
Abstract
Background
The deletion of a short arm fragment on chromosome 8 is a rare cause of Kallmann syndrome and spherocytosis due to deletion of the FGFR1 and ANK1 genes.
Case presentation
This case study describes a 4-month-old child with growth and psychomotor retardation, auricle deformity, microcephaly, polydactyly, a heart abnormality, and feeding difficulties. An approximately 12.00 MB deletion was detected in the 8p11.22-p21.2 region of chromosome 8. After sequencing, we found that 65 protein genes had been deleted, including FGFR1, which resulted in Kallmann syndrome. There was no deletion of the ANK1 gene associated with spherocytosis, consistent with the phenotype.
Conclusion
This patient is a new case of short arm deletion of chromosome 8, resulting in novel and previously unreported clinical features.
Similar content being viewed by others

Background
Short arm deletions of chromosome 8 in people are rare. Since the first report of a short arm deletion of chromosome 8 related to spherocytosis [1], ten additional cases have been reported globally. White et al. [2] noted that deletion of the ANK1 gene on chromosome 8 was associated with spherocytosis. Dodé et al. [3] reported that deletion of the FGFR1 gene resulted in Kallmann syndrome. Mu et al. [4] described a more extensive range of heterozygous deletions on chromosome 8, including the SLC20A2 and THAP1 genes, which led to dystonia. Tham et al. [5] proposed that deletion of KAT6A resulted in a syndrome that included heart defects, intellectual disability, difficulty eating, craniocerebral protrusion, and unique facial features. This case study reports a new 12 MB deletion in the short arm 8p11.22-p21.2 of chromosome 8.
Case presentation
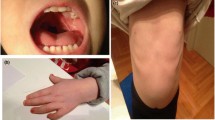
A 4-month, 20-day-old girl was admitted to the University of Chinese Academy of Science-Shenzhen Hospital on February 8, 2021, with a cough that had lasted six days and a fever for three h. She was a full-term child whose parents were not close relatives. There was no history of radiation exposure, smoking, or drinking during pregnancy. Additionally, there was no history of cold, fever, medication, nontoxic substances, or radiation exposure during early pregnancy. The child was the fourth fetus and second birth in the family. She was delivered vaginally at a gestational age of 39 + 3 weeks with a birth weight of 2.7 kg. During delivery, there was no asphyxia, umbilical cord around the neck, or premature rupture of membranes; the amniotic fluid was clear, and the placenta was normal. Her Apgar scores 1 and 5 min after birth were both 10. She had six fingers on her right hand (Fig. 1). Two days after birth, after feeding poorly, moaning for half a day, and developing a fever for 1 h, she was diagnosed with neonatal purulent meningitis, sepsis, pneumonia, and hyperbilirubinemia. Nineteen days after birth, a swollen, dark tumor developed on the child's right thumb. Due to evident ischemia and necrosis, a multi-finger resection was performed, and she was discharged from the hospital 22 days later. She was hospitalized 98 days after birth because she was not eating. Before admission, she ate 20–30 mL per meal, six times a day. On the day of admission, her total milk volume was 50 mL. She was eating 40 mL per meal every 3–4 h 12 days later and discharged.

The patient has six congenital fingers
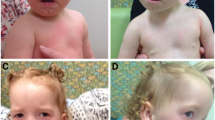
Before the present admission, she ate 50 mL per meal every 3–4 h. Physical examination indicated the following: head circumference 34 cm (unchanged from birth), body temperature 37.8 °C, pulse 146 per min, respirations 42 per min, blood oxygen saturation 95%, weight 4 kg (< 3rd percentile), and body length 53 cm (< 3rd percentile) (Fig. 2). The fontanels were open. Her development was delayed (neck muscle weakness; could stand for 1 min, head height < 3rd percentile, could not support chest and abdomen for one minute on elbows). The pursuit of sounds and objects was normal. She was conscious, had an auricle deformity, warm limbs without cyanosis, and CRT 1.5 s. The patient has no cleft lip/ palate. Her sucking force was inferior, and muscle strength in both arms was slightly reduced, but both legs were normal and she could move her legs freely. The physiological reflexes were present, and no pathological reflexes were elicited. Brain magnetic resonance imaging (MRI) indicated an abnormal signal in the bilateral occipital subarachnoid space, suggestive of leptomeningitis or subarachnoid hemorrhage. Considering the possibility of an abscess, an enhanced MRI was performed. There were no apparent abnormalities in brain ultrasound or MRI of the head (Fig. 3). Color Doppler echocardiography showed a continuous 2.7 mm interruption in the middle of the atrial septum (Fig. 4). Re-examination was recommended after one year of age to rule out a patent foramen ovale. The systolic and diastolic functions of the heart were normal.

