
Abstract
Background
Glioblastoma multiforme (GBM) is a type of fast-growing brain glioma associated with a very poor prognosis. This study aims to identify key genes whose expression is associated with the overall survival (OS) in patients with GBM.
Methods
A systematic review was performed using PubMed, Scopus, Cochrane, and Web of Science up to Journey 2024. Two researchers independently extracted the data and assessed the study quality according to the New Castle Ottawa scale (NOS). The genes whose expression was found to be associated with survival were identified and considered in a subsequent bioinformatic study. The products of these genes were also analyzed considering protein-protein interaction (PPI) relationship analysis using STRING. Additionally, the most important genes associated with GBM patients’ survival were also identified using the Cytoscape 3.9.0 software. For final validation, GEPIA and CGGA (mRNAseq_325 and mRNAseq_693) databases were used to conduct OS analyses. Gene set enrichment analysis was performed with GO Biological Process 2023.
Results
From an initial search of 4104 articles, 255 studies were included from 24 countries. Studies described 613 unique genes whose mRNAs were significantly associated with OS in GBM patients, of which 107 were described in 2 or more studies. Based on the NOS, 131 studies were of high quality, while 124 were considered as low-quality studies. According to the PPI network, 31 key target genes were identified. Pathway analysis revealed five hub genes (IL6, NOTCH1, TGFB1, EGFR, and KDR). However, in the validation study, only, the FN1 gene was significant in three cohorts.
Conclusion
We successfully identified the most important 31 genes whose products may be considered as potential prognosis biomarkers as well as candidate target genes for innovative therapy of GBM tumors.
Similar content being viewed by others


Introduction
Glioblastoma multiforme (GBM) is one of the most malignant gliomas of the central nervous system (CNS) [1]. The disappointing outcome of GBM treatment is a median survival of only 15 months despite multi-modalities of treatments [2, 3]. Based on the literature, GBM has special biological characteristics presenting high heterogeneity, diffusing invasiveness, and capacity to resist conventional therapies. In addition, the existence of biological barriers, e.g., BBB, makes this tumor difficult to treat [4]. Hence, the development of new methods for the clinical treatment of GBM may be facilitated by identifying the key genes associated with GBM prognosis [5].
Over the last decade, an increased focus has been on clarifying the origin, genomic landscape, and gene expression profile of GBM by identifying specific molecular markers and pathways involved in this pathology [6]. The advent of large-scale transcriptomic analyses in various cancers has tremendously increased our understanding of tumor biology and possible cancer therapy approaches [4]. Accordingly, in recent years, an increasing number of studies have focused on gene expression patterns to propose biomarkers and GBM tumor treatment strategies [7]. However, most of this information has not been translated into clinical practice for GBM patients [7].
The vast quantities of genomic data are now being deposited in public database repositories, such as Array Express (https://www.ebi.ac.uk/arrayexpress/), The Cancer Genome Atlas (TCGA, https://portal.gdc.cancer.gov), Chinese Glioma Genome Atlas (CGGA, http://www.cgga.org.cn) and Gene Expression Omnibus (GEO, https://www.ncbi.nlm.nih.gov/geo/). These genomic data are used by researchers around the world for the discovery of new genes of interest in GBM tumors. Several studies considered numerous mRNA expression datasets and identified gene signature panels to estimate prognosis in GBM tumors to improve the prognostic and predictive assessment of the tumors [8,9,10,11,12,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40,41,42,43,44,45,46,47,48,49,50,51,52,53,54,55,56,57,58,59,60,61,62,63,64,65,66,67,68,69,70,71,72,73,74,75,76,77,78,79,80,81,82,83,84,85,86,87,88,89,90,91,92,93,94,95,96,97,98,99,100,101,102,103,104,105,106,107,108,109,110,111,112,113,114,115,116,117,118,119,120,121,122,123,124,125,126,127,128,129,130,131,132,133,134,135,136,137,138,139,140,141,142,143,144,145,146,147,148,149,150,151,152,153,154,155,156,157,158,159,160,161,162,163,164,165,166,167,168,169,170,171,172,173,174,175,176,177,178,179,180,181,182,183,184,185,186,187,188,189,190,191,192,193,194,195,196,197,198,199,200,201,202,203,204,205,206,207,208,209,210,211,212,213,214,215,216,217,218,219,220,221,222,223,224,225,226,227,228,229,230,231,232,233,234,235,236,237,238,239,240,241,242,243,244,245,246,247,248,249,250,251,252,253,254,255,256,257,258,259,260,261,262]. However, there is no consensus in the literature on the top gene sets that could be eventually used in clinical practice.
Considering the current state of our knowledge, we sought that a systematic survey of the literature is urgently required to identify genes whose expression could be predictive of GBM survival. Subsequently, to determine the top genes whose expression could be of interest in clinical practice, we assess biological pathways and protein-protein interaction (PPI) networks associated with these genes via bioinformatic analyses.
Materials and methods
The Preferred Reporting Items for Systematic Reviews and Meta-Analysis (PRISMA) guideline [263] was followed to conduct the review (Supplementary File 1, Table S1.). PubMed, Scopus, Cochrane, and Web of Science databases were used to search for relevant studies published between 24th February 2003 and 1st January 2024. The search was conducted by the terms “gene expression” or “expressed genes” or “mRNA” or “RNA-Seq” and “survival” or “prognostic” or “biomarker” and “Glioblastoma multiforme (GBM)” or “high-grade glioma”. The full search strategy is reported in the supplementary File 1, Table S2.
