
Abstract
Background
Joubert syndrome is a genetically heterogeneous autosomal recessive ciliopathy characterized by the combination of hypoplasia/aplasia of the cerebellar vermis, thickened and elongated superior cerebellar peduncles and a deep interpeduncular fossa, known as “molar tooth sign” associated with hypotonia, respiratory control disturbances and abnormal eye movements. To date, pathogenic variants in over 35 genes are known to cause autosomal recessive Joubert Syndrome, while one gene is associated with X-linked recessive inheritance.
Case presentation
We describe here a non-consanguineous Vietnamese family with Joubert syndrome, a fetus and 10-year-old developmentally delayed boy. Ultrasonography showed ventriculomegaly at 26 + 6 weeks of gestation in the fetus. The 10-year-old-boy was diagnosed with cerebral palsy of unknown origin. Clinical physical examination at the age of 10, he showed clinical features of Joubert syndrome including typical facial dysmorphism, ataxia, severe psychomotor delay, oculomotor apraxia and molar tooth sign on brain MRI. Whole exome sequencing analysis identified a novel compound heterozygous c.725A > G p.Asn242Ser and c.313-3 T > G p.Lys105Valfs*16 TMEM67 variant in the proband and the affected fetus. These two variants were inherited from each parent and confirmed by Sanger sequencing. The variant c.725A > G p.Asn242Ser was previously documented in patients with JS, the novel splice-site c.313-3 T > G p.Lys105Valfs*16 TMEM67 variant produced an aberrant transcript with the loss of four nucleotides of exon 03.
Conclusion
This study confirms the diagnosis of Joubert syndrome in a Vietnamese family and expands the mutational spectrum of TMEM67 sequence variations. We also highlight the importance of molecular approaches to unravel underlying mechanisms of human genetic disorders. Early precise diagnosis could help provide further accurate genetic counseling for recurrence-risk assessment, future diagnostic option, management as well as treatment guidance for rare disorders.
Similar content being viewed by others

Background
Joubert syndrome (JS, MIM 213300) is an autosomal recessive ciliopathy characterized by specific midbrain-hindbrain malformations, recognizable on axial brain magnetic resonance imaging, known as the “Molar Tooth Sign”, hypotonia and developmental delays. Ciliopathies are a group of genetic disorders that are caused by abnormal formation or function of cellular primary cilia in many organs of the human body. To date, over 35 genes are known to cause autosomal recessive JS and one gene causes X-linked JS. Typical clinical features of JS include neonatal hypotonia, abnormal breathing patterns and eye movements, ataxia and developmental delays. Additionally, patients with JS also exhibit variable associated clinical features including retinal dystrophy, ocular colobomas, congenital heart disease, microcystic kidney disease, liver fibrosis, polydactyly, cleft clip and palate [1]. TMEM67 (MIM *609884) encodes the Frizzled-like Wnt receptor, a transmembrane protein (meckelin) that regulates canonical Wnt/β-catenin signaling pathway in the developing cerebellum. Tmem67−/− mutant mice display cerebellar vermis hypoplasia/aplasia, deep interpeduncular fossa and posterior fossa defects compatible with JS phenotype [2]. Biallelic TMEM67 sequence variations cause a wide range of clinical features observed in ciliopathies with multiorgan involvement and different clinical outcomes including JS (MIM 610688), Meckel-Gruber syndrome (MIM 607361), COACH syndrome (Cerebellar vermis hypoplasia, Oligophrenia, Ataxia, Coloboma and Hepatic fibrosis) (MIM 216360), RHYNS syndrome (Retinitis pigmentosa, Hypopituitarism, Nephronophthisis, Skeletal dysplasia), polycystic kidney disease, nephronophthisis-associated ciliopathy (MIM 613550) [3,4,5,6,7] (Table 1). Moreover, TMEM67-mutated patients with JS are also at increased risk for liver disease development complicated by probable portal hypertension in the second or third decades of life. The most relevant genotype-phenotype correlation has been established between TMEM67 sequence variations and the subtypes of JS with liver disease [29]. We here describe the clinical characteristics and mutational analysis of the first Vietnamese family presenting clinical features of JS. Whole exome sequencing identified a novel compound heterozygous TMEM67 variant. This study expands the mutational spectrum of TMEM67 in JS as well as underscores the importance of molecular diagnosis and genetic counseling in patients initially diagnosed with cerebral palsy in Viet Nam.
