
Abstract
Purpose
This study aims to analyze the Norrie disease gene (NDP) variants in patients with familial exudative vitreoretinopathy (FEVR) and their clinical features.
Methods
Thirty-three Chinese patients (22 familial and 11 simplex) who were diagnosed as FEVR underwent detailed ocular examinations in Beijing Tongren Hospital. Peripheral venous blood was drawn from the patients and their family members for the extraction of genomic DNA. All exons of NDP gene were analyzed by direct sequencing of PCR-amplified DNA fragments.
Results
Four novel mutations in NDP gene were identified in four X-linked FEVR families: a C → T transversion, c. 625C → T, in exon 3, resulting in a serine-to-proline change in codon 73 (S73P); a C → G transition, c. 751C → G, in exon 3, resulting in an arginine-to-glycine change in codon 115 (R115G); a T → C transversion of nucleotide 331 at 5’UTR in exon 2 (c.331 T → C); and a C → T transversion of the nucleotide 5 in intron 1 (IVS1 + 5C → T). The mutations were not present in the control group (n = 100).
Conclusions
Our results extend the spectrum of NDP gene mutations. The mutations in the non-coding region of NDP may play a crucial role in the pathogenesis of FEVR.
Similar content being viewed by others

Introduction
Familial exudative vitreoretinopathy (FEVR) is an inherited vitreous retinal disease characterized by incomplete vascularization of the peripheral retina with secondary complications including dragged retina, exudation, vitreous hemorrhage, and retinal detachment [1, 2]. The severity of the disease varies from no apparent visual symptoms or impairment to complete blindness [3].
The inheritance pattern of FEVR is autosomal dominant, recessive, and X-linked. To date, genes involved in the pathogenesis of FEVR include FZD4, NDP, LRP5, TSPAN12, ZNF408, KIF11, and CTNNB1 [4,5,6,7,8,9]. The Norrie disease gene (NDP) is located on chromosome Xp11.2–11.3 and encodes for the 133-amino-acid protein Norrin. Norrin and FZD4 (Frizzled-4) function as a high-affinity ligand-receptor pair, and Norrin together with the auxiliary component tetraspanin-12 (TSPAN12) induces FZD4 and lipoprotein receptor-related protein-5 (LRP5) dependent activation of classical Wnt pathway via promoting the recruitment of β-catenin (encoded by CTNNB1) into the nucleus [10,11,12]. Defects in Wnt signaling cascade affect ocular growth and development and play an important role in the pathological processes underlying both X-linked FEVR and Norrie disease (ND, MIM#310620) [4], which may present as pseudoglioma, mental retardation, or hearing loss. KIF11 encodes a mitotic kinesin known as Eg5, while ZNF409 encodes a zinc-finger homeobox protein, but their function in FEVR remains unclear.
Over 160 mutations of the NDP gene have been reported so far, most of which are missense mutations (about 50%), while others are deletion mutations (about 6%)(HGMD Professional 2017.3, http://www.hgmd.cf.ac). Some nonsense, splicing site, insertion, deletion-insertion, and regulatory region mutations are also reported in the NDP gene. Most patients with NDP gene mutations show the Norrie disease phenotype; only 5% of patients show FEVR. Mutation screening of the NDP gene in our Chinese patients with FEVR could, therefore, determine further enrich the spectrum and frequency of mutations in NDP-causing FEVR.
Methods
Study participants
Thirty-three patients with FEVR and their family members were recruited for this study at the Beijing Tongren Hospital, Department of Ophthalmology. Of the 33 patients, 22 had familial FEVR and 11 had sporadic FEVR (with no apparent family history). All the patients were Chinese and were born at term with normal weight. The diagnosis of FEVR was based on the presence of at least one of the typical clinical signs including peripheral retinal avascularization with abnormal retinal vascular formation, severe retinal exudates, retinal neovascularization, peripheral fibrovascular mass, macular ectopia, retinal folds, retinal detachment, and vitreous hemorrhage. Fundus fluorescein angiography was performed in the selected cases to confirm the diagnosis [13]. Genetically unrelated individuals from the Beijing Tongren Hospital were recruited as controls for conditions such as senile cataract, floaters, and itchy eyes. They underwent the same ophthalmic examinations and were diagnosed not to have FEVR or other major eye diseases [13]. The study protocol was approved by the Ethics Committee for Human Research of the Beijing Tongren Hospital Capital Medical University and adhered to the tenets of the Declaration of Helsinki. Informed consent was obtained from all the volunteers after providing a detailed explanation of the nature and possible consequences of the study. Venous blood was obtained from the participants and stored at − 20 °C for less than 2 months before DNA extraction.