Patient's face and height. Paper is the size of an A4 Sheet

MRI of the child's head

Image of Color Doppler echocardiography
Due to the postnatal growth and psychomotor retardation, auricle deformity, microcephaly, polydactyly, cardiac abnormality, and feeding difficulties, low-depth genome sequencing was performed at Shenzhen Huada Gene Research Institute with the consent of her family (Fig. 5). This showed SEQ (grch37) del (8p21.2p11.22) chr8. A 12 MB pathogenic fragment, G.27228261–39230720del, was from an unknown source. The comprehensive ClinGen CNV score was ≥ 0.99, indicating a pathogenic lesion. The parents' and older brothers’ chromosomes were normal.

Detection of chromosome defects by high-throughput sequencing
As a result of the chromosomal disease, the patient's postnatal growth and development were delayed, and she had muscle weakness, insufficient food intake, and severe pneumonia. As she was weakly positive for Mycoplasma, she was given erythromycin and atomization treatment. Her cough improved with rehabilitation and physiotherapy. At discharge, she could hold her head up, remain upright for 2–3 min, and support her chest and abdomen on her elbows for a few minutes. She consumed 50 mL of milk every 3–4 h.
Discussion
Short arm deletion of chromosome 8 is rare. Since 1975, there have been 15 reports of deletion in 8p11pter (including 6 of deletion in 8p11.1–8p21.1) (Table 1). The common clinical manifestations of chromosome 8 short arm deletion are microcephaly, developmental delay, gonadal hypoplasia, difficulty eating, unique facial features, and spherocytosis. In addition, our patient had six fingers on one hand and external ear malformations. Similarly to this case, only one previously reported case had an abnormal ear [6] due to a chromosome 8p short arm deletion. To the best of our knowledge, it is the first report that the patient with chromosome 8 short arm deletion has polydactyly as one of the clinical manifestations.
This case study describing the deletion of chromosome 8p11.22-p21.2 is the first report of this occurrence, although there are several reported cases of 8p11.1-p21.1 or 8p11.22-p21.1. Kitatani et al. [7] reported an 8p11.22-p21.1 deletion mutation in a 13-month-old boy who presented with postnatal growth and psychomotor retardation, microcephaly, a high arch back, epicanthus, penile cryptorchidism, hypoplasia of two fingernails, sacral depression, and spherocytosis. Okamoto et al. [8] reported a deletion of 8p11.23-p21.1 in a 30-month-old boy who was 48 cm long at birth, with a head circumference of 33.5 cm, stunted development, a heart defect, difficulty feeding, multiple deformities, and spherocytosis. [9] detected an 8p12-p21.2 deletion mutation in a 2.5-year-old girl with developmental delays, facial deformity, and feeding difficulties, but without spherocytosis.
The missing region was found to contain 65 protein-coding genes using whole-genome sequencing, one of which was FGFR1 (Table 2). The FGFR1 deletion is associated with Kallmann syndrome [3], hypogonadotropic hypogonadism with anosmia, or hypo-olfaction. In this case, the patient was assisted in smelling milk, vinegar, and soy sauce, respectively, and her facial expression was monitored. From her facial expressions, it was ascertained that she could distinguish different tastes. Other common features of the FGFR1 deletion include microcephaly, growth retardation, and facial features. The gonadal growth of our patient cannot yet be assessed. However, microcephaly, growth retardation, and facial features are consistent with Kallmann syndrome.