Inclusion criteria were: (a) clinical study with human participants, (b) bioinformatics analysis study, (c) full-text articles, (d) published in the English language, (e) published in peer-reviewed journals, and (f) only genes related to GBM were considered. The exclusion criteria were as follows: (a) reviews, letters to the editor, and abstracts, (b) duplicate publications, (c) Plasma biomarker study, (d) participants with immunohistochemical (IHC) and Western blot analysis, (e) cell line study, (f) studies that did not observe a significant correlation between mRNA expression and overall survival, (g) recurrent glioblastoma, (h) pseudogene, and (i) animal study and progression-free survival (PFS) were not considered.
Data extraction and quality assessment
Two independent authors (PA and TA) assessed and extracted all relevant articles. For each study, the following items were extracted: first author, publication year, country, mRNA, increased expression, decreased expression, public gene databases, detection method, short survival, long survival, and area under curve (AUC) for gene panel. The Newcastle–Ottawa Scale (NOS) was used to evaluate the quality of the eligible articles for case-control studies. NOS involves three perspectives: study group selection, group comparability, and whether the exposure or the outcome of interest for a case–control study is listed in the scale. Each study can obtain a maximum of nine stars [264]. Studies scoring above the median NOS value were considered as high quality (low risk of bias) and those scoring below the median value were considered as low quality (high risk of bias). A summary of the method of quality evaluation is presented in Table 1.
Bioinformatic and statistical analysis
Protein–protein interaction (PPI) network and signaling pathways analysis
All 613 genes (with p < 0.05) obtained from this review study were considered in the bioinformatic analysis. The PPI network was constructed by Cytoscape software (version 3.9.0; https://cytoscape.org/). The top important nodes of the PPI network were obtained based on the Cytohubba plug-in. The 5 well-known central indices, including degree, stress, betweenness, closeness, and radiality of nodes, were considered to rank the network nodes. The top 10% of genes were determined in each metric. Then, common genes were identified between five metrics. Finally, between common genes, proteins with a high degree of centrality were selected and were considered the most important ones to investigate their association with survival in GBM patients. Moreover, the top 10 genes ranked by degree are calculated.
A pathway analysis using the GO Biological Process (GOBP) 2023 database through the ENRICHR package (https://maayanlab.cloud/Enrichr/, accessed on 23 March 2024) was then performed for further specified related mechanisms involved in cancer such as cell proliferation, differentiation, apoptosis, mitosis, angiogenesis, and stemness. Only GOBP terms with adjusted p-value < 0.01 by ENRICHR analysis were used.
Survival analysis and validation of the gene expression in the GEPIA2 and CGGA datasets
To confirm the reliability of the identified gene from the PPI network, Kaplan-Meier curves were created according to the GEPIA2 (http://gepia2.cancer-pku.cn) and the CGGA (http://www.cgga.org.cn) databases. CGGA contained two glioma data sets, namely, mRNAseq_325 and mRNAseq_693. Primary GBM of CGGA (mRNAseq_325) and CGGA (mRNAseq_693) data were considered. To determine differences in overall survival for patients with a low and high gene-expressing GBM, OS Kaplan-Meier analysis was performed by the GEPIA2 using the TCGA gene expression dataset and CGGA online applications. Kaplan–Meier curves were generated with a 50% median expression cutoff for high- and low-expressing groups. The estimation of hazard ratios was done by Cox proportional hazards model regression analysis. A 95% confidence interval was set and used. P < 0.05 was a statistically significant difference in validation cohorts from GEPIA2 and CGGA.
Results
Descriptive statistics
The workflow of the literature selection process is shown in Fig. 1. In brief, 4104 articles were found via an initial literature search of the databases, and 1296 studies were excluded owing to duplication. After screening the titles and abstracts, 2371 studies were not considered relevant to the purpose of this systematic review based on method. Subsequently, 255 studies were enrolled, the characteristics of each study were shown in Supplementary Table 2. Among these, 161 studies were conducted in China [16, 18, 20–21, 23–29, 31, 34, 36–37, 40–44, 47–48, 50, 53, 55, 64, 66–69, 71–74, 80, 82–87, 89, 92–95, 97–98, 102–105, 107, 109–113, 115–118, 120–124, 126–127, 135–137, 139–144, 146–152, 154–155, 157, 160–164, 166, 168–170, 172–173, 177–179, 184–190, 193–195, 197–197, 206–208, 212–213, 215–221, 223–226, 229, 231–246, 244, 246–257, 259–261 ], 23 studies were conducted in USA [10, 13–14, 32, 45–46, 58, 63, 75, 77, 88, 106, 108, 114, 133–134, 159, 165, 182, 192, 228, 243, 245]; besides that, India [9, 19, 30, 33, 59, 101, 130, 132, 196, 205], Taiwan [61, 79, 96, 145, 156, 175–176, 242], Germany [38, 51, 65, 90, 180, 230], Japan [22, 57, 76, 129, 258], Republic of Korea [62, 167, 171, 209, 241], UK [39, 60, 131, 191], Spain [15, 119, 153, 227], Lithuania [49, 99, 125, 138], Italy [100, 183, 203], France [ 8,12,158], Slovenia [11, 81, 128], Switzerland [35, 52], Sweden [78, 174], Turkey [211, 214], Russia [222], Finland [17], Netherland [204], Hungary [54], Canada [70], Brazil [210] Iran [262], and Austria [181]. Among 255 studies, 192, and 37 of them used the dataset of the TCGA and the CGGA, respectively.

Flowchart of the selection process
In the 720 genes studies, 613 unique genes were identified whose expression was associated with overall survival in GBM, of which 107 were described in two or more studies. See Supplementary Table 2 for details about the number of studies that described each gene, and whether or not it was found to be upregulated, downregulated, and the databases used.
NOS assessment
The risk of bias evaluation of the included studies for case-control studies according to the NOS is shown in Supplementary Tables 2 and Supplementary File 1, Table S4. Based on the NOS, the median score of the included studies was 7. Among the 255 studies, 131 studies that scored ≥ 7 were considered to present a low risk of bias. 124 of the studies were considered with a high risk of bias since they scored b < 7.
