondrial homeostasis disruption in tumorigenesis. Metabolic alterations including mitochondrial dysfunction and oxidative phosphorylation disturbances, among others, are one of the hallmark features of OSCC [14]. Research has shown that TIMELESS (Timeless circadian regulator) influences cell proliferation and metabolic pathways by enhancing glycolysis and reducing oxidative phosphorylation through the SIRT1 pathway, thereby exacerbating the progression of OSCC [15]. Zhang et al.‘s study indicates that ALT inhibits the proliferation of oral squamous cell carcinoma cells and promotes apoptosis by impairing mitochondrial homeostasis and regulating Drp1 [16]. Swarnendu Bag and colleagues, based on NMR serum metabolomics studies, found that the levels of glutamine, propionate, acetone, and choline can accurately differentiate OSCC patients from healthy controls [17]. Research concerning mitochondrial damage in OSCC remains notably deficient and necessitates immediate attention.
Mitochondrial damage-related genes (MDGs) are a group of genes involved in maintaining mitochondrial function and structure [18]. Their aberrant expression or dysfunction has been closely associated with the development of many diseases, particularly in tumor biology. In the context of OSCC, MDGs play a crucial role as they are not only involved in the metabolic reprogramming of tumor cells but also associated with tumor aggressiveness and drug resistance [19]. Current research indicates a significant association between mitochondrial damage and overall survival as well as the risk of OSCC [20, 21]. Nonetheless, the prognostic potential of other MDGs in OSCC requires further investigation. This study aimed to analyze the differentially expressed MDGs in OSCC. A clinical prognosis model for OSCC patients, based on MDGs, was constructed using Cox analysis to calculate risk scores. Furthermore, we validated our findings using a dataset obtained from the Gene Expression Omnibus (GEO) database. This research provides a novel perspective on understanding the potential mechanisms underlying OSCC onset and progression, as well as identifying new therapeutic targets for OSCC.
Materials and methods
Data retrieval
The RNA-seq data of 370 cases were downloaded from The Cancer Genome Atlas (TCGA) database (https://portal.gdc.cancer.gov/). Our study included 338 OSCC samples and 32 control samples. Relevant clinical information is shown in Supplementary Table 1 . The mean survival time of the patient cohort was approximately 894.7 days. The cancerous sites included oral tongue, oral cavity, floor of the mouth, palate, buccal mucosa, and oropharynx. A total of 731 MDGs were extracted from GeneCards (https://www.genecards.org) [22] with a relevance score ≥ 10 (Supplementary Table 2). Subsequently, differential expression analysis was conducted in the R software, with a cutoff criterion of |log2 (fold change) | > 0.585 and adjusted P-value < 0.05.
GO and KEGG analysis
The “ClusterProfiler” package in R software was utilized for gene ontology (GO) analysis of these differentially expressed mitochondrial damage-related genes. Additionally, Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis was employed to annotate their functions [23, 24]. A significance level of P < 0.05 was set.
Cox regression analysis
Initially, all MDGs were subjected to univariate Cox regression to identify prognostic genes with a significance level below 0.05. Subsequently, a stepwise regression analysis was employed to construct the Cox risk model based on these prognostic genes. Finally, seven MDGs were incorporated into the risk Cox regression, and the Cox model was visualized in R software based on risk scores assigned to each patient [25]. Patients’ risk scores were sorted in ascending order, and the median was determined as the threshold to stratify patients into high-risk and low-risk groups. Furthermore, receiver operating characteristic (ROC) analysis was used to assess the predictive capability of the features.
Both univariate and multivariate Cox regression analyses were employed to analyze clinical features, including age, gender, grading, staging, T stage, N stage, and risk score. Due to the presence of a substantial amount of missing data, the M stage was excluded.
Construction and validation of nomogram
The integration of feature data such as survival status or survival time was carried out using the R software’s rms package. Subsequently, the total score was calculated to predict the 1-year, 3-year, and 5-year survival rates of OSCC cases. The scores for each factor were summed and integrated into the constructed nomogram, allowing for the prediction of the 1-year, 3-year, and 5-year survival rates of OSCC cases. Subsequently, a calibration curve was plotted to evaluate the accuracy of the nomogram’s predictive performance, with the 45° line representing the optimal prediction. The clinical value of various OSCC indicators was evaluated using DCA.