NDP screening
Genomic DNA was extracted from 200 μl of whole blood using a Qiamp Blood Kit (Qiagen, Hilden, Germany). To detect any possible novel disease-causing mutations in the NDP gene, the three coding exons and adjacent sequences of NDP were screened by polymerase chain reaction followed by direct DNA sequencing with the BigDye Terminator DNA sequencing kit on a 3130XL analyzer (Applied Biosystems, Foster City, CA, USA), using the same set of primers previously described by Chen et al. [4] Reference sequence of NDP (NM_012193.2; GenBank, http://www.ncbi.nlm.nih.gov/Genbank; provided in the public domain by the National Center for Biotechnology Information) was used for the identification of the variants.
Results
The DNAs from the 33 FEVR patients were analyzed for mutations in the three exons and flanking exon-intron boundaries of NDP gene. Four different novel mutations were identified in four FEVR families (Table 1, and Figs. 1 and 2). All probands were men, and therefore, these changes were interpreted as homozygous. These changes in the sequence were not found in the 100 unrelated and unaffected individuals, all females, in the Chinese population. All the changes were co-segregated as the X-linked recessive form of FEVR in the four families in this study.

Schematic diagram of the NDP gene and locations of the mutations

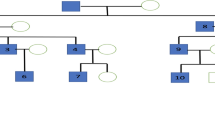
Schematic pedigrees of the families with NDP mutations. Arrows indicate probands; open symbols, clinically unaffected; solid symbols, clinically affected; diagonal lines through symbols, deceased; vertical lines in symbols, affected by hearsay; circles inside symbols, asymptomatic for familial exudative vitreoretinopathy; +/−, heterozygous with familial mutation; and −/−, wild type. For the NDP mutations, the nomenclature is based on GenBank NM_012193.2
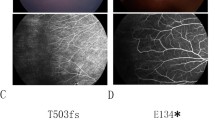
Four members of Family 1 were studied. The proband was an 11-month-old boy who showed bilateral retinal folds resembling persistent hyperplastic primary vitreous (Fig. 3a). His maternal uncle was diagnosed with FEVR because of bilateral retinal folds at 7 years of age. Direct DNA sequencing of these two cases revealed a C → T transversion, c. 625C → T, in exon 3 of the NDP gene, where serine was replaced by proline at codon 73 (S73P; Fig. 2). The proband’s mother was heterozygous for the S73P mutation.

Fundus images of proband in FEVR families
In Family 2, the proband was a 4-month-old boy who showed a retrolental mass with total retinal detachment in the right eye (Fig. 3b). The typical features of FEVR, such as peripheral avascularization temporal to the macula and neovascularization of the retinal vessels, were observed in the left eye where laser photocoagulation had been performed. Sequence analyses demonstrated that he carried a C → G transition, c. 751C → G, in exon 3, resulting in an arginine-to-glycine change in codon 115 (R115G; Fig. 2). None of the family members had FEVR, however, the mother was heterozygous for the R115G mutation.
Family 3 was a three-generation family with affected individuals presenting X-linked exudative vitreoretinopathy without deafness or mental retardation. A T → C transversion of nucleotide 331 at 5’UTR in exon 2 (c.331 T → C) of NDP gene was identified in all the affected family members. The proband was a 6-month-old boy who showed bilateral retinal folds, macular ectopia, retrolental fibroplasia, and retinal detachment (Fig. 3c). The proband’s maternal cousin (son of his mother’s sister) was diagnosed with FEVR resulting from retrolental fibroplasias and retinal detachment at the age of 7 years. His vision was restricted to light perception in the left eye but no light perception in the right eye, and he could not follow a moving finger with either eye. Furthermore, the proband’s maternal uncle, who was blind since the age of five, presented with bilateral retrolental fibroplasias, retinal detachment, and a flat anterior chamber.
