
Abstract
Background
Rubinstein–Taybi syndrome (RSTS) is an extremely rare autosomal dominant inheritable disorder caused by CREBBP and EP300 mutations, while atypical RSTS harbouring variant from the same genes but not obvious resembling RSTS. There are only a few cases of Menke–Hennekam syndrome (MKHK) with variant of exon 30 or 31 of CREBBP or EP300 gene have been reported that not resembling RSTS recent years. Atypical RSTS cannot be accurately classified as MKHK, nor is it easy to identify the obvious classic characteristics of RSTS. The clinical manifestations and genetic variation of atypical RSTS are not fully understood.
Case presentation
We present a Chinese core family with a girl had recurrent respiratory tract infection and developmental delay. The patient with language and motor mild development retardation, she has slight abnormal facial features, mild hirsutism and post-axial hexadactylia of left foot. Her cisterna magna is enlarged to connect with the fourth ventricle, and the ventricular system is enlarged. She has a malacia beside the posterior horn of the left lateral ventricle. The patient has primary low immunoglobulin G and A, but her level of immunoglobulin M content in blood is normal. The patient harbors a novel heterozygous frameshift variant of c.2499dupG in exon 14 of EP300 gene, that it is proved to de novo origin. The mutation is judged to be a pathogenic mutation, and it has high-grade pathogenic evidence.
Conclusion
The clinical and genetic evaluation of this case corroborates that clinical features caused by c.2499dupG in exon 14 of EP300 are less marked than RSTS2 patient although it is difficult to establish an accurate genotype–phenotype correlation. Our additional case also helps to deepen the clinical and genetic spectrum in this disorder. The case provides a novel mutation of EP300 and enriches the phenotypes related with the gene. We have contributed new variation and disease information for guardians and doctors to broaden the knowledge about EP300-RSTS genotype and phenotype, this may contribute to ameliorate the health management of patients and improve the genetic counseling to the families.
Similar content being viewed by others

Background
Rubinstein–Taybi syndrome (RSTS; OMIM #180849, #613684) is a known disorder characterized by a distinctive face, broad thumbs, broad halluces and big toes, short stature, and variable degree of intellectual disability [1, 2]. It is a rare autosomal dominant genetic pluri-malformative syndrome, which well-defined facial features include down slanting palpebral fissures, convex nasal bridge, columella below alae nasi, and a characteristic grimacing smile [3]. The other aspects of manifestation include growth retardation, microcephaly and behavioural problems [3]. Broad and angulated thumbs and halluces are usually considered as hallmarks for clinical diagnosis [4]. RSTS1 (OMIM #180849) is caused by variants of CREBBP that coding CREB-binding protein acted as transcriptional co-activators, and RSTS2 (OMIM #613684) is caused by variants of its paralog EP300 that coding E1A associated protein p300. CREBBP and EP300 mutations have been identified in majority (50–60%) and minority (3–5%) of RSTS affected individuals, respectively [4,5,6], most, if not all, of them occur de novo.
Recently Menke–Hennekam syndrome (MKHK, OMIM #618332, #618333) is put forward as an entity caused by the same gene CREBBP and EP300 mutations but not resembling Rubinstein–Taybi syndrome [7, 8]. The manifestations of MKHK include variable impairment of intellectual development and specific facial appearance, but patients with MKHK do not resemble the striking phenotype of RSTS. The other symptoms of MKHK are also consist of feeding difficulties, autistic behavior, recurrent upper airway infections, hearing impairment, and short stature, microcephaly are also frequently seen. Current evidence suggests that MKHK2 (OMIM #618333) is caused by heterozygous mutations in exon 30 or 31 of the EP300 gene, which symptom is milder than MKHK1 (OMIM #618332) caused by heterozygous mutations in exon 30 or 31 of the CREBBP gene [9, 10]. Here we report one further child with one frameshift variant, although outside exon 30 or 31 of EP300, without the typical RSTS features.