The ANK1 mutation or deletion in the proximal region of 8p11.2 has been linked with spherocytosis [10]. Sequencing revealed no ANK1 mutation or deletion in this case and no anemia caused by spherocytosis, similar to a case of 8p11.1p21 deletion reported by [11], which had the Kallmann syndrome phenotype but no spherocytosis.
Four genes have been reported among the 65 missing genes: GSR (glutathione disulfide reduction), NRG1 (neuregulin 1), EXTL3 (exostatin-like glycosyltransferase 3), and WRN (WRN RecQ like helicase). In some reports, the complete absence of GSR [12] or homozygous mutations in the gene for GSR [9] are a cause of hemolytic anemia related to low levels of glutathione. Conversely, the level of GSR activity in patients reported by Chilcote et al. [13] and Okamoto et al. [8] was not considered low enough to cause hemolysis through GSR deficiency. The patient in this case study also did not develop hemolytic anemia. NRG1 plays an essential role in the nervous system and heart development [14]. Neurological symptoms such as muscle hypotonia or absent/decreased reflexes were described in four other patients with interstitial 8p deletions [6, 8, 13]. In this case study, her sucking force was inadequate, and the muscle strength of both arms was slightly reduced, but that of both legs was normal. Okamoto et al. [8] described one patient with a deletion containing the EXTL3 gene with retinal dysplasia who was virtually blind. In the current case study, the patient's vision is normal, and there are no other reports correlating the EXTL3 gene with loss of vision. The mutation of WRN is often associated with Werners’ syndrome, an autosomal recessive disorder characterized by the premature onset of a number of age-related diseases [15]. Compared with other reports detailing patients lacking these genes (GSR, NRG1 and EXTL3), the clinical manifestations are different and the mechanisms needs to be closely investigated. In this case, the patient is too young to assess whether Werners syndrome will emerge.
The chromosomes of both parents and older brother were normal, and they displayed no symptoms related to a chromosome deletion. Therefore, the patient's disease is likely due to a de novo mutation in meiosis and needs further investigation.
This case study describes the first patient experiencing the deletion of chromosome 8p11.22-p21.2, affecting 65 protein-coding genes. The limitation of this study is the challenge of confirming whether the specific gene deletion was directly associated with the newly reported phenotypes. The symptoms observed in the female newborn are microcephaly, developmental delays, gonadal hypoplasia, difficulty eating, polydactyly, unique facial features and external ear malformations. On the other hand, due to the discharged young age, some symptoms cannot be fully characterized, such as primary failure of sexual development and visual acuity. Due to this disease's rarity, clinical experience in diagnosing and treating this disease is lacking, with no detailed analysis of bone abnormalities, renal agenesis, and multiple developmental defects.
In conclusion, we report a new 12 MB deletion in the short arm 8p11.22-p21.2 of chromosome 8. Clinically, the prenatal diagnosis should be straightforward due to the many missing fragments on chromosome 8. To date, there is no effective treatment for diseases caused by chromosome deletion, leaving prenatal screening as the primary strategy to prevent genetic defects.
Availability of data and materials
The raw data have been deposited in the SRA database under the accession number PRJNA719751. The rest of the data that support the conclusions of this study are available from the corresponding author upon request.
Abbreviations
- kg:
-
Kilogram
- min:
-
Minute
- h:
-
Hour
- mL:
-
Milliliter
- s:
-
Second
- °C:
-
Degrees Celsius
- mm:
-
Millimeter
References
Kimberling WJ, Fulbeck T, Dixon L, Lubs HA. Localization of spherocytosis to chromosome 8 or 12 and report of a family with spherocytosis and a reciprocal translocation. Am J Hum Genet. 1975;27(5):586–94.
White RA, Birkenmeier CS, Lux SE, Barker JE. Ankyrin and the hemolytic anemia mutation, nb, map to mouse chromosome 8: presence of the nb allele is associated with a truncated erythrocyte ankyrin. Proc Natl Acad Sci U S A. 1990;87(8):3117–21.