In Family 4, the proband was a 2-year-old boy. He was referred to our hospital at 7 months of age, and FEVR was suspected because of bilateral retinal folds, macular ectopia, retrolental fibroplasias, and persistent hyaloid remnant (Fig. 3d). Photocoagulation was performed on both eyes at the age of 1 year, after which he could follow a moving object. Sequence analyses demonstrated that he carried a C → T transversion of nucleotide five in intron 1 (IVS1 + 5C → T) (Fig. 2). Although both parents did not have any ocular abnormalities, the mother was heterozygous for the IVS1 + 5C → T mutation.
Discussion
We identified four novel disease-causing mutations in the NDP gene in four different Chinese families. Of the four mutations, one was a novel splicing mutation, one was a novel 5’UTR mutation, while the other two were novel missense mutations. All of these mutations were responsible for X-linked FEVR and were not found in normal individuals from the Chinese population. These results suggest that the four mutations are pathogenic. Furthermore, our data indicate that the NDP mutations cause about 12.1% (4/33) of FEVR in the Chinese population. The prevalence of NDP mutation in our patients is slightly higher than that reported in previous studies, which may be due to ethnic difference.
NDP gene-related diseases are X-linked recessive genetic diseases [4, 14]. NDP-related retinal diseases, caused by abnormalities in the NDP gene, include various progressive diseases with retinal vascular hypoplasia, such as Norrie disease, Coat’s disease, FEVR, persistent hyperplastic primary vitreous, and retinopathy of prematurity [14,15,16,17].
The NDP gene spans 2.8 kBP, and the cDNA contains three exons. Exon 1 does not encode and may promote gene transcription [18]. The region encoding the protein is located in the second half of the second exon and the first part of the third exon. Previous studies have shown that most of the NDP gene mutations leading to the FEVR phenotype are missense mutations, while the nonsense, splice-site, and large deletion mutations mainly lead to Norrie disease. To date, nearly 20 mutations in the NDP gene have been identified in individuals with X-linked familial and sporadic exudative vitreoretinopathy, however, all of them were missense mutations. The missense mutations may lead to drastic amino acid changes and often affect one of the many cysteine residues immediately adjacent to a cysteine. Mutations in these regions may disrupt protein folding or directly interfere with the norrin-receptor interactions [5, 14, 18, 19]. Schubak et al. detected NDP gene mutations in 24 families, most of which were located in or near cysteine residues [20]. Furthermore, Wu et al. [19]. analyzed 11 patients with NDP gene mutations, of whom, four with Norrie disease had NDP mutations involving cysteine residues, while four with FEVR, one with retinopathy of prematurity, and one with persistent primary vitreous hyperplasia did not [21]. However, in our study, two novel missense mutations (S73P and R115G) did not involve cysteine.
Two patients with the novel missense mutations, S73P and R115G, had typical manifestations of FEVR, including peripheral retinal vascular anomalies, fibrous proliferation, and dragged macula. This finding is consistent with that in an mice model with an NDP mutation [22]. Multiple sequence alignment of human Norrin against the available species sequences revealed that codons S73 and R115 were conserved in rats and mice [data not shown]. S73P and R115G were the non-conserved mutations located within the cystine knot domain. Moreover, Walker et al. [23] previously reported one family with Norrie disease with a 626C → A transversion of the NDP gene, resulting in an S73X substitution. The affected family members had a severe ocular phenotype, but no mental retardation or deafness. In addition, the R115L mutation was previously identified by Kondo and coworkers in a Japanese patient with typical FEVR [24]. This finding is consistent with that of Wu et al. [19] who reported the mutations in the non-cysteine residues of the Norrin protein that led to abnormal vascular and retinal development and phenotypes consistent with FEVR. Using a Norrin-based reporter assay to analyze the effects of the FEVR-causing mutations, Qin et al. [25] demonstrated that Norrin mutants (K54N and R115L) demonstrated variable effects on signal transduction and impaired cell surface binding. Although the identification of a second point mutation affecting amino acid 73 and 115 in these FEVR families made the proposed pathogenic nature of the S73P and R115G robust, the functional relevance of each mutation should be explored in further detail.