Bioinformatic analysis
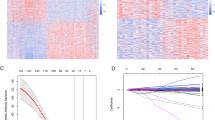
After removing duplicates, 613 genes were included in the bioinformatic analysis (Supplementary File 2). A PPI network was built using the STRING database and Cytoscape application, with an input of 613 genes (Fig. 2). The network was analyzed, and the nodes were ranked based on centrality parameters. The PPI network contains 602 nodes and 5570 edges. Top genes based on the degree value, betweenness centrality, closeness centrality, and stress were selected and organized into 5 groups (Table 2). By considering the degree of connectivity in the PPI network, as described in the materials and methods section, 31 important genes including (IL6, EGFR, STAT3, MMP9, CD44, FN1, CD4, TGFB1, CXCL8, CCL2, IL10, ICAM1, IL1A, CD274, KDR, SPP1, ITGB2, CDKN2A, PARP1, MYD88, AGT, NOTCH1, SERPINE1, TNFRSF1A, CDK1, CAV1, ITGB3, CDK4, FOXO3, MDM2, PROM1), were introduced (Table 2). In addition, the top 10 genes with the highest node degree score were identified as hub genes, as shown in Fig. 3.

The 613 differentially expressed genes were input into STRING database for PPI network analysis, and achieved a PPI network of 602 nodes and 5570 edges, with PPI enrichment p-value < 1.0 × 10–16. The network was constructed by Cytoscape based on the PPI correlations from the STRING database

The top 10 genes in the PPI network, in terms of degree ranking, were regarded as hub genes. The node color changes gradually from yellow to red in ascending order according to the degree of the genes
In the validation step as shown in Fig. 4, and Table 2, genes (FN1, CXCL8, and TNFRSF1A) from the GEPIA2 dataset, genes (IL6, STAT3, MMP9, FN1, CD4, CCL2, IL10, ICAM1, KDR, MYD88, MDM2) from the mRNA_seq325 of the CCGA, and genes (FN1, NOTCH1, CDKN2A) from the mRNA_seq693 of the CCGA cohort were associated significantly with overall survival in GBM patients.

(a–n) Kaplan-Meier analysis of overall survival for GBM patients in the GEPIA2 using the TCGA cohort (a. FN1; b. CXCL8; and c. TNFRSF1A), the mRNA_seq325 of the CCGA (d. IL6; e. STAT3; f. MMP9; g. FN1; h. CD4; i. CCL2; j. IL10; k. ICAM1; l. KDR; m. MYD88; and n, MDM2), and the mRNA_seq693 of the CCGA (o, FN1, p, NOTCH1, q, CDKN2A) based on low- and high-expression of genes. The red line represents samples with high expression of the genes, and the blue line represents the samples with low expression of genes. Among 31 genes, p < 0.05 was considered to be statistically different
The list of the top 31 genes is used as input for computing enrichment. As a result, 1271 GOBP terms were found and 11 GOBP terms were considered. The complete list of significantly enriched GOBP terms and related genes is given in Table 3.
Discussion
To the best of our knowledge, this systematic literature review is the most comprehensive review of gene expression for predicting GBM overall survival outcomes. The most 31 important genes including IL6, EGFR, STAT3, MMP9, CD44, FN1, CD4, TGFB1, CXCL8, CCL2, IL10, ICAM1, IL1A, CD274, KDR, SPP1, ITGB2, CDKN2A, PARP1, MYD88, AGT, NOTCH1, SERPINE1, TNFRSF1A, CDK1, CAV1, ITGB3, CDK4, FOXO3, MDM2, and PROM1, respectively, were considered as candidate biomarkers for GBM survival. Our analyses showed that in fact they all could be considered biomarkers. Nevertheless, based on the search strategy (Supplementary File 1, Table S2.), this review aimed to conduct a comprehensive, systematic literature review to identify all relevant studies that have significantly reported genes related to overall survival in GBM patients. However, some impact reports on this topic might have been missed due to limitations in the search strategy [265–266]. In the study future, given the well-established heterogeneity of GBM, the assessment of the prognostic value of specific genes must be conducted with consideration for GBM molecular subtypes, to ensure a comprehensive understanding of their impact, and would pave the way for precision medicine [266].
Detection of a specific gene expression in GBM tumors may be used to diagnose the existence of a GBM disease or enable clinicians to select the most effective treatment. As there was heterogeneity among the studied genes, bioinformatic analyses were performed to compile these data. The results identified 31 key genes, which had high weight and good topological properties (degree, stress, betweenness, closeness, and radiality) in the pathogenic networks. In addition, these genes were validated by RT-qPCR assays or bioinformatic analysis of datasets. In this study according to 5 typical nodal metrics, we found the most 31 important genes related to the survival of patients with GBM. However, there is currently no consensus on how to use these metrics for the interpretation of biological networks [267]. Therefore, these findings require further investigation.