Case presentation
A Chinese family from central China was recruited for this study. The patient was a girl at 4 years of age, she was hospitalized for recurrent respiratory infections and pneumonia in the past half 1 year. The sufferer's parents are 33 years old and healthy. The patient sought medical and genetic consultations in the hospital from September 2020 to February 2021. Written informed consent has been signed for the collection and use of clinical materials. In this research, we chose a family-based strategy to determine the exact inheritance pattern and candidate disease causing gene mutation. Using such a family-based strategy, we can also determine whether phenotype and genotype co-segregate in the family, which helps to estimate the pathogenicity of candidate mutation and explore the relationship between phenotype and genotype. The proband was screened by whole exome sequencing (WES), then, the parents were tested by Sanger sequencing to detect and verify the carrying status of candidate mutation screened through WES.
The patient is the first child of nonconsanguineous Chinese parents born at 41 weeks gestation, following an unremarkable pregnancy with a birth weight of 3.1 kg. At birth, her left foot was noted to be hexadactylia. She was low neonatal response. Her parents and family feel that she has a peculiar appearance, neither like father nor mother. There are no concerns about her hearing and vision, but eye-tracking and listen-tracking was poor during infancy. From 3 to 8 months, she received a 6-month rehabilitation training for movement development retardation. She rolled over at aged 7 months, sat unsupported at aged 9 months, crawled at aged 12 months and walked independently at aged 16 months. At 2 years of age, she received rehabilitation training for language delay. She is mild growth retardation. She had no self-injurious behavior. She often pushes other children, but is sociable and interactive. At present, she is height of 101 cm, weight of 15.5 kg and head circumference of 49 cm, which are at low level among the normal range (103.1 ± 3.9 cm, 16.17 ± 1.73 kg, 49.4 ± 1.3 cm) of girls aged 4 years. She can ride a tricycle independently and freely (Additional file 1: Short Video S1). She also builds blocks and draws with no problem (Additional file 1: Short Videos S2 and S3). Her language comprehension is somewhat poor. She can now express long sentences or recite simple enlightening poems of the Tang dynasty such as singing goose, spring dawn and so on. But when she said long sentences, she sometimes would miss one word. She is now in kindergarten middle shift.
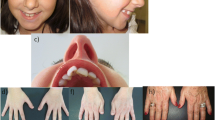
Patient’s facial and physical features are shown in Fig. 1 and Additional file 1: Fig. S1a–g. She at 4 years of age, has slightly arched eyebrows, synophridia, long eyelashes, a square tip to his nose, normal columella, prominent two front teeth and normal tooth number. She is absence of microcephaly, beaked nose and characteristic grimacing smile like RSTS. The fine hairs on the front of the ear and on the cheek form a hair whorl. The child has hirsute back. She has no broad or angulated thumbs, nor broad distal phalanges of the fingers, as seen in patients with RSTS. Her brain plain scan of MRI shows as follows. Punctate long T1 and long T2 signal shadow was seen beside the posterior horn of left lateral ventricle; FLAIR showed high signal outside and low signal inside; DWI showed low signal with high b value, and the lesion had no space occupying effect. It indicated that softening lesion of the posterior horn of left lateral ventricle. The enlarged cisterna magna was connected with the fourth ventricle, the ventricular system was enlarged, and there was no widening and deepening phenomenon in the cerebral sulcus, cisterna and cerebral fissure. The morphology, structure and signal intensity of cerebellum and brainstem were normal. The midline structure did not deviate.

Phenotypic features of patient described in this study. a and b The patient 4 years old. Note slightly arched eyebrows and synophridia, a square tip to his nose, normal columella, prominent two front teeth, normal tooth number and absence of characteristic grimace of Rubinstein–Taybi syndrome. c The fine hairs on the front of the ear and on the cheek are hair whorl. d and e The child has heavy fine hair on her back and opisthenar. f Patient has no broad or angulated thumbs, nor broad distal phalanges of the fingers, as seen in patients with Rubinstein–Taybi syndrome. g Girl has a sixth toe of her left foot, that hexadactyly
The patient has been hospitalized for 5 times due to recurrent respiratory infections with or without pneumonia in the last half year. The patient was found to have primary low immunity, transient EB virus infection and splenomegaly. Her examination results of immune globulins content in peripheral blood or urine are presented in Table 1. It indicates that she suffers primary low immunoglobulin G/A. She has normal level of immunoglobulin M.