Immune infiltration analysis
CIBERSORT is a deconvolution algorithm used to calculate the proportions of 22 immune cell types in each sample, along with their corresponding p-values. Samples with a p-value < 0.05 were deemed statistically significant. Spearman correlation analysis was conducted between these genes and the relative proportions of immune cells. The results were visualized using the heatmap R package [26].
Quantitative real-time PCR (qPCR)
A total of 5 patients (Clinical features are shown in Supplementary Table 3) aged 50–72 years old were selected from Yijishan Hospital of Wuhu City. This study was approved by the Ethics Committee of Wannan Medical College, Wuhu City, Anhui Province, and all patients or their families gave informed consent and signed informed consent (No:2022-9). Healthy oral tissue samples, serving as the control group, were obtained from areas adjacent to the tumor samples. Total RNA extraction using TRIzol reagent (Yeasen, Shanghai, China) was performed as previously described [27]. Total RNA was then converted to cDNA by Hifair® II 1st Strand cDNA Synthesis SuperMix (Yeasen, Shanghai, China) and subjected to qRT-PCR with Hieff® qPCR SYBR Green Master Mix (Yeasen, Shanghai, China). In total, three duplicates of each sample were analyzed. Once the expression levels of GAPDH were analyzed, the results were calculated in 2−ΔΔCt. The sequences of the PCR primers were as follows:
SLC25A20-forward 5′- ACCGAGTTTGCCTGGACAACCT - 3′;
SLC25A20-reverse 5′- CCCAAAGAAGCACACGGCAAAC -3′.
BID-forward 5′- TGGGACACTGTGAACCAGGAGT - 3′;
BID -reverse 5′- GAGGAAGCCAAACACCAGTAGG -3′.
GAPDH-forward 5′- GTCTCCTCTGACTTCAACAGCG - 3′;
GAPDH-reverse 5′- ACCACCCTGTTGCTGTAGCCAA -3′.
Statistical analysis
Statistical analysis was conducted using R software (version 4.3.1). Kaplan-Meier survival curves and log-rank tests were utilized to assess differences in survival rates between two patient groups. p-values were determined via Student’s t-test. In addition to the specified statistical criteria, all statistically significant results were required to meet a threshold of P < 0.05.
Results
Analysis of differential expression of MDGs
To initiate our investigation, we employed the Limma software package to perform differential analysis on MDGs. This volcano plots led to the identification of 85 upregulated genes and 87 downregulated genes among the MDGs (Fig. 1). While we understand the involvement of these genes in mitochondrial dysfunction, it is essential to delve deeper into their specific pathways and biological functions through GO and KEGG analyses. The KEGG pathway analysis unveiled associations between differentially expressed MDGs and pathways such as oxidative phosphorylation and carbon metabolism (Fig. 2A). Furthermore, GO analysis highlighted the enrichment of differentially expressed MDGs in various biological processes, including cellular respiration, energy derivation by oxidation of organic compounds, and aerobic respiration. The cellular component section revealed that these MDGs predominantly localized in the mitochondrial inner membrane, mitochondrial protein-containing complexes, and the mitochondrial matrix. Molecular function analysis indicated that the selected MDGs were notably enriched in electron transfer activity, acidoreduction-driven active transmembrane transporter activity, and proton transmembrane transporter activity (Fig. 2B).