Case presentation
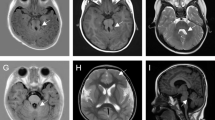
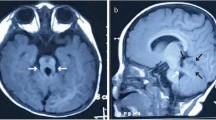
This 38 year-old female first came to our clinic because of family history of developmental delay. The fetus (II:2) was the second child of a non-consanguineous Vietnamese healthy couple, mid-trimester prenatal ultrasound at 26 + 6 weeks of gestation showed an abnormal enlarged fourth ventricle with abnormalities of the ventricle floor (Fig. 1a). Moreover, renal hypoplasia and polycystic kidney were also noticed. She already had a first child (II:1) with developmental delay. This 10-year-old-boy was born at term after an uneventful pregnancy. His birth weight, height and head circumference were respectively 3400 g (50-90th centile), 53 cm (90th percentile), 34 cm (50th centile). Hypotonia and abnormal breathing pattern were noted at birth. He was diagnosed with cerebral palsy of unknown origin at 2 years old. Neither genetic counseling nor molecular genetic testing was provided. According to the clinical evaluation conducted at the age of 10, his weight, height and head circumference were respectively 23 kg (5th), 129 cm (5-10th) and 52 cm (10th–25th). The proband had severe psychomotor and language delay, began to walk at the age of 72 months and spoke the first word at the age of 60 months. Clinical physical evaluation showed a prominent forehead with bitemporal narrowing, high arched eyebrows, bilateral ptosis, hypertelorism, lower lip eversion, mild clinodactyly of the fifth finger and tapered fingers (Fig. 1b). The patient also displayed ataxic gait and oculomotor apraxia. He had mild intellectual disability. Family history was unremarkable. His MRI showed a pathognomonic finding of molar tooth sign (Fig. 1c). The couple was concerned about their pregnancy, and thus requested genetic counseling regarding her second pregnancy.

a Ultrasound at 26 + 6 weeks of gestation showing an abnormal enlarged fourth ventricle (red asterisk) and the ventricle floor is abnormal. b Photographs of the face (left image) and right profile (middle image) showing typical facial features of Joubert syndrome including prominent forehead, high arched eyebrows, bilateral ptosis, hypertelorism, lower lip eversion (left and middle image), mild clinodactyly of the fifth finger and tapered fingers (right image). c Axial MRI through the cerebellum and brain stem showing cerebellar vermis hypoplasia, thick and elongated superior cerebellar peduncles (red arrows, left image) and absence of the cerebellar vermis, deep interpeduncular fossa and the fourth ventricle has a bat-wing configuration (red arrows, right image)
Genetic analysis
Conventional cytogenetics and array-CGH
Blood lymphocytes were cultured in RPMI 1640 supplemental with PHA (Gibco, USA) and chromosomes metaphases were harvested according to the laboratory standard protocol. Conventional cytogenetics showed a normal male karyotype 46,XY. 180 K array-CGH (Agilent Technologies, Santa Clara, USA) with a median probe spacing of 13 kb was carried out according to the manufacturer’s instructions and required at least three consecutive probes to make a call. Array-CGH analysis identified no chromosomal imbalances arr(1–22)×2,(X,Y)×1.