Candidate disease causing mutation detected in the patient through WES, and it was verified by Sanger sequencing. The parents were also tested by Sanger sequencing to detect the carrying status of candidate mutation (Fig. 2). The patient harbors a heterozygous mutation of c.2499dupG in exon 14 of EP300 gene, which encodes the adenovirus E1A-associated cellular p300 transcriptional co-activator protein. NM_001429 was chosen as transcript reference sequence of EP300 gene during NGS data analysis. The genetic relationship between parents and daughter was confirmed by paternity test. Subsequent Sanger sequencing tests displayed that none of the parents carried the EP300 c.2499dupG mutation, and it proved to be de novo origin.

Sanger sequencing results of EP300 c.2499dupG mutation in the core family. Sample P denotes the patient, sample F and M denote the father and mother of the patient, respectively
EP300 c.2499dupG is a frameshift mutation in exon 14, which is presumed to cause amino acid sequence changes of p.Pro834Alafs*4. It results in substitution of amino acid 834th and termination of translation after four codons shift, which is sited in a region, not domain, of the protein with still uncharacterized function (Fig. 3). The c.2499dupG p.(Pro834Alafs*4) variant in our patient is a novel that not been related to disease recorded in HGMD database. The variant is not found in all populations or the East Asian population in gnomAD database, so minor allele frequency of the rare mutation is unknown (extremely low). Due to nonsense mediated decay of frameshift mutations, truncated nonfunctional product proteins may be generated, and such mutation will be deleterious. According to ACMG guidelines, the novel frameshift mutation of EP300 c.2499dupG p.(Pro834Alafs*4) should be considered pathogenic due to its high evidence grade of pathogenicity (PVS1 + PS2 + PM2).

Distribution of EP300 domains and mutations in our patient (in red arrow) versus in previous patient cohort (in black arrow). Schematic representation of the EP300 protein and functional domains excerpted from http://www.ebi.ac.uk/interpro/ at March 26th 2021. Previous patient cohort refers to the cohort published [7, 14,15,16]. Previous patients marked by red box accorded with the diagnosis of MKHK, the other previous patients marked by green box accorded with the diagnosis of RSTS
Discussion
Mutations in CREBBP and EP300 caused RSTS has been well-known, while the patients with missense variant in exon 30/31 of CREBBP and EP300 didn't have typical characteristics of RSTS that reported in recently publications [7, 8, 10]. CREBBP is originally isolated as a nuclear protein with intrinsic histone or non-histone acetyltransferase activity, which is ubiquitously expressed and involved in the transcriptional co-activation of a variety of transcription factors through binding to cAMP-response element binding protein (CREB) [11]. The protein of CREBBP also functions as a scaffold to stabilize additional protein interactions with the transcription complex [12]. EP300 encodes the adenovirus E1A-associated cellular p300 transcriptional co-activator protein that is significant in the processes of cell proliferation and differentiation. Its product protein acts as histone acetyltransferase participating in chromatin remodeling to regulate the process of transcription. And EP300 also mediates cAMP-gene regulation through specifically binding to phosphorylated CREB protein [12]. There is very high sequence similarity in homologous area of bromo-domain, cysteine-histidine-rich regions, and histone acetyltransferase domain of the proteins of EP300 and CREBBP. They play important roles in embryonic development, growth control, and homeostasis, which implements through coupling chromatin remodeling to transcription factor recognition.
There are 25 patients reported in the medical literature without the typical RSTS features and with exon 30/31 missense or in-frame deletion CREBBP variants so far [7, 8, 10]. The main features of MKHK1 caused by CREBBP mutations include developmental delay, distinctive facial features, autistic behavior, feeding difficulties, recurrent upper airway infections, and brain abnormalities revealed by MRI. An additional Chinese case with one CREBBP in-frame deletion variant in the beginning of exon 30 has atypical RSTS phenotypes [13]. Now it seems that she is consistent with the diagnosis of MKHK1, although the mutation in the HAT domain where no pathogenic variants have been previously reported to be responsible for MKHK. The 2-year-7-month-old Chinese girl presented facial dysmorphism of MKHK included telecanthus, a depressed nasal ridge, short nose, anteverted nares, short columella, and long philtrum. Her other symptoms were mild cognitive impairments, developmental delay, short stature, recurrent upper airway infections, and mild thinning of corpus callosum revealed by MRI.