Dodé C, Levilliers J, Dupont JM, De Paepe A, Le Dû N, Soussi-Yanicostas N, Coimbra RS, Delmaghani S, Compain-Nouaille S, Baverel F, et al. Loss-of-function mutations in FGFR1 cause autosomal dominant Kallmann syndrome. Nat Genet. 2003;33(4):463–5.
Mu W, Tochen L, Bertsch C, Singer HS, Barañano KW. Intracranial calcifications and dystonia associated with a novel deletion of chromosome 8p11.2 encompassing SLC20A2 and THAP1. BMJ Case Rep. 2019;12(5):e228782.
Tham E, Lindstrand A, Santani A, Malmgren H, Nesbitt A, Dubbs HA, Zackai EH, Parker MJ, Millan F, Rosenbaum K, et al. Dominant mutations in KAT6A cause intellectual disability with recognizable syndromic features. Am J Hum Genet. 2015;96(3):507–13.
Cohen H, Walker H, Delhanty JD, Lucas SB, Huehns ER. Congenital spherocytosis, B19 parvovirus infection and inherited interstitial deletion of the short arm of chromosome 8. Br J Haematol. 1991;78(2):251–7.
Kitatani M, Chiyo H, Ozaki M, Shike S, Miwa S. Localization of the spherocytosis gene to chromosome segment 8p112.2––8p21. Hum Genet. 1988;78(1):94–5.
Okamoto N, Wada Y, Nakamura Y, Nakayama M, Chiyo H, Murayama K, Inoue T, Kanzaki A, Yawata Y, Hirono A, et al. Hereditary spherocytic anemia with deletion of the short arm of chromosome 8. Am J Med Genet. 1995;58(3):225–9.
Klopocki E, Fiebig B, Robinson P, Tönnies H, Erdogan F, Ropers HH, Mundlos S, Ullmann R. A novel 8 Mb interstitial deletion of chromosome 8p12-p212. Am J Med Genet A. 2006;140(8):873–7.
Eber SW, Gonzalez JM, Lux ML, Scarpa AL, Tse WT, Dornwell M, Herbers J, Kugler W, Ozcan R, Pekrun A, et al. Ankyrin-1 mutations are a major cause of dominant and recessive hereditary spherocytosis. Nat Genet. 1996;13(2):214–8.
Stratton RF, Crudo DF, Varela M, Shapira E. Deletion of the proximal short arm of chromosome 8. Am J Med Genet. 1992;42(1):15–8.
Loos H, Roos D, Weening R, Houwerzijl J. Familial deficiency of glutathione reductase in human blood cells. Blood. 1976;48(1):53–62.
Chilcote RR, Le Beau MM, Dampier C, Pergament E, Verlinsky Y, Mohandas N, Frischer H, Rowley JD. Association of red cell spherocytosis with deletion of the short arm of chromosome 8. Blood. 1987;69(1):156–9.
Falls DL. Neuregulins: functions, forms, and signaling strategies. Exp Cell Res. 2003;284(1):14–30.
Lebel M. Werner syndrome: genetic and molecular basis of a premature aging disorder. Cell Mol Life Sci. 2001;58(7):857–67.
Acknowledgements
Not applicable.
Funding
This study is supported by Basic research on science and technology projects in Shenzhen (No. JCYJ20180507183428877, JCYJ20180504165657443). The funding body had no role in the study's design, collection, analysis, and interpretation of data or writing of the manuscript.
Author information
Authors and Affiliations
Contributions
JCD performed the experiments and wrote the manuscript. HXT and JZ collected and analysed the clinical data. XSC revised the manuscript and conducted experiments. BQW designed the study and reviewed the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
This study was approved by the Ethics Committee of the University of Chinese Academy of Science-Shenzhen Hospital. Written informed consent was obtained from all participants and the legal guardian of the participants under the age of 16.
Consent to publish
Written informed consent for publication of identifying images or other personal or clinical details was obtained from all participants and the legal guardian of the participants under the age of 18.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Dai, J., Zeng, J., Tan, H. et al. Novel 12 Mb interstitial deletion of chromosome 8p11.22-p21.2: a case report. BMC Med Genomics 15, 126 (2022). https://doi.org/10.1186/s12920-022-01274-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12920-022-01274-0