Identification of survival-associated genes in GBMs has been ongoing over the past decade. However, the gene lists identified by researchers [8,9,10,11,12,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40,41,42,43,44,45,46,47,48,49,50,51,52,53,54,55,56,57,58,59,60,61,62,63,64,65,66,67,68,69,70,71,72,73,74,75,76,77,78,79,80,81,82,83,84,85,86,87,88,89,90,91,92,93,94,95,96,97,98,99,100,101,102,103,104,105,106,107,108,109,110,111,112,113,114,115,116,117,118,119,120,121,122,123,124,125,126,127,128,129,130,131,132,133,134,135,136,137,138,139,140,141,142,143,144,145,146,147,148,149,150,151,152,153,154,155,156,157,158,159,160,161,162,163,164,165,166,167,168,169,170,171,172,173,174,175,176,177,178,179,180,181,182,183,184,185,186,187,188,189,190,191,192,193,194,195,196,197,198,199,200,201,202,203,204,205,206,207,208,209,210,211,212,213,214,215,216,217,218,219,220,221,222,223,224,225,226,227,228,229,230,231,232,233,234,235,236,237,238,239,240,241,242,243,244,245,246,247,248,249,250,251,252,253,254,255,256,257,258,259,260,261,262] differ considerably; only 107 common genes from 720 genes could be identified in studies. These differences can be attributed to two major factors. First, researchers have analyzed GBM datasets from various cohorts worldwide. Second, studies have analyzed different types of datasets, obtained using different approaches such as PCR or next-generation sequencing data. Due to technical limitations and cohorts’ specificity, the expression profiles of similar genes identified from different datasets may be inconsistent.
To improve prognostic and predictive survival power in GBM patients, researchers [69, 91–92, 94–95, 97, 109, 131, 137–138, 144, 147, 172, 186, 189, 195, 197, 200, 222, 242, 251, 253], identified a panel of 2, 3, 4, 6,7, 8, 13, or 14 genes using mRNA expression datasets. They established a risk score model that performed well in survival prediction. High-risk group patients had significantly poorer survival as compared with those in the low-risk group. In this study, AUC for the 1-year overall survival predictions was reported between o.587 [195], to 0.905 [69]. This difference may be due to the use of different databases and cohorts’ specificities. The obtained 31 mRNA panel in this study, is suggested to predict OS in glioblastoma in various cohorts.
The present study showed that ten hub genes (IL6, EGFR, STAT3, MMP9, CD44, FN1, CD4, TGFB1, CXCL8, CCL2) with higher node degree in PPI networks have been predicted to be survival biomarkers for GBM patients and some have been experimentally validated. These hub genes can be offered to the candidate biomarkers of future research for therapeutic targets in patients with GBM. In addition, this study showed that five hub genes (IL6, NOTCH1, TGFB1, EGFR, and KDR) were involved in most of the pathways, and they can be further investigated for biological discoveries. Moreover, we noticed that cell proliferation, apoptotic process, cell migration, and cell differentiation contain many of these genes (see Table 3). In addition, various studies showed that overexpression of these five genes leads to increased cell proliferation and invasion, and inhibition of apoptosis in glioblastoma tumors and was associated with poor patient survival [268,269,270,271]. Therefore, these GOBP terms may exert a synergistic effect on the survival of GBM, which could be clues to therapeutic strategies for this disease.
One might inquire about the hub genes obtained from this study. Certainly, various computer modeling algorithms and prediction methods have been and are being developed and used to predict outcomes in medical research. It is noted that each modeling approach has its strengths and weaknesses and there is no best one for all cases [272]. The best modeling approach is uncertain, and may be obtained by combining more than one model, and research in this field continues [272]. By changing the method of prediction, the most important variables will be changed to predict outcomes [272]. We identified two hub gene groups that were associated with overall survival in GBM patients. The ten and five hub genes are ranked by degree and pathways analysis methods, respectively. It is noted that, when a different gene selection criterion is applied, the number of genes in the two top-ranking lists of the two methods will also change [273]. In this study, the algorithms yielded different top-ranking gene lists due to their different approach. Interestingly, the two lists of hub genes have three in common, that were selected as the most important genes for the prediction of survival in methods and can be considered as three hub genes (IL6, TGFB1, and EGFR).
In this study, the five and ten lists of hub genes were selected based on two different methods, therefore, the two groups are different [272–273]. On the other hand, as we all know, the TCGA and the CGGA databases are the world’s largest and most comprehensive gene expression public databases in GBM patients. Hence, these databases were used for validation of our study results. In the validation analysis, we used the GEPIA2, the mRNA_seq325, and the mRNA_seq693 of the CCGA. Only, the FN1 gene was significant in three cohorts. Although the mRNA_seq693 includes more patients with Grade 4 glioma compared to the mRNA_seq325, only three genes were significant compared to eleven genes observed in the other cohort of the CGGA (Table 3). The differences seen between the three databases can be due to the differences in genetics between the different populations.
Study consistency
Of the 255 manuscripts, all studies were prospective. No randomized trial was found. 107 mRNAs (14.9%; 107/720) were common in all studies. However, a large number of studies have not been validated; hence, there was a lack of high-quality evidence in this study. 124 studies were rated as fair quality; 131 studies were considered to be of high quality. Types of studies and datasets were not consistently reported, resulting in a potential bias.
Among studies in this systematic review, genes with a significant correlation between gene expression and overall survival were considered. Previous studies have found that gene expression levels are associated with prognosis and some genes can be applied to predict the survival risk of GBM patients. However, some studies have conflicts regarding significant differences in gene expression and overall survival. These conflicts seem to depend on the GBM sample size, the heterogeneity of GBM, the datasets used, and the methodologies employed. All the above- mentioned may explain why the validation step did not yield significant results.
Study quality
The small sample size in PCR-based studies, the high number of single-center studies, various databases, and the high number of studies from China (161/255), the USA (23/155), and India (10/255), which may affect the quality of studies. The heterogeneity of the studies reduced the quality of the data. In some of these datasets and GBM samples, the type and severity of GBM disease were not specified. In addition, some studies lacked validation of their candidate genes in a GBM patient cohort. Therefore, further research with large sample sizes and validation in GBM patients is warranted.
Strengths, limitations, and future perspectives
To the best of our knowledge, the current study is the first that systematically reviewed published data on gene expression related to the survival of GBM patients. Of note, the major strength of the current systematic review is that bioinformatic analyses were performed, which added new information to the previously studied gene expression on this topic. The findings reported here provide a better view of gene expression biomarkers in predicting the prognosis of patients with GBM.