Whole exome sequencing
In order to find causative gene variants, we performed whole exome sequencing in the proband (II:1). Genomic DNA was extracted from whole blood from the family using standard methods (QIAGEN, Germany). Five hundred ng DNA was fragmented with a Biorupter (Diagenode, Seraing, Belgium) and the fragmented DNA quality was checked on a bioanalyser MultiNA (Shimadzu Corporation, Kyoto, Japan). Libraries were performed using the Ultra DNA library preparation kit (NewEngland, Biolabs, UK), exome enrichment was performed using TruSeq Exome Library Prep Kit (Illumina, USA) and IDT’s xGen® Exome Research Panel (Integrated DNA Technologies, USA). Finally, sequencing was done on an Illumina HiSeq® 2500. The exome was covered to a mean depth of 100, data with >10X mean coverage accounted for 95% of the whole data. Variants were validated by Sanger DNA sequencing using the ABI BigDye Terminator v3.1 Cycle Sequencing kit and the ABI PRISM 3130xl genetic analyzer (Applied Biosystems, CA, USA). Sequence alterations were reported according to Human Genome Variation Society guidelines (HGVS) and mapped to Human Genome Build GRCh37/UCSC hg19. Whole exome sequencing showed novel compound heterozygous c.725A > G p.Asn242Ser and c.313-3 T > G p.Lys105Valfs*16 TMEM67 variants in the proband (II:1) (Fig. 2a). No other potentially pathogenic variants in other genes associated with developmental delay were identified. These two variants were interpreted as likely pathogenic according to standards and guidelines from the American College of Medical Genetics and Genomics. The variant c.725A > G has a MAF < 0.01 (PM2: Extremely low frequency). It was previously reported in patients with JS (PM3: For recessive disorders, detected in trans with a pathogenic variant and PP1:Co-segregation with disease in multiple affected family members) and predicted to be deleterious by in silico prediction (PP3: Multiple lines of computational evidence support a deleterious effect on the gene or gene product). The novel splice-site variant has never been documented (PM2: Absent from controls) and produces an aberrant splicing transcript (PVS1: Null variant). Sanger sequencing confirmed that the variant c.725A>G p.Asn242Ser was paternally inherited and the splice-site variant c.313-3T>G p.Lys105Valfs*16 was of maternal origin (Fig. 2b, c). Prenatal diagnosis was performed on genomic DNA extracted from amniocytes and Sanger sequencing identified the same compound heterozygous variant c.725A>G p.Asn242Ser and c.313-3T>G p.Lys105Valfs*16 in the fetus (II:2).

a Familial pedigree with Joubert syndrome shows a novel compound heterozygous TMEM67 variant in the case-index and the fetus while the parents were heterozygous. b, c Sang sequencing showing the proband and his father were heterozygous for TMEM67 c.725A > G p.Asn242Ser variant. Moreover, the proband and his mother were heterozygous for a novel TMEM67 splice-site variant c.313-3 T > G p.Leu105Valfs*16. d Reverse transcriptase PCR showing alternative splicing effect with the deletion of 4 bps in exon 03 in the mRNA resulting in an aberrant transcript with premature codon stop. The last nucleotide in exon 2 is yellow highlighted
Reverse transcriptase PCR for alternative splicing study (RT-PCR)
Total RNA from blood sample was extracted using Qiagen RNeasy blood mini kit according to the manufacturer’s instruction (Qiagen, Germany). One microgram of RNA was used for Reverse-Transcriptase PCR (RT-PCR) (Quantabio, USA). RT-PCR was performed with primers spanning exons 1 and 5. PCR products were separated on agarose gel 2% and sequenced using the ABI BigDye Terminator v.3.1 Cycle Sequencing kit and the ABIPRISM 3500 XL genetic analyzer (Applied Biosystems, CA, USA). Splicing study demonstrated that the novel splice-site variant produced an aberrant transcript with the loss of four nucleotides of exon 03 (Fig. 2d).