Some scholars call the atypical RSTS phenotypes with EP300 variant Menke–Hennekam syndrome 2 [7]. The main typical characteristics of previous patient E1 and E2 regarded as MKHK2 include variable impairment of intellectual development and facial dysmorphisms, and feeding difficulties, autistic behavior, recurrent infections are also frequently seen. Combined with the analysis of clinical manifestations of previous MKHK1 cases caused by CREBBP exon 30/31 mutations, Menke–Hennekam syndrome is described as a congenital disorder characterized by variable impairment of intellectual development and facial dysmorphisms that do not resemble the striking phenotype of RSTS [7, 8], and MKHK2 is milder than MKHK1. Patient E1 with EP300 c.5471A > C p.(Gln1824Pro) had recurrent otitis, airway infections and urinary tract infections owing to low immunoglobulins, and was diagnosed with autism spectrum disorder. She necessitated a percutaneous gastrostomy (PEG) when as a neonate. Patient E2 with EP300 c.5492_5494del p.(Arg1831del) continued to have recurrent otitis, and autism spectrum disorder was also diagnosed (Fig. 3). Her language and motor delayed. Brain MRI was not performed in these patients.
The number of MKHK2 cases caused by EP300 gene mutation is still small. The previously reported patients [14] with an EP300 variant in exon 31 could reconsider the name of the disease according to the new concept. Individual 8 with EP300 c.6915_6918del p.(Asn2305Lysfs*47) was male, his striking phenotypes included mild specific facial appearance, varying degrees of speech and motor retardation, reactive airway disease, undescended testicle and severely restricted growth, without broad thumb and radial deviation of the hand. Individual 8 was mild RSTS2, but cannot be attributed to MKHK2. His demise caused by severe cardiac involvement, which may be a few and isolated phenotype. While Individual 12 with EP300 frameshift variant of c.5720delC p.(Pro1907Leufs*53) had classical characteristics of RSTS. These two patients also did not do brain MRI. There were five persons with EP300 exon 30/31 variants previously reported as RSTS2 patients (Fig. 3) [15]. The Patient #42 was a 9 years old boy with an EP300 in-frame deletion variant of c.6627_6638del p.(Asn2209_Gln2213delinsLys) in exon 31, but he had the typical manifestations of RSTS. The Patient #11 was a 18 years old male youth with an EP300 frameshift variant of c.4954_4957dup p.(Cys1653Tyrfs*21) in exon 30, and both of the Patients #45 of mother and daughter had EP300 frameshift variant of c.7222_7223del p.(Gln2408Glufs*39) in exon 31. They were all RSTS2 patients with variable disease symptoms, respectively. Patients #45 have milder clinical manifestation than Patient #11, and even Patients #45 with the same mutation in one same family also realized the heterogeneity of clinical features of the disease.
We also compare our case with the other 2 cases harbouring a nonsense variant of the same exon 14 reported in Fergelot's paper [16]. The case #7 harbours c.2554C > T (p.Gln852*) heterozygous variation and the case #34 harbours c.2437C > T (p.Gln813*) heterozygous variation. Since there are no facial photos of these two male patients, we mainly refer to the descriptive information in the literature. These two cases of #7 and #34 have more typical facial RSTS characteristics than our case to some extent, such as microcephaly, columella below alae nasi and low-set ears, etc. They have more noticeable specific facial appearance, but no hypertrichosis. The most significant difference between our case and the #7 and #34 cases is that histories of recurrent respiratory infections and primary low immunity, the two previous cases have no recurrent respiratory infections and immune problems.
These above investigations have expanded our knowledge about the spectrum of diverse phenotypes driven by dissimilar molecular and cellular consequences resulting from different class of variants in the same gene [10]. However, by comparison, we can see that it is not exactly the same as Menke and Hennekam proposed that ‘Menke–Hennekam syndrome is caused by missense mutations in the last part of exon 30 and beginning of 31 of the CREBBP/EP300 gene resulted in a gain of function. Mutations elsewhere in the gene causing Rubinstein–Taybi syndrome, result in haplo-insufficiency or perturb the function of a domain, specifically the HAT’. The previous Chinese case with, paralog of EP300, one CREBBP in-frame deletion variant in the 5′ end of HAT domain of beginning exon 30 has phenotypes more similar to typical MKHK rather than RSTS, and while EP300 in-frame deletion variant in exon 31 not always cause MKHK2 (previous Patient #42). Disease caused by mutations outside the exon 30/31 of EP300 sometimes is not always typical Rubinstein–Taybi syndrome [3, 14, 15].