There are some limitations in our work. Firstly, the search strategy was restricted to the English language literature only, hence, there is a possibility of excluding qualified studies published in other languages. Secondly, the study showed a high level of heterogeneity in the methods used among the included studies. In particular, there were heterogeneities in (1) Variety in disease severity; and (2) Age- and gender-related changes in GBM patients were not considered. Thirdly, the overall survival has been associated with multiple factors such as poor immune response, which was not considered in this study. Additionally, the role of gene expression was not completely clarified in various biological processes and the potential application of these molecules as gene therapies. Hence, future studies are required to clarify the biological roles of the mRNAs to investigate the possibility of their clinical utilization in GBM patients.
Conclusion
Our review suggests that the current evidence for gene expression associated with GBM survival is highly variable. At present, no clear decisions can be made from this systematic review for application into clinical practice. The key recommendation from this study is that genetic data sharing develops strategies and guidelines in this field that can be used to answer important questions. Moreover, in future a combination of significant genes expression signatures can be applied to identify a powerful and independent predictor for outcome in GBM patients.
Data availability
All data analyzed during this study are included in this published article. GEPIA2 and CGGA databases are publicly available at (http://gepia2.cancer-pku.cn) and (http://www.cgga.org.cn), respectively.
Abbreviations
- GBM:
-
Glioblastoma Multiforme
- CNS:
-
Central Nervous System
- OS:
-
Overall Survival
- TCGA:
-
The Cancer Genome Atlas
- CGGA:
-
Chinese Glioma Genome Atlas
- GEPIA:
-
Gene Expression Profiling Interactive Analysis
- PPI:
-
Protein-Protein Interaction
- AUC:
-
Area under curve
- GEO:
-
Gene Expression Omnibus
- WHO:
-
World Health Organization
References
Grochans S, Cybulska AM, Simińska D, Korbecki J, Kojder K, Chlubek D, et al. Epidemiol Glioblastoma Multiforme-Literature Rev Cancers (Basel). 2022;14(10):2412.
Das P, Puri T, Jha P, Pathak P, Joshi N, Suri V, et al. A clinicopathological and molecular analysis of glioblastoma multiforme with long-term survival. J Clin Neurosci. 2011;18(1):66–70.
De Vleeschouwer S, editor. Glioblastoma [Internet]. Brisbane (AU): Codon; 2017.
Tang X, Zhang S, Fu R, Zhang L, Huang K, Peng H, et al. Therapeutic prospects of mRNA-Based gene therapy for Glioblastoma. Front Oncol. 2019;9:1208.
Mao H, Lebrun DG, Yang J, Zhu VF, Li M. Deregulated signaling pathways in glioblastoma multiforme: molecular mechanisms and therapeutic targets. Cancer Invest. 2012;30(1):48–56.
Brandner S, Jaunmuktane Z. Neurological update: gliomas and other primary brain tumours in adults. J Neurol. 2018;265(3):717–27.
Zheng Y, Luo Y, Chen X, Li H, Huang B, Zhou B, et al. The role of mRNA in the development, diagnosis, treatment, and prognosis of neural tumors. Mol Cancer. 2021;20(1):49.
Tchirkov A, Rolhion C, Kémény JL, Irthum B, Puget S, Khalil T, et al. Clinical implications of quantitative real-time RT-PCR analysis of hTERT gene expression in human gliomas. Br J Cancer. 2003;88(4):516–20.
Santosh V, Arivazhagan A, Sreekanthreddy P, Srinivasan H, Thota B, Srividya MR, et al. Grade-specific expression of insulin-like growth factor-binding proteins-2, -3, and – 5 in astrocytomas: IGFBP-3 emerges as a strong predictor of survival in patients with newly diagnosed glioblastoma. Cancer Epidemiol Biomarkers Prev. 2010;19(6):1399–408.
Kuan CT, Wakiya K, Herndon JE 2nd, Lipp ES, Pegram CN, Riggins GJ, et al. MRP3: a molecular target for human glioblastoma multiforme immunotherapy. BMC Cancer. 2010;10:468.
Ardebili SY, Zajc I, Gole B, Campos B, Herold-Mende C, Drmota S, et al. CD133/prominin1 is prognostic for GBM patient’s survival, but inversely correlated with cysteine cathepsins’ expression in glioblastoma derived spheroids. Radiol Oncol. 2011;45(2):102–15.
Metellus P, Nanni-Metellus I, Delfino C, Colin C, Tchogandjian A, Coulibaly B, et al. Prognostic impact of CD133 mRNA expression in 48 glioblastoma patients treated with concomitant radiochemotherapy: a prospective patient cohort at a single institution. Ann Surg Oncol. 2011;18(10):2937–45.
Rutkowski MJ, Sughrue ME, Kane AJ, Kim JM, Bloch O, Parsa AT. Epidermal growth factor module-containing mucin-like receptor 2 is a newly identified adhesion G protein-coupled receptor associated with poor overall survival and an invasive phenotype in glioblastoma. J Neurooncol. 2011;105(2):165–71.
Jarboe JS, Anderson JC, Duarte CW, Mehta T, Nowsheen S, Hicks PH, et al. MARCKS regulates growth and radiation sensitivity and is a novel prognostic factor for glioma. Clin Cancer Res. 2012;18(11):3030–41.
Leone PE, González MB, Elosua C, Gómez-Moreta JA, Lumbreras E, Robledo C, et al. Integration of global spectral karyotyping, CGH arrays, and expression arrays reveals important genes in the pathogenesis of glioblastoma multiforme. Ann Surg Oncol. 2012;19(7):2367–79.