Analysis of MDGs. Volcano plot displaying MDGs from the TCGA database. In the plot, red dots represent significantly upregulated genes, while blue indicates significantly downregulated genes

Functional enrichment analysis. (A) KEGG Pathway Analysis. (B) GO Analysis. BP: Biological Process. CC: Cellular Component. MF: Molecular Function
Establishment of cox regression model
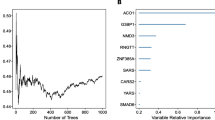
According to univariate Cox regression analysis, 15 MDGs emerged as potential prognostic factors for OSCC. These MDGs were further subjected to multivariate Cox regression and stepwise Cox regression analyses. Ultimately, seven variables were integrated into the Cox model (Fig. 3A), comprising SLC25A20, NLRX1, ETFDH, BID, ALDH2, HADHB, and DDIT4. The risk score was calculated as follows: Risk Score = -(0.1635 * SLC25A20) - (0.0329 * NLRX1) + (0.2832 * ETFDH) + (0.038 * BID) + (0.04154 * ALDH2) + (0.01772 * HADHB) + (0.00628 * DDIT4). Subsequently, based on the median of the Cox formula, OSCC patients from the TCGA database were categorized into high-risk and low-risk groups. Survival analysis revealed a significantly lower overall survival rate in the high-risk group compared to the low-risk group (Fig. 3B). The ROC curves for 1 year, 3 years, and 5 years (Fig. 3C) yielded AUC values of 0.687, 0.704, and 0.70, respectively, indicating that our model based on these seven mitochondrial dysfunction-associated genes is a valuable prognostic tool with satisfactory accuracy and sensitivity for long-term prediction.

Construction of Cox regression models. (A) Multivariate cox regression based on 15 MDGs. (B) KM survival analyses were performed on high-risk and low-risk samples. Patients in the high-risk group had shorter survival times compared to the low-risk group. (C) The ROC curve analyses demonstrated the accuracy and reliability of the prognostic characteristics in determining 1, 3, and 5 years survival outcomes
Clinical value of prognostic features
To investigate whether this prognostic model is correlated with other clinical characteristics of OSCC patients, we constructed nomograms for 1-year, 3-year, and 5-year survival based on the aforementioned predictive factors (Fig. 4A). The results revealed that older patients and those with higher N classifications tend to have higher risk scores. The calibration curves for 1-year, 3-year, and 5-year survival (Fig. 4B) closely approximate the 45-degree line, indicating the accuracy of the nomograms in predicting OSCC survival rates at 1, 3, and 5 years. DCA analysis results indicate that treatment decisions based on risk scores provide a higher net benefit compared to those based on common single clinical parameters such as TN stage and Age. (Fig. 4C).

Clinical relevance of prognostic markers (A) Nomograms predicting the survival period of OSCC patients. The nomograms assess the survival time of OSCC patients at 1, 3, and 5 years by analyzing clinical features (N and Age) along with risk scores. (B) Calibration curves for the columnar charts predicting 1-year, 3-year, and 5-year overall survival periods. (C) Decision curve analysis for the evaluation of the net benefits
Immune infiltration analysis
Subsequently, we employed the CIBERSORT algorithm to assess immune cell infiltration based on gene expression profiles, revealing the proportions of all 22 subtypes of immune cells in each sample (Fig. 5A). These findings indicate differences in immune infiltration between high-risk and low-risk samples. Subsequently, further analysis through Spearman correlation demonstrates the relationship between immune cell infiltration and MDGs expression (Fig. 5B). Notably, the most positively correlated immune cell-MDGs pairing is ETFDH-Dendritic cells (R = 0.35), while the most negatively correlated pairing is SLC25A20-Macrophages M0 cell (R=-0.33).

Correlation between immune infiltration and MDGs. (A) Proportion of infiltrating 22 immune cell types in OSCC patient tissues. (B) Correlation between infiltrating immune cells and seven key MDGs
Identification of relevant MDGs and validation
We conducted LASSO regression on the seven OSCC-related MDGs, performing feature selection and dimensionality reduction to eliminate insignificant regulatory factors (Fig. 6A-B). Subsequently, we established a classifier through multivariate logistic regression to distinguish healthy samples from OSCC samples (Fig. 6C). This classifier comprises two MDGs, and the ROC curve demonstrates excellent performance of these two MDGs in classifying health and OSCC, underscoring the pivotal role of MDGs in OSCC development (Fig. 6D).