Our patient has slightly arched eyebrows and synophridia, mild hirsutism, post-axial hexadactylia of left foot, and with no beaked nose or other striking features of RSTS. She has mild development retardation of language and motor, abnormality of brain structure was seen by MRI. The patient had a history of multiple respiratory tract infections and primary low immunity. All these clinical manifestations suggest that our patient is consistent with the phenotype of atypical RSTS, and she harbours a de novo frameshift EP300 variant of c.2499dupG p.(Pro834Alafs*4) in exon 14 that not involved HAT domain. EP300 related RSTS phenotype is often less classical than that related to pathogenic CREBBP variants, so it would be more difficult to differentiate between RSTS and MKHK. On the other hand, taking into account that only two patients with EP300 variants and MKHK have been officially described, it is appropriate for this Chinese girl to be diagnosed as atypical RSTS. Looking back at these patients with EP300 mutations, we found that a history of recurrent infection or even primary low immunity is a notable feature of the disorder. Some scholars believe that variants causing RSTS in other parts of CREBBP/EP300 would result in haplo-insufficiency of functions or perturb the function of the HAT domain, typically [7]. But not all functions of CREBBP/EP300 are dose-dependent. The disease caused by EP300 mutation shows phenotype heterogeneity and likely constitutes at least two different entities, such as RSTS and MKHK. Due to the relatively small number of patients with EP300 mutations reported, there are still a lot of details about these two entities that need to be constructed. This case contributes one novel de novo EP300 variant and novel phenotypes to the atypical RSTS. It further extends the borders of the EP300 variant resulting in RSTS-like disease and expands its clinical spectrum.
Conclusion
This case report demonstrates that clinical features with c.2499dupG p.(Pro834Alafs*4) in exon 14 of EP300 are less marked than in RSTS2 patient, although previous studies have suggested that mutations beyond the exons 30 and 31 of EP300 gene can lead to RSTS. Our additional case could help to deepen the clinical and genetic spectrum in this disorder, so as to establish the correlationship between phenotypes and genotypes to some extent. Our case provides a novel EP300 mutation for atypical RSTS and enriches the phenotypes. This could contribute to improve the health management and genetic counselling for patients, and facilitate clinical research in a certain extent.
Availability of data and materials
The raw datasets used and analyzed during the current study are not deposited in publicly available repositories because of considerations about the security of human genetic resources. Any questions should be directed to the corresponding author. We provide conclusive variant information without identifying/confidential patient data in the paper or its appendix. For other details of the availability of data and material, please refer to the methods section of the article.
Abbreviations
- NGS:
-
Next-generation sequencing
- gnomAD:
-
Genome aggregation database
- HGMD:
-
The human gene mutation database at the institute of medical genetics in Cardiff
- ACMG:
-
The American College of Medical Genetics and Genomics
- MRI:
-
Magnetic resonance imaging
- FLAIR:
-
Fluid attenuated inversion recovery
- DWI:
-
Diffusion weighted imaging
References
Hennekam RC. Rubinstein–Taybi syndrome. Eur J Hum Genet. 2006;14(9):981–5.
Rubinstein JH, Taybi H. Broad thumbs and toes and facial abnormalities. A possible mental retardation syndrome. Am J Dis Child. 1963;105:588–608.
Negri G, et al. Clinical and molecular characterization of Rubinstein–Taybi syndrome patients carrying distinct novel mutations of the EP300 gene. Clin Genet. 2015;87(2):148–54.
Bartholdi D, et al. Genetic heterogeneity in Rubinstein–Taybi syndrome: delineation of the phenotype of the first patients carrying mutations in EP300. J Med Genet. 2007;44(5):327–33.
Roelfsema JH, et al. Genetic heterogeneity in Rubinstein–Taybi syndrome: mutations in both the CBP and EP300 genes cause disease. Am J Hum Genet. 2005;76(4):572–80.