Xu T, Qin R, Zhou J, Yan Y, Lu Y, Zhang X, et al. High bone sialoprotein (BSP) expression correlates with increased tumor grade and predicts a poorer prognosis of high-grade glioma patients. PLoS ONE. 2012;7(10):e48415.
Haapa-Paananen S, Kiviluoto S, Waltari M, Puputti M, Mpindi JP, Kohonen P, et al. HES6 gene is selectively overexpressed in glioma and represents an important transcriptional regulator of glioma proliferation. Oncogene. 2012;31(10):1299–310.
Bao ZS, Zhang CB, Wang HJ, Yan W, Liu YW, Li MY, et al. Whole-genome mRNA expression profiling identifies functional and prognostic signatures in patients with mesenchymal glioblastoma multiforme. CNS Neurosci Ther. 2013;19(9):714–20.
Arimappamagan A, Somasundaram K, Thennarasu K, Peddagangannagari S, Srinivasan H, Shailaja BC, et al. A fourteen gene GBM prognostic signature identifies association of immune response pathway and mesenchymal subtype with high risk group. PLoS ONE. 2013;8(4):e62042.
Fan S, Meng D, Xu T, Chen Y, Wang J, Li X, et al. Overexpression of SLC7A7 predicts poor progression-free and overall survival in patients with glioblastoma. Med Oncol. 2013;30(1):384.
Han S, Xia J, Qin X, Han S, Wu A. Phosphorylated SATB1 is associated with the progression and prognosis of glioma. Cell Death Dis. 2013;4(10):e901.
Kawaguchi A, Yajima N, Tsuchiya N, Homma J, Sano M, Natsumeda M, et al. Gene expression signature-based prognostic risk score in patients with glioblastoma. Cancer Sci. 2013;104(9):1205–10.
Bao Z, Zhang C, Yan W, Liu Y, Li M, Zhang W, et al. BMP4, a strong better prognosis predictor, has a subtype preference and cell development association in gliomas. J Transl Med. 2013;11:100.
Chen QR, Hu Y, Yan C, Buetow K, Meerzaman D. Systematic genetic analysis identifies Cis-eQTL target genes associated with glioblastoma patient survival. PLoS ONE. 2014;9(8):e105393.
Turtoi A, Blomme A, Bianchi E, Maris P, Vannozzi R, Naccarato AG, et al. Accessibilome of human glioblastoma: collagen-VI-alpha-1 is a new target and a marker of poor outcome. J Proteome Res. 2014;13(12):5660–9.
Cheng YS, Lin C, Cheng YP, Yu YL, Tang CT, Hueng DY. Epithelial cell transformation sequence 2 is a potential biomarker of unfavorable survival in human gliomas. Neurol India. 2014;62(4):406–9.
Hua C, Zhao G, Li Y, Bie L. Minichromosome Maintenance (MCM) family as potential diagnostic and prognostic tumor markers for human gliomas. BMC Cancer. 2014;14:526.
Bao Z, Feng Y, Wang H, Zhang C, Sun L, Yan Z, et al. Integrated analysis using methylation and gene expression microarrays reveals PDE4C as a prognostic biomarker in human glioma. Oncol Rep. 2014;32(1):250–60.
Xing ZY, Sun LG, Guo WJ. Elevated expression of Notch-1 and EGFR induced apoptosis in glioblastoma multiforme patients. Clin Neurol Neurosurg. 2015;131:54–8.
Sibin MK, Bhat DI, Narasingarao KV, Lavanya Ch, Chetan GK. CDKN2A (p16) mRNA decreased expression is a marker of poor prognosis in malignant high-grade glioma. Tumour Biol. 2015;36(10):7607–14.
Cai J, Zhang W, Yang P, Wang Y, Li M, Zhang C, et al. Identification of a 6-cytokine prognostic signature in patients with primary glioblastoma harboring M2 microglia/macrophage phenotype relevance. PLoS ONE. 2015;10(5):e0126022.
Zhou P, Erfani S, Liu Z, Jia C, Chen Y, Xu B, et al. CD151-α3β1 integrin complexes are prognostic markers of glioblastoma and cooperate with EGFR to drive tumor cell motility and invasion. Oncotarget. 2015;6(30):29675–93.
Sibin MK, Lavanya CH, Bhat DI, Rao N, Geethashree N, Vibhuti W, et al. CD133 and BMI1 expressions and its prognostic role in primary glioblastoma. J Genet. 2015;94(4):689–96.
Liu Q, Zhang C, Yuan J, Fu J, Wu M, Su J, et al. PTK7 regulates Id1 expression in CD44-high glioma cells. Neuro Oncol. 2015;17(4):505–15.
Frei K, Gramatzki D, Tritschler I, Schroeder JJ, Espinoza L, Rushing EJ, et al. Transforming growth factor-β pathway activity in glioblastoma. Oncotarget. 2015;6(8):5963–77.
Bache M, Rot S, Keßler J, Güttler A, Wichmann H, Greither T, et al. mRNA expression levels of hypoxia-induced and stem cell-associated genes in human glioblastoma. Oncol Rep. 2015;33(6):3155–61.
Delic S, Thuy A, Schulze M, Proescholdt MA, Dietrich P, Bosserhoff AK, et al. Systematic investigation of CMTM family genes suggests relevance to glioblastoma pathogenesis and CMTM1 and CMTM3 as priority targets. Genes Chromosomes Cancer. 2015;54(7):433–43.
Stegen B, Butz L, Klumpp L, Zips D, Dittmann K, Ruth P, et al. Ca2+-Activated IK K + Channel Blockade Radiosensitizes Glioblastoma cells. Mol Cancer Res. 2015;13(9):1283–95.