To validate the differential expression of the two crucial MDG regulators between OSCC and control groups, we employed an external validation dataset (GSE30784, clinicopathological information has been included in Supplementary Table 4) consisting of 212 samples for further verification. As depicted in Fig. 7A-B, we observed significant downregulation of SLC25A20 (p < 0.01) and substantial upregulation of BID (p < 0.01) in OSCC patients.
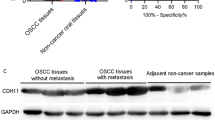
Finally, we performed qPCR by 5 pairs of tissues from tumor and control (the adjacent tumor area) of OSCC patients. The results showed that BID was up-regulated (Fig. 8A) in OSCC while SLC25A20 was down-regulated (Fig. 8B), which is consistent with the results of bioinformatic analysis.

Distinguishing MDGs between control and OSCC samples. (A) Minimum absolute shrinkage and selection operator (LASSO) coefficient curve for the seven OSCC-related MDGs. (B) Partial likelihood deviance plotted against log (λ) values. (C) Establishment of distinguishing features of MDGs through multivariate logistic regression. (D) ROC curve analysis assessing the discriminative ability of MDGs between healthy and OSCC samples, evaluated by AUC values

The predictive roles of two MDGs in OSCC (A-B) Comparison of gene expression of the two MDGs between patients with and without OSCC occurrence in GSE30784.

Expression of SLC25A20 (A) and Bid (B) in tumor tissues and control tissues
Discussion
OSCC presents diagnostic challenges, often remaining undetected during its latent period and frequently diagnosed in advanced stages. Patients diagnosed with OSCC at later stages usually experience distant metastasis at the time of diagnosis [28]. Therefore, the exploration of effective therapeutic targets and sensitive diagnostic biomarkers holds significant importance in understanding the development of OSCC. Some studies have already established OSCC prognostic models using polyamine-related and autophagy-related genes to investigate their prognostic capabilities and potential for targeted therapy [25, 29]. Mitochondrial dysfunction and damage are closely associated with the initiation and progression of cancer [30]. However, research on the impact of MDGs on OSCC prognosis prediction remains limited. Hence, it is essential to explore key MDGs that can serve as prognostic biomarkers and therapeutic targets for OSCC.
This study initially included 731 MDGs for analysis. Through differential analysis, we identified 172 genes exhibiting differential expression in OSCC. KEGG pathway analysis revealed that these MDGs were primarily associated with pathways like oxidative phosphorylation and carbon metabolism. GO analysis further indicated that these differentially expressed MDGs were closely related to mitochondrial composition and function. In fact, some MDGs were identified as potential biomarkers for OSCC. Research has suggested that HADHB may be associated with overall survival and risk in OSCC [20, 21]. However, the relationship between HADHB and OSCC remains unclear. HADHB is associated with fatty acid metabolism and has been reported to be elevated in clear cell renal carcinoma and colorectal cancer [31]. Currently, research on the prognostic value of other MDGs in oral squamous cell carcinoma is still very limited.
Subsequently, the gene signature composed of seven MDGs that we identified demonstrated precise prognostic capabilities for OSCC. According to the KM survival curves from the training cohort, high-risk patients exhibited significantly reduced overall survival compared to low-risk patients. The ROC curves for risk scores at 1-year, 3-year, and 5-year intervals had AUC values of 0.687, 0.704, and 0.70, respectively. The predictive model constructed from seven mitochondrial damage-related genes demonstrated satisfactory performance in the long-term prediction of overall survival in OSCC. Through univariate and multivariate Cox regression analyses of patient clinical characteristics and risk scores, it was established that risk scores serve as independent prognostic factors. Subsequently, a nomogram was devised by integrating the risk model with clinical case features. Calibration curves indicated the commendable predictive capacity of our nomogram for oral squamous cell carcinoma prognosis. These findings underscore that integrating risk scores with other clinical factors contributes to accurate predictions of patient survival rates.