In this study, in Family 3, a T → C transversion of nucleotide 331 at 5’UTR in exon 2 (c.331 T → C) of the NDP gene was identified in all the three affected members, whereas, in Family 4, a C → T transversion of nucleotide five in intron 1 (IVS1 + 5C → T) was identified in a 2-years-old boy with FEVR. In addition, no pathogenic mutation was found in the subsequent screening of other genes related to FEVR (FZD4, LRP5, TSPAN12, ZNF408, and CTNNB1) [data not shown]. Although the two mutations (c.331 T → C and IVS1 + 5C → T) belonged to the non-coding region of the NDP gene, we believe that they are pathogenic. Ghislaine et al. [14] previously reported two French families with Norrie disease with a c.1A → G in exon 1 in an 18-month-old boy and a c.23479–1G → C in intron 2 in the families of five patients. These data indicate that the mutations in the non-coding region may still lead to protein misfolding, resulting in functional abnormalities that may lead to various diseases.
In summary, we identified four novel mutations that will extend spectrum on NDP mutations, especially in Chinese FEVR patients. The sequence changes of NDP gene that do not cause changes in the amino acid sequence may play a role in the pathogenesis of FEVR. Patients with NDP mutations have the characteristics of younger onset age and serious eye disease.
Availability of data and materials
All data generated or analysed during this study are included in this published article.
References
Criswick VG, Schepens CL. Familial exudative vitreoretinopathy. Am J Ophthalmol. 1969;68:578–94.
Benson WE. Familial exudative vitreoretinopathy. Trans Am Ophthalmol Soc. 1995;93:473–521.
Canny CL, Oliver GL. Fluorescein angiographic findings in familial exudative vitreoretinopathy. Arch Ophthalmol. 1976;94:1114–20.
Chen ZY, Battinelli EM, Fielder A, Bundey S, Sims K, Breakefield XO, et al. A mutation in the Norrie disease gene (NDP) associated with X-linked familial exudative vitreoretinopathy. Nat Genet. 1993;5:180–3.
Robitaille J, MacDonald ML, Kaykas A, Sheldahl LC, Zeisler J, Dubé MP, et al. Mutant frizzled-4 disrupts retinal angiogenesis in familial exudative vitreoretinopathy. Nat Genet. 2002;32:326–30.
Toomes C, Bottomley HM, Jackson RM, Towns KV, Scott S, Mackey DA, et al. Mutations in LRP5 or FZD4 underlie the common familial exudative vitreoretinopathy locus on chromosome 11q. Am J Hum Genet. 2004;74:721–30.
Nikopoulos K, Gilissen C, Hoischen A, van Nouhuys CE, Boonstra FN, Blokland EA, et al. Next-generation sequencing of a 40 Mb linkage interval reveals TSPAN12 mutations in patients with familial exudative vitreoretinopathy. Am J Hum Genet. 2010;86:240–7.
Collin RW, Nikopoulos K, Dona M, Gilissen C, Hoischen A, Boonstra FN, et al. ZNF408 is mutated in familial exudative vitreoretinopathy and is crucial for the development of zebrafish retinal vasculature. Proc Natl Acad Sci U S A. 2013;110:9856–61.
Panagiotou ES, Sanjurjo Soriano C, Poulter JA, Lord EC, Dzulova D, Kondo H, et al. Defects in the cell signaling mediator β-catenin cause the retinal vascular condition FEVR. Am J Hum Genet. 2017;100:960–8.
Shen G, Ke J, Wang Z, Cheng Z, Gu X, Wei Y, et al. Structural basis of the Norrin-frizzled 4 interaction. Cell Res. 2015;25:1078–81.
Chang TH, Hsieh FL, Zebisch M, Harlos K, Elegheert J, Jones EY. Structure and functional properties of Norrin mimic Wnt for signalling with Frizzled4, Lrp5/6, and proteoglycan. Elife. 2015;4:e06554.