Zhang SD, Leung KL, McCrudden CM, Kwok HF. The Prognostic significance of combining VEGFA, FLT1 and KDR mRNA expressions in brain tumors. J Cancer. 2015;6(9):812–8.
Cheng W, Ren X, Zhang C, Cai J, Liu Y, Han S, et al. Bioinformatic profiling identifies an immune-related risk signature for glioblastoma. Neurology. 2016;86(24):2226–34.
Li Q, Chen B, Cai J, Sun Y, Wang G, Li Y, et al. Comparative analysis of Matrix Metalloproteinase Family members reveals that MMP9 predicts survival and response to Temozolomide in patients with primary glioblastoma. PLoS ONE. 2016;11(3):e0151815.
Gao YF, Zhu T, Mao CX, Liu ZX, Wang ZB, Mao XY, et al. PPIC, EMP3 and CHI3L1 are novel prognostic markers for high Grade Glioma. Int J Mol Sci. 2016;17(11):1808.
Lin L, Wang G, Ming J, Meng X, Han B, Sun B, et al. Analysis of expression and prognostic significance of vimentin and the response to temozolomide in glioma patients. Tumour Biol. 2016;37(11):15333–9.
Wang Z, Zhang C, Liu X, Wang Z, Sun L, Li G, et al. Molecular and clinical characterization of PD-L1 expression at transcriptional level via 976 samples of brain glioma. Oncoimmunology. 2016;5(11):e1196310.
Nduom EK, Wei J, Yaghi NK, Huang N, Kong LY, Gabrusiewicz K, et al. PD-L1 expression and prognostic impact in glioblastoma. Neuro Oncol. 2016;18(2):195–205.
Bayin NS, Frenster JD, Kane JR, Rubenstein J, Modrek AS, Baitalmal R, et al. GPR133 (ADGRD1), an adhesion G-protein-coupled receptor, is necessary for glioblastoma growth. Oncogenesis. 2016;5(10):e263.
Cheng W, Li M, Jiang Y, Zhang C, Cai J, Wang K, et al. Association between small heat shock protein B11 and the prognostic value of MGMT promoter methylation in patients with high-grade glioma. J Neurosurg. 2016;125(1):7–16.
Wang Z, Zhang C, Sun L, Liang J, Liu X, Li G, et al. FGFR3, as a receptor tyrosine kinase, is associated with differentiated biological functions and improved survival of glioma patients. Oncotarget. 2016;7(51):84587–93.
Steponaitis G, Skiriutė D, Kazlauskas A, Golubickaitė I, Stakaitis R, Tamašauskas A, et al. High CHI3L1 expression is associated with glioma patient survival. Diagn Pathol. 2016;11:42.
Sun L, Zhang C, Yang Z, Wu Y, Wang H, Bao Z, et al. KIF23 is an independent prognostic biomarker in glioma, transcriptionally regulated by TCF-4. Oncotarget. 2016;7(17):24646–55.
Kolodziej MA, Weischer C, Reinges MH, Uhl E, Weigand MA, Schwarm FP, et al. NDRG2 and NDRG4 expression is altered in Glioblastoma and influences Survival in patients with MGMT-methylated tumors. Anticancer Res. 2016;36(3):887–97.
Codó P, Weller M, Kaulich K, Schraivogel D, Silginer M, Reifenberger G, et al. Control of glioma cell migration and invasiveness by GDF-15. Oncotarget. 2016;7(7):7732–46.
Wang HY, Li JY, Liu X, Yan XY, Wang W, Wu F, et al. A three ion channel genes-based signature predicts prognosis of primary glioblastoma patients and reveals a chemotherapy sensitive subtype. Oncotarget. 2016;7(46):74895–903.
Murnyák B, Kouhsari MC, Hershkovitch R, Kálmán B, Marko-Varga G, Klekner Á, et al. PARP1 expression and its correlation with survival is tumour molecular subtype dependent in glioblastoma. Oncotarget. 2017;8(28):46348–62.
Xu R, Han M, Xu Y, Zhang X, Zhang C, Zhang D, et al. Coiled-coil domain containing 109B is a HIF1α-regulated gene critical for progression of human gliomas. J Transl Med. 2017;15(1):165.
Zhu C, Kros JM, van der Weiden M, Zheng P, Cheng C, Mustafa DA. Expression site of P2RY12 in residential microglial cells in astrocytomas correlates with M1 and M2 marker expression and tumor grade. Acta Neuropathol Commun. 2017;5(1):4.
Ohtaki S, Wanibuchi M, Kataoka-Sasaki Y, Sasaki M, Oka S, Noshiro S, et al. ACTC1 as an invasion and prognosis marker in glioma. J Neurosurg. 2017;126(2):467–75.
Zhai L, Ladomersky E, Lauing KL, Wu M, Genet M, Gritsina G, et al. Infiltrating T cells increase IDO1 expression in Glioblastoma and contribute to decreased patient survival. Clin Cancer Res. 2017;23(21):6650–60.
Soni P, Qayoom S, Husain N, Kumar P, Chandra A, Ojha BK, et al. CD24 and nanog expression in stem cells in Glioblastoma: correlation with response to Chemoradiation and overall survival. Asian Pac J Cancer Prev. 2017;18(8):2215–9.
Haynes HR, White P, Hares KM, Redondo J, Kemp KC, Singleton WGB, et al. The transcription factor PPARα is overexpressed and is associated with a favourable prognosis in IDH-wildtype primary glioblastoma. Histopathology. 2017;70(7):1030–43.
Huang SP, Chang YC, Low QH, Wu ATH, Chen CL, Lin YF, et al. BICD1 expression, as a potential biomarker for prognosis and predicting response to therapy in patients with glioblastomas. Oncotarget. 2017;8(69):113766–91.