Discussion and conclusion
In the era of clinical genomics in Viet Nam, clinical genetics testing is still relatively new and clinicians have very little knowledge about current approaches to genetic disorders. Many developmentally delayed children suffering from genetic disorders were diagnosed with cerebral palsy and they have not benefited from a modern multi-disciplinary care model. We report a Vietnamese family including a 10-year-old child diagnosed with cerebral palsy without etiologic diagnosis and a fetus with central nervous system malformations. Physical examination of the age of the 10-year old showed typical clinical features of JS and an MRI showing the pathognomonic finding of a molar tooth sign, which confirms the JS diagnosis. Whole exome sequencing identified compound heterozygous TMEM67 variants in the proband (II:1). The variant c.725A > G p.Asn242Ser was previously documented in patients with JS and predicted to be pathogenic by SIFT, Polyphen-2 and Mutation Taster [21]. This variant was considered as founder mutation in Eastern Iranian population. The novel splice-site variant c.313-3 T > G p.Lys105Valfs*16 has never been documented in the medical literature and the nucleotide T at this position is highly conserved across multiple species (Additional file 1: Figure S1). Moreover, alternative splicing study demonstrated that the variant c.313-3 T > G p.Lys105Valfs*16 produced an aberrant transcript with the loss of the first four nucleotides of exon 03 leading to a premature stop codon.
TMEM67 sequence variations were associated with a large clinical spectrum and sequence variants were distributed throughout the entire coding region (Table 1). However, several peculiar phenotypes might be predicted, i.e., TMEM67 missense variants falling in exon 8 to 15, especially combined with a truncating variant would predict to give rise to Meckel-Gruber syndrome. In addition, most of TMEM67 sequence variants were predominantly located in 8 of 28 exons (2, 6, 8, 11, 13, 15, 18, 24) [10]. Based on the review of TMEM67 sequence variations previously recorded in the medical literature, our report also showed several mutational hotspots, which were consistent with the result documented by Lannicelli et al., 2010. The most TMEM67 frequently mutated hotspot was exon 8, followed by exons 24, 18, 6, 13, 11, 2, 15 (Table 1).
Prenatal molecular diagnosis was carried out on DNA extracted from amniocytes and the fetus (II:2) harbored the same compound heterozygous TMEM67 variants found in the 10-year-old sibling. The parents were referred for genetic counseling for the current pregnancy and the pregnancy was terminated at 32 weeks of gestation. Furthermore, the proband was recommended to follow annual surveillance of hepatic functions as well as evaluation of kidney function [30]. The couple was offered pre-implantation genetic diagnosis or prenatal diagnosis options for the future pregnancy. In conclusion, our study reports the first Vietnamese family of JS and expands the TMEM67 mutational spectrum in JS. Furthermore, we also stress the important role of molecular approaches in order to identify the causative gene. Accurate diagnosis would further help in genetic counseling, early management of genetic disorders as well as offer prenatal diagnostic options for future pregnancy.
Availability of data and materials
The data was submitted in LOVD (Leiden Open Variation Database): https://databases.lovd.nl/shared/login. Individual number #00265632 in LOVD database.
Abbreviations
- Array-CGH:
-
Array-comparative genomic hybridization
- JS:
-
Joubert syndrome
- MRI:
-
Magnetic resonance imaging
- RT-PCR:
-
Reverse-transcriptase polymerase chain reaction
- SIFT:
-
Sorting intolerant from tolerant
- TMEM67:
-
Transmembrane protein 67
References
Bachmann-Gagescu R, Dempsey JC, Phelps IG, O’Roak BJ, Knutzen DM, Rue TC, Ishak GE, Isabelle CR, Gorden N, Adkins J, Boyle EA, de Lacy N, O’Day D, Alswaid A, Ramadevi AR, Lingappa L, Lourenço C, Martorell L, Garcia-Cazorla A, Ozyurek H, Haliglolu G, Tuysuz B, Topcu M, et al. Joubert syndrome: a model for untangling recessive disorders with extreme genetic heterogeneity. J Med Genet. 2015;52:514–22.
Abdelhamed ZA, Abdelmottaleb DI, EI-Asrag ME, Natarajan S, Wheway G, Inglehearn CF, Toomes C, Johnson CA. The ciliary frizzled-like receptor Tmem67 regulates canonical Wnt/β-catenin signaling in the developing cerebellum via Hoxb5. Sci Rep. 2019;9:5446.