Bartsch O, et al. Two patients with EP300 mutations and facial dysmorphism different from the classic Rubinstein–Taybi syndrome. Am J Med Genet A. 2010;152a(1):181–4.
Menke LA, et al. Further delineation of an entity caused by CREBBP and EP300 mutations but not resembling Rubinstein–Taybi syndrome. Am J Med Genet A. 2018;176(4):862–76.
Menke LA, et al. CREBBP mutations in individuals without Rubinstein–Taybi syndrome phenotype. Am J Med Genet A. 2016;170(10):2681–93.
Angius A, et al. Confirmation of a new phenotype in an individual with a variant in the last part of exon 30 of CREBBP. Am J Med Genet A. 2019;179(4):634–8.
Banka S, et al. Genotype-phenotype specificity in Menke–Hennekam syndrome caused by missense variants in exon 30 or 31 of CREBBP. Am J Med Genet A. 2019;179(6):1058–62.
Ramos YF, et al. Genome-wide assessment of differential roles for p300 and CBP in transcription regulation. Nucleic Acids Res. 2010;38(16):5396–408.
Dyson HJ, Wright PE. Role of intrinsic protein disorder in the function and interactions of the transcriptional coactivators CREB-binding protein (CBP) and p300. J Biol Chem. 2016;291(13):6714–22.
Wang Q, et al. A novel CREBBP in-frame deletion variant in a Chinese girl with atypical Rubinstein–Taybi syndrome phenotypes. J Mol Neurosci. 2021;71(3):607–12.
Cohen JL, et al. EP300-related Rubinstein–Taybi syndrome: highlighted rare phenotypic findings and a genotype–phenotype meta-analysis of 74 patients. Am J Med Genet A. 2020;182(12):2926–38.
López M, et al. Rubinstein–Taybi 2 associated to novel EP300 mutations: deepening the clinical and genetic spectrum. BMC Med Genet. 2018;19(1):36.
Fergelot P, et al. Phenotype and genotype in 52 patients with Rubinstein–Taybi syndrome caused by EP300 mutations. Am J Med Genet A. 2016;170(12):3069–82.
Acknowledgements
We would like to thank all the participants for their help and willingness to participate in this study. We thank the reviewers for their comments. We also thank the editors of this manuscript.
Funding
This work was funded in part by the National Key R&D Program of China (2018YFC1002206-2). The role of the funding body—this study is the authors’ independent work, and the funding agency only provides relevant financial support. The design of the study and collection, analysis, and interpretation of data and writing of the manuscript were completed by all authors independently.
Author information
Authors and Affiliations
Contributions
ZB and XK contributed to the conception and design of the study. ZB carried out the experiments. ZB and GL performed the data analysis. ZB wrote and revised the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The clinical investigation was conducted according to the Declaration of Helsinki, and the study was approved by the institutional review board of the Medical Ethics Committee of the First Affiliated Hospital of Zhengzhou University(Grant No: KS-2018-KY-36). The Medical Ethics Committee of the First Affiliated Hospital of Zhengzhou University authorized our team to access the clinical patient data used in this research. The patient and her parents consented to participate in this study, and written INFORMED CONSENT was obtained.
Consent for publication
Written informed consent for publication of clinical details and/or clinical images was obtained from the parents of juvenile patient.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Fig. S1a–g.
Phenotypic features of patient described in this study. a and b The patient 4 years old. Note slightly arched eyebrows and synophridia, a square tip to his nose, normal columella, prominent two front teeth, normal tooth number and absence of characteristic grimace of Rubinstein–Taybi syndrome. c The fine hairs on the front of the ear and on the cheek are hair whorl. d and e The child has heavy fine hair on her back and opisthenar. f Patient has no broad or angulated thumbs, nor broad distal phalanges of the fingers, as seen in patients with Rubinstein–Taybi syndrome. g Girl has a sixth toe of her left foot, that hexadactyly. Short Video S1. She can ride a tricycle independently and freely. Short Video S2 and S3. She can build blocks and draw with no problem.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Bai, Z., Li, G. & Kong, X. Case report: a Chinese girl like atypical Rubinstein–Taybi syndrome caused by a novel heterozygous mutation of the EP300 gene. BMC Med Genomics 16, 24 (2023). https://doi.org/10.1186/s12920-022-01424-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12920-022-01424-4