Xu Q, Wang Y, Dabdoub A, Smallwood PM, Williams J, Woods C, et al. Vascular development in the retina and inner ear: control by Norrin and Frizzled-4, a high-affinity ligand-receptor pair. Cell. 2004;116:883–95.
Jia LY, Li XX, Yu WZ, Zeng WT, Liang C. Novel frizzled-4 gene mutations in chinese patients with familial exudative vitreoretinopathy. Arch Ophthalmol. 2010;128(10):1341–9.
Royer G, Hanein S, Raclin V, Gigarel N, Rozet JM, Munnich A, et al. NDP gene mutations in 14 French families with Norrie disease. Hum Mutat. 2003;22:499.
Robitaille JM, Zheng B, Wallace K, Beis MJ, Tatlidil C, Yang J, et al. The role of Frizzled-4 mutations in familial exudative vitreoretinopathy and coats disease. Br J Ophthalmol. 2011;95:574–9.
Aponte EP, Pulido JS, Ellison JW, Quiram PA, Mohney BG. A novel NDP mutation in an infant with unilateral persistent fetal vasculature and retinal vasculopathy. Ophthalmic Genet. 2009;30:99–102.
Rathi S, Jalali S, Musada GR, Patnaik S, Balakrishnan D, Hussain A, et al. Mutation spectrum of NDP, FZD4 and TSPAN12 genes in Indian patients with retinopathy of prematurity. Br J Ophthalmol. 2018;102:276–81.
Meitinger T, Meindl A, Bork P, et al. Molecular modelling of the Norrie disease protein predicts a cystine knot growth factor tertiary structure. Nat Genet. 1993;5:376–80.
Wu WC, Drenser K, Trese M, Capone A Jr, Dailey W. Retinal phenotype-genotype correlation of pediatric patients expressing mutations in the Norrie disease gene. Arch Ophthalmol. 2007;125:225–30.
Schuback DE, Chen ZY, Craig IW, Breakefield XO, Sims KB. Mutations in the Norrie disease gene. Hum Mutat. 1995;5:285–92.
Shastry BS. Persistent hyperplastic primary vitreous: congenital malformation of the eye. Clin Experiment Ophthalmol. 2009;37:884–90.
Berger W, van de Pol D, Bächner D, Oerlemans F, Winkens H, Hameister H, et al. An animal model for Norrie disease (ND): gene targeting of the mouse ND gene. Hum Mol Genet. 1996;5:51–9.
Walker JL, Dixon J, Fenton CR, Hungerford J, Lynch SA, Stenhouses SA, et al. Two new mutations in exon 3 of the NDP gene: S73X and S101F associated with severe and less severe ocular phenotype, respectively. Hum Mutat. 1997;9:53–6.
Kondo H, Qin M, Kusaka S, Tahira T, Hasebe H, Hayashi H, et al. Novel mutations in Norrie disease gene in Japanese patients with Norrie disease and familial exudative vitreoretinopathy. Invest Ophthalmol Vis Sci. 2007;48:1276–82.
Qin M, Kondo H, Tahira T, Hayashi K. Moderate reduction of Norrin signaling activity associated with the causative missense mutations identified in patients with familial exudative vitreoretinopathy. Hum Genet. 2008;122:615–23.
Acknowledgements
We thank all patients and their families for their cooperation in this study.
Funding
National Natural Science Foundation of China. 30901638.
Author information
Authors and Affiliations
Contributions
Li-yun JIA: Responsible for design and carry out of the work, analyze and interpret the data; drafted the work. Kai MA: Responsible for collecting patients and guiding clinical diagnosis of phenotype. The authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The study protocol was approved by the Ethics Committee for Human Research of the Beijing Tongren Hospital Capital Medical University and adhered to the tenets of the Declaration of Helsinki. Informed consent was obtained from all the volunteers after providing a detailed explanation of the study.
Consent for publication
The informed consent for the study is attached, and the paper was approved by the parents of the participants. All the original documents were kept and managed by the hospital.
Competing interests
Non-financial competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Jia, LY., Ma, K. Novel Norrie disease gene mutations in Chinese patients with familial exudative vitreoretinopathy. BMC Ophthalmol 21, 84 (2021). https://doi.org/10.1186/s12886-021-01852-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12886-021-01852-3