Increasing evidence suggests that OSCC behaves as an immunosuppressive disease [32, 33]. Within the immune tumor microenvironment of OSCC, immune cells such as T cells are often found in an inactivated state, bolstering the cancer cells’ immunosuppressive defenses [34]. Throughout the progression of OSCC, mast cells accumulate near blood vessels surrounding tumors, secreting several factors that promote angiogenesis and inhibit immune responses [35]. Our CIBERSORT analysis results reveal that, correlation demonstrates the relationship between immune cell infiltration and seven MDGs expression. In summary, our study reveals that immune cell infiltration and MDG expression are intricately linked in OSCC.
Lastly, to mitigate potential biases stemming from a single training dataset, we incorporated an additional dataset, GSE30784, for independent validation of the two prognostic MDGs (SLC25A20 and BID). BID serves as a p53 effector, and its cleavage promotes conformational changes in mitochondrial-associated BAX, thereby facilitating mitochondrial dysfunction and cytochrome c release [36]. Research suggests that dimethyl fumarate (DMF) can upregulate BID expression, promoting apoptosis in oral squamous cell carcinoma [37]. SLC25A20 is a recognized carnitine transporter, and alterations in its expression have been associated with the development of inflammation and inflammation-mediated cancers [38].
Conclusion
In this study, we performed a comprehensive analysis to identify seven MDGs associated with the prognosis of OSCC. Subsequently, we successfully constructed a risk model for predicting the prognosis of OSCC patients, with the risk score emerging as a potential independent prognostic biomarker for OSCC. Additionally, we identified SLC25A20 and BID as two promising independent prognostic biomarkers with potential applications in OSCC diagnosis and treatment. These findings contribute to more accurate prognosis assessment for OSCC and offer the possibility of personalized therapeutic approaches for these patients.
Data availability
The datasets presented in this study can be found in online repositories TCGA database (https://portal.gdc.cancer.gov/) and GEO database (GSE30784).
References
Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, Bray F. Global Cancer statistics 2020: GLOBOCAN estimates of incidence and Mortality Worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021;71(3):209–49.
Coletta RD, Yeudall WA, Salo T. Grand challenges in oral cancers. Front Oral Health. 2020;1:3.
Chamoli A, Gosavi AS, Shirwadkar UP, Wangdale KV, Behera SK, Kurrey NK, Kalia K, Mandoli A. Overview of oral cavity squamous cell carcinoma: risk factors, mechanisms, and diagnostics. Oral Oncol. 2021;121:105451.
Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. CA Cancer J Clin. 2019;69(1):7–34.
Pfister DG, Spencer S, Adelstein D, Adkins D, Anzai Y, Brizel DM, Bruce JY, Busse PM, Caudell JJ, Cmelak AJ, et al. Head and Neck cancers, Version 2.2020, NCCN Clinical Practice guidelines in Oncology. J Natl Compr Cancer Network: JNCCN. 2020;18(7):873–98.
Li H, Zhang Y, Xu M, Yang D. Current trends of targeted therapy for oral squamous cell carcinoma. J Cancer Res Clin Oncol. 2022;148(9):2169–86.
Zhang M, Liang J, Yang Y, Liang H, Jia H, Li D. Current trends of targeted drug delivery for oral Cancer therapy. Front Bioeng Biotechnol. 2020;8:618931.
Greenberg JS, El Naggar AK, Mo V, Roberts D, Myers JN. Disparity in pathologic and clinical lymph node staging in oral tongue carcinoma. Implication for therapeutic decision making. Cancer. 2003;98(3):508–15.
Rivera C, Venegas B. Histological and molecular aspects of oral squamous cell carcinoma (review). Oncol Lett. 2014;8(1):7–11.
Georgaki M, Theofilou VI, Pettas E, Stoufi E, Younis RH, Kolokotronis A, Sauk JJ, Nikitakis NG. Understanding the complex pathogenesis of oral cancer: a comprehensive review. Oral Surg Oral Med Oral Pathol Oral Radiol. 2021;132(5):566–79.
Carew JS, Huang P. Mitochondrial defects in cancer. Mol Cancer. 2002;1:9.
Boese AC, Kang S. Mitochondrial metabolism-mediated redox regulation in cancer progression. Redox Biol. 2021;42:101870.