Kim Y, Do IG, Hong M, Suh YL. Negative prognostic effect of low nuclear GLI1 expression in glioblastomas. J Neurooncol. 2017;133(1):69–76.
Wang Y, Wu S, Zheng S, Wang S, Wali A, Ezhilarasan R, et al. APOBEC3G acts as a therapeutic target in mesenchymal gliomas by sensitizing cells to radiation-induced cell death. Oncotarget. 2017;8(33):54285–96.
Han MZ, Xu R, Xu YY, Zhang X, Ni SL, Huang B, et al. TAGLN2 is a candidate prognostic biomarker promoting tumorigenesis in human gliomas. J Exp Clin Cancer Res. 2017;36(1):155.
Feldheim J, Kessler AF, Schmitt D, Wilczek L, Linsenmann T, Dahlmann M, et al. Expression of activating transcription factor 5 (ATF5) is increased in astrocytomas of different WHO grades and correlates with survival of glioblastoma patients. Onco Targets Ther. 2018;11:8673–84.
Jia D, Li S, Li D, Xue H, Yang D, Liu Y. Mining TCGA database for genes of prognostic value in glioblastoma microenvironment. Aging. 2018;10(4):592–605.
Yue H, Xu Q, Xie S. High EMP3 expression might independently predict poor overall survival in glioblastoma and its expression is related to DNA methylation. Med (Baltim). 2018;97(1):e9538.
Wang W, Zhao Z, Wu F, Wang H, Wang J, Lan Q, et al. Bioinformatic analysis of gene expression and methylation regulation in glioblastoma. J Neurooncol. 2018;136(3):495–503.
Xu P, Yang J, Liu J, Yang X, Liao J, Yuan F, et al. Identification of glioblastoma gene prognosis modules based on weighted gene co-expression network analysis. BMC Med Genomics. 2018;11(1):96.
Roy LO, Poirier MB, Fortin D. Differential expression and clinical significance of transforming growth factor-Beta isoforms in GBM tumors. Int J Mol Sci. 2018;19(4):1113.
Chen D, Chen D, Cao D, Hu J, Yao Y. A signature based on survival-related genes identifies high-risk glioblastomas harboring immunosuppressive and aggressive ECM characteristics. Zhong Nan Da Xue Xue Bao Yi Xue Ban. 2018;43(4):368–82.
Shu C, Wang Q, Yan X, Wang J. Whole-genome expression microarray combined with machine learning to identify prognostic biomarkers for high-Grade Glioma. J Mol Neurosci. 2018;64(4):491–500.
Liu S, Xu Y, Zhang S. LINGO1, C7orf31 and VEGFA are prognostic genes of primary glioblastoma: analysis of gene expression microarray. Neoplasma. 2018;65(4):532–41.
Zhang Y, Xu J, Zhu X. A 63 signature genes prediction system is effective for glioblastoma prognosis. Int J Mol Med. 2018;41(4):2070–8.
Gilder AS, Natali L, Van Dyk DM, Zalfa C, Banki MA, Pizzo DP, et al. The urokinase receptor induces a mesenchymal gene expression signature in Glioblastoma Cells and promotes Tumor Cell Survival in Neurospheres. Sci Rep. 2018;8(1):2982.
Takashima Y, Kawaguchi A, Kanayama T, Hayano A, Yamanaka R. Correlation between lower balance of Th2 helper T-cells and expression of PD-L1/PD-1 axis genes enables prognostic prediction in patients with glioblastoma. Oncotarget. 2018;9(27):19065–78.
Han J, Puri RK. Analysis of the cancer genome atlas (TCGA) database identifies an inverse relationship between interleukin-13 receptor α1 and α2 gene expression and poor prognosis and drug resistance in subjects with glioblastoma multiforme. J Neurooncol. 2018;136(3):463–74.
Vasaikar S, Tsipras G, Landázuri N, Costa H, Wilhelmi V, Scicluna P, et al. Overexpression of endothelin B receptor in glioblastoma: a prognostic marker and therapeutic target? BMC Cancer. 2018;18(1):154.
Cheng YC, Tsai WC, Sung YC, Chang HH, Chen Y. Interference with PSMB4 expression exerts an Anti-tumor Effect by decreasing the Invasion and Proliferation of Human Glioblastoma cells. Cell Physiol Biochem. 2018;45(2):819–31.
Zhang Y, Xia Q, Lin J. Identification of the potential oncogenes in glioblastoma based on bioinformatic analysis and elucidation of the underlying mechanisms. Oncol Rep. 2018;40(2):715–25.
Breznik B, Limback C, Porcnik A, Blejec A, Krajnc MK, Bosnjak R, et al. Localization patterns of cathepsins K and X and their predictive value in glioblastoma. Radiol Oncol. 2018;52(4):433–42.
Cho HR, Jeon H, Park CK, Park SH, Choi SH. Radiogenomics Profiling for Glioblastoma-related Immune cells reveals CD49d expression correlation with MRI parameters and prognosis. Sci Rep. 2018;8(1):16022.
Gao M, Lin Y, Liu X, Li Y, Zhang C, Wang Z, et al. ISG20 promotes local tumor immunity and contributes to poor survival in human glioma. Oncoimmunology. 2018;8(2):e1534038.
Zhou L, Tang H, Wang F, Chen L, Ou S, Wu T, et al. Bioinformatics analyses of significant genes, related pathways and candidate prognostic biomarkers in glioblastoma. Mol Med Rep. 2018;18(5):4185–96.
Guan X, Luo L, Begum G, Kohanbash G, Song Q, Rao A, et al. Elevated Na/H exchanger 1 (SLC9A1) emerges as a marker for tumorigenesis and prognosis in gliomas. J Exp Clin Cancer Res. 2018;37(1):255.
Zhang C, Zhang