Abdelhamed ZA, Wheway G, Szymanska K, Natarajan S, Toomes C, Inglehearn C, Johnson CA. Variable expressivity of ciliopathy neurological phenotypes that encompass Meckel-Gruber syndrome and Joubert syndrome is caused by complex de-regulated ciliogenesis, Shh and Wnt signaling defects. Hum Mol Genet. 2013;22:1358–72.
Doherty D, Parisi MA, Finn LS, Gunay-Aygun M, Al-Mateen M, Bates D, Clericuzio C, Demir H, Dorschner M, van Essen AJ, Gahl WA, Gentile M, Gorden NT, Hikida A, Knutzen D, Ozyurek H, Phelps I, Rosenthal P, Verloes A, Weigand H, Chance PF, Dobyns WB, Glass IA. Mutations in 3 genes (MKS3, CC2D2A and RPGRIP1L) cause COACH syndrome (Joubert syndrome with congenital hepatic fibrosis). J Med Genet. 2010;47:8–21.
Brancati F, Camerota L, Colao E, Vega-Warner V, Zhao X, Zhang R, Bottillo I, Castori M, Caglioti A, Sangiuolo F, Novelli G, Perrotti N, Otto EA, Undiagnosed Disease Network Italy. Biallelic variants in the ciliary gene TMEM67 cause RHYNS syndrome. Eur J Hum Genet. 2018;26:1266–71.
Rasmussen M, Sunde L, Nielsen ML, Ramsing M, Petersen A, Hjortshøj TD, Olsen TE, Tabor A, Hertz JM, Johnsen I, et al. Targeted gene sequencing and whole-exome sequencing in autopsied fetuses with prenatally diagnosed kidney anomalies. Clin Genet. 2018;93:860–9.
Otto EA, Ramaswami G, Janssen S, Chaki M, Allen SJ, Zhou W, Airik R, Hurd TW, Ghosh AK, Wolf MT, Hoppe B, Neuhaus TJ, Bockenhauer D, Milford DV, Soliman NA, Antignac C, Saunier S, Johnson CA, Hildebrandt F, GPN study group. Mutation analysis of 18 nephronophthisis associated ciliopathy disease genes using a DNA pooling and next generation sequencing strategy. J Med Genet. 2011;48:105–16.
Kroes HY, Monroe GR, van der Zwaag B, Duran KJ, de Kovel CG, van Roosmalen MJ, Harakalova M, Nijiman IJ, Kloosterman WP, Giles RH, Knoers NV, van Haaften G. Joubert syndrome: genotyping a northern European cohort. Eur J Hum Genet. 2016;24:214–20.
Vilboux T, Doherty DA, Glass IA, Parisi MA, Phelps IG, Cullinane AR, Zein W, Brooks BP, Heller T, Soldatos A, Oden NL, Yildirimli D, Vemulapalli M, Mullikin JC, et al. Molecular genetic findings and clinical correlations in 100 patients with Joubert syndrome and related disorders prospectively evaluated at a single center. Genet Med. 2017;19:875–82.
Lannicelli M, Brancati F, Mougou-Zerelli S, Mazzotta A, Thomas S, Elkhartoufi N, Travaglini L, Gomes C, Ardissino GL, Bertini E, Boltshauser E, Castorina P, D’Arrigo S, Fischetto R, Leroy B, Loget P, Bonnière M, Starck L, Tantau J, Gentilin B, Majore S, et al. Novel TMEM67 mutations and genotype-phenotype correlates in meckelin-related ciliopathies. Hum Mutat. 2010;31:E1319–31.
Szymanska K, Berry I, Logan CV, Cousins SR, Lindsay H, Jafri H, Raashid Y, Malik-Sharif S, Castle B, Ahmed M, Bennet C, Carlton R, Johnson. Founder mutations and genotype-phenotype correlations in Meckel-Gruber syndrome and associated ciliopathies. Cilia. 2012;1:18.