Zhu Y, Dean AE, Horikoshi N, Heer C, Spitz DR, Gius D. Emerging evidence for targeting mitochondrial metabolic dysfunction in cancer therapy. J Clin Invest. 2018;128(9):3682–91.
Vitorio JG, Duarte-Andrade FF, Dos Santos Fontes Pereira T, Fonseca FP, Amorim LSD, Martins-Chaves RR, Gomes CC, Canuto GAB, Gomez RS. Metabolic landscape of oral squamous cell carcinoma. Metabolomics. 2020;16(10):105.
Chen Y, Han Z, Zhang L, Gao C, Wei J, Yang X, Han Y, Li Y, Zhang C, Wei Y, et al. TIMELESS promotes reprogramming of glucose metabolism in oral squamous cell carcinoma. J Translational Med. 2024;22(1):21.
Zhang Y, Yang B, Tu C, Ping Y, Chen S, Wu T, Zhao Z, Mao Y, Yang Z, Cao Z, et al. Mitochondrial impairment and downregulation of Drp1 phosphorylation underlie the antiproliferative and proapoptotic effects of alantolactone on oral squamous cell carcinoma cells. J Translational Med. 2023;21(1):328.
Bag S, Banerjee DR, Basak A, Das AK, Pal M, Banerjee R, Paul RR, Chatterjee J. NMR ((1)H and (13)C) based signatures of abnormal choline metabolism in oral squamous cell carcinoma with no prominent Warburg effect. Biochem Biophys Res Commun. 2015;459(4):574–8.
Lin MM, Liu N, Qin ZH, Wang Y. Mitochondrial-derived damage-associated molecular patterns amplify neuroinflammation in neurodegenerative diseases. Acta Pharmacol Sin. 2022;43(10):2439–47.
Zong Y, Li H, Liao P, Chen L, Pan Y, Zheng Y, Zhang C, Liu D, Zheng M, Gao J. Mitochondrial dysfunction: mechanisms and advances in therapy. Signal Transduct Target Ther. 2024;9(1):124.
Zhang J, Ma C, Qin H, Wang Z, Zhu C, Liu X, Hao X, Liu J, Li L, Cai Z. Construction and validation of a metabolic-related genes prognostic model for oral squamous cell carcinoma based on bioinformatics. BMC Med Genomics. 2022;15(1):269.
Huang ZD, Yao YY, Chen TY, Zhao YF, Zhang C, Niu YM. Construction of prognostic risk prediction model of oral squamous cell Carcinoma Based on Nine Survival-Associated metabolic genes. Front Physiol. 2021;12:609770.
Stelzer G, Rosen N, Plaschkes I, Zimmerman S, Twik M, Fishilevich S, Stein TI, Nudel R, Lieder I, Mazor Y et al. The GeneCards suite: from Gene Data Mining to Disease Genome sequence analyses. Curr Protoc Bioinf 2016, 54:1 30 31–31 30 33.
Kanehisa M. Toward understanding the origin and evolution of cellular organisms. Protein Sci. 2019;28(11):1947–51.
Kanehisa M, Furumichi M, Sato Y, Kawashima M, Ishiguro-Watanabe M. KEGG for taxonomy-based analysis of pathways and genomes. Nucleic Acids Res. 2023;51(D1):D587–92.
Huang GZ, Lu ZY, Rao Y, Gao H, Lv XZ. Screening and identification of autophagy-related biomarkers for oral squamous cell carcinoma (OSCC) via integrated bioinformatics analysis. J Cell Mol Med. 2021;25(9):4444–54.
Newman AM, Liu CL, Green MR, Gentles AJ, Feng W, Xu Y, Hoang CD, Diehn M, Alizadeh AA. Robust enumeration of cell subsets from tissue expression profiles. Nat Methods. 2015;12(5):453–7.
Wen W, Huang B, Ye S. Metformin ameliorates epithelial-mesenchymal transition of renal tubular epithelial cells in diabetes by increasing vitamin D receptor expression. Diabetes Metab Syndr Obes. 2022;15:4001–10.