Suzuki T, Miyake N, Tsurusaki Y, Okamoto N, Alkindy A, Inaba A, Sato M, Ito S, Muramatsu K, Kimura S, Leda D, Saitoh S, Hiyane M, Suzumura H, Yagyu K, et al. Molecular genetic analysis of 30 families with Joubert syndrome. Clin. Genet. 2016;90:526–35.
Tsurusaki Y, Kobayashi Y, Hisano M, Ito S, Doi H, Nakashima M, Saitsu H, Matsumoto N, Miyake N. The diagnostic utility of exome sequencing in Joubert syndrome and related disorders. J Hum Genet. 2013;58:113–5.
Lee SH, Nam TS, Li W, Kim JH, Yoon W, Choi YD, Kim KH, Cai H, Kim MJ, Kim C, Choy HE, Kim N, Chay KO, Kim MK, Choi SY. Functional validation of novel MKS3/TMEM67 mutations in COACH syndrome. Sci Rep. 2017;7:10222.
Sanjeev RK, Kapoor S, Goyal M, Kapur R, Gleeson JG. Molar tooth sign with deranged liver function tests: an Indian case with COACH syndrome. Case Rep Pediatr. 2015;385910. https://doi.org/10.1155/2015/385910 Epub 2015 May 17.
Huynh JM, Galindo M, Laukaitis CM. Missense variants in TMEM67 in a patient with Joubert syndrome. Clin Case Rep. 2018;6:2189–92.
Vora NL, Powell B, Brandt A, Strande N, Hardisty E, Gilmore K, Foreman AKM, Wihelmsen K, Bizon C, Reilly J, Owen P, Powell CM, Skinner D, Rini C, Lyerly AD, Boggess KA, Weck K, Berg JS, Evans JP. Prenatal exome sequencing in anomalous fetuses: new opportunities and challenges. Genet Med. 2017;19:1207–16.
Vogel I, Ott P, Lildballe D, Hamilton-Dutoit S, Vilstrup H, Gronbaek H. Isolated congenital hepatic fibrosis. Clin Case Rep. 2017;5:1098–102.
Habritter J, Diaz K, Chaki M, Porath JD, Tarrier B, Fu C, Innis JL, Allen SJ, Lyons RH, Stefanidis CJ, Omran H, Soliman NA, Otto EA. High-throughput mutation analysis in patients with a nephronophthisis-associated ciliopathy applying multiplexed barcoded array-based PCR amplification and next-generation sequencing. J Med Genet. 2012;12:756–67.
Khaddour R, Smith U, Baala L, Martinovic J, Clavering D, Shaffiq R, Ozilou C, Cullinane A, Kyttala M, Shalev S, Audollent S, d’Humières C, et al. Spectrum of MKS1 and MKS3 mutations in Meckel syndrome: a genotype-phenotype correlation. Mutation in brief #960. Online. Hum Mutat. 2007;28:523–4.
Dehghani M, Mojarad M, Ghayoor Karimiani E, Vahidi Mehrjardi MY, Sahebalzamani A, Ashrafzadeh F, Beiraghi Toosi M, Eslahi A, Ahangari N, Yassini SM, Hassanbeigi A, Rasti A, Kalantar SM, Maroofian R. A common ancestral Asn242Ser mutation in TMEM67 identified in multiple Iranian families with Joubert syndrome. Public Health Genomics. 2017;20:188–93.
Kang HG, Lee HK, Ahn YH, Joung JG, Nam J, Kim NK, Ko JM, Cho MH, Shin JL, Kim J, Park HW, Park YS, Ha IS, Chung WY, Lee DY, Kim SY, Park WY, Cheong HI. Targeted exome sequencing resolves allelic and the genetic heterogeneity in the genetic diagnosis of nephronophthisis-related ciliopathy. Exp Mol Med. 2016;48:e251.
Otto EA, Tory K, Attanasio M, Zhou W, Chaki M, Paruchuri Y, Wise EL, Wolf MT, Utsch B, Becker C, Nurnberg G, Nurnberg P, Nayir A, Saunier S, Antignac C, Hildebrandt F. Hypomorphic mutations in meckelin (MKS3/TMEM67) cause nephronophthisis with liver fibrosis (NPHP11). J Med Genet. 2009;46:663–70.
Baala L, Romano S, Khaddour R, Saunier S, Smith UM, Audollent S, Ozilou C, Faivre L, Laurent N, Foliguet B, Munnich A, Lyonnet S, Salomon R, et al. The Meckel-Gruber syndrome gene, MKS3, is mutated in Joubert syndrome. Am J Hum Genet. 2007;80:186–94.
Zhang M, Cheng J, Liu A, Wang L, Xiong L, Chen M, Sun Y, Li J, Lu Y, Yuan H, Li Y, Lu Y. A missense mutation in TMEM67 causes Meckel-Gruber syndrome type 3 (MKS3): a family from China. Int J Clin Exp Pathol. 2015;8:5379–86.
Szabo T, Orosz P, Balogh E, Javorszky E, Mattyus I, Bereczki C, Maroti Z, Kalmar T, Szabo AJ, Reusz G, Varkonyi I, Marian E, Gombos E, Orosz O, Madar L, Balla G, Kappelmayer J, Tory K, Balogh I. Comprehensive genetic testing in children with a clinical diagnosis of ARPKD identifies phenocopies. Pediart Nephrol. 2018;33:1713–21.
Arrigoni F, Romaniello R, Peruzzo D, De Luca A, Parazzini C, Valente EM, Borgatti R, Triulzi F. Anterior mesencephalic cap dysplasia: novel brain stem malformative features associated with Joubert syndrome. AJNR Am J Neuroradiol. 2017;38:2385–90.
Komatsu Y, Suzuki T, Tsurusaki Y, Miyake N, Matsumoto N, Yan K. TMEM67 mutations found in a case of Joubert syndrome with renal hypoplasia. CEN Case Rep. 2016;5:137–40.
Strongin A, Heller T, Doherty D, Glass IA, Parisi MA, Bryant J, Choyke P, Turkbey B, Daryanani K, Yildirimli D, Vemulapalli M, Mullikin JC, Malicdan MC, Vilboux T, Gahl WA, Gunay-Aygun M, NISC Comparative Sequencing Programe. Characteristics of liver disease in 100 individuals with Joubert syndrome prospectively evaluated at a single center. J Pediatr Gastroenterol Nutr. 2018;66:428–35.
Bachmann-Gagescu R, Dempsey JC, Bulgheroni S, Chen ML, D’Arrigo S, Glass IA, Heller T, Héon E, Hildebrandt F, Joshi N, Knutzen D, Kroes HY, Mack SH, Nuovo S, Parisi MA, Snow J, Summers AC, Symons JM, Zien WM, Boltshauser E, Sayer JA, Gunay-Aygun M, Valente EM, Doherty D. Healthcare recommendations for Joubert syndrome. Am J Med Genet A. 2019. https://doi.org/10.1002/ajmg.a.61399 [Epub ahead of print].
Acknowledgements
We thank the patient and his parents for taking part in this study.
Funding
The authors declare that they did not receive any funding.
Author information
Authors and Affiliations
Contributions
MTH designed the study and wrote the manuscript. BTPH, NTN, VDN collected and analyzed the data. TTHL, DHD, HNN performed the molecular genetic experimental. All authors have read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This study was reviewed and approved by the Ethics Committee of Vinmec International Hospital. Written informed consent was obtained from the patient’s parents for participation in this study. This study was carried out following the Principles of the Declaration of Helsinki.
Consent for publication
Written informed consent of this case report was obtained from the patient’s parents.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1:
Figure S1. Multiple sequence alignment of TMEM67 sequences across species showing that the nucleotide c.313-3 T variant is well conserved throughout evolution (red box).
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Bui, T.P.H., Nguyen, N.T., Ngo, V.D. et al. Novel compound heterozygous TMEM67 variants in a Vietnamese family with Joubert syndrome: a case report. BMC Med Genet 21, 18 (2020). https://doi.org/10.1186/s12881-020-0962-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12881-020-0962-0