Gonzalez-Moles MA, Aguilar-Ruiz M, Ramos-Garcia P. Challenges in the Early Diagnosis of Oral Cancer, Evidence Gaps and Strategies for Improvement: A Scoping Review of Systematic Reviews. Cancers (Basel) 2022, 14(19).
Tang J, Wu X, Cheng B, Lu Y. Identification of a polyamine-related signature and six novel prognostic biomarkers in oral squamous cell carcinoma. Front Mol Biosci. 2023;10:1073770.
Boland ML, Chourasia AH, Macleod KF. Mitochondrial dysfunction in cancer. Front Oncol. 2013;3:292.
Li M, Jin C, Xu M, Zhou L, Li D, Yin Y. Bifunctional enzyme ATIC promotes propagation of hepatocellular carcinoma by regulating AMPK-mTOR-S6 K1 signaling. Cell Communication Signaling: CCS. 2017;15(1):52.
Kouketsu A, Haruka S, Kuroda K, Hitoshi M, Kensuke Y, Tsuyoshi S, Takahashi T, Hiroyuki K. Myeloid-derived suppressor cells and plasmacytoid dendritic cells are associated with oncogenesis of oral squamous cell carcinoma. J Oral Pathol Med. 2023;52(1):9–19.
Li S, Mai Z, Gu W, Ogbuehi AC, Acharya A, Pelekos G, Ning W, Liu X, Deng Y, Li H, et al. Molecular subtypes of oral squamous cell Carcinoma based on immunosuppression genes using a Deep Learning Approach. Front Cell Dev Biol. 2021;9:687245.
Lafuente Ibanez de Mendoza I, Maritxalar Mendia X, Garcia de la Fuente AM, Quindos Andres G, Aguirre Urizar JM. Role of Porphyromonas gingivalis in oral squamous cell carcinoma development: a systematic review. J Periodontal Res. 2020;55(1):13–22.
Hemmerlein B, Reinhardt L, Wiechens B, Khromov T, Schliephake H, Brockmeyer P. Is CCL2 an important mediator of mast cell-tumor cell interactions in oral squamous cell carcinoma? Int J Mol Sci 2023, 24(4).
Warren CFA, Wong-Brown MW, Bowden NA. BCL-2 family isoforms in apoptosis and cancer. Cell Death Dis. 2019;10(3):177.
Basilotta R, Lanza M, Filippone A, Casili G, Mannino D, De Gaetano F, Chisari G, Colarossi L, Motta G, Campolo M et al. Therapeutic potential of Dimethyl Fumarate in Counteract oral squamous cell carcinoma progression by modulating apoptosis, oxidative stress and epithelial-mesenchymal transition. Int J Mol Sci 2023, 24(3).
Rashid K, Ahmad A, Liang L, Liu M, Cui Y, Liu T. Solute carriers as potential oncodrivers or suppressors: their key functions in malignant tumor formation. Drug Discov Today. 2021;26(7):1689–701.
Acknowledgements
Not applicable.
Funding
This work was supported by the Doctoral Research Initiation Fund of Wannan medical college (WYRCQD2023028), Major Projects of Natural Science Research in Colleges and Universities in Anhui Province (KJ2021ZD0103), Supported by Anhui Province Engineering Research Center for dental materials and application (Grant No: 2024AMCD06), Program for Excellent Sci-tech innovation Teams of Universities in Anhui Province (Grant No: 2023AH010073).
Author information
Authors and Affiliations
Contributions
Data curation and Formal analysis: WWJ; Funding acquisition, Project administration and Supervision: LR; Visualization: WDY; Writing—original draft: WWJ; Writing—review and editing: CL. All authors have read and agreed to the published version of the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This study was approved by the Ethics Committee of Wannan Medical College, Wuhu City, Anhui Province, and all patients or their families gave informed consent and signed informed consent (No:2022-9).
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Wenjie, W., Rui, L., Dongyong, W. et al. Exploring the prognostic landscape of oral squamous cell carcinoma through mitochondrial damage-related genes. BMC Med Genomics 17, 208 (2024). https://doi.org/10.1186/s12920-024-01985-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12920-024-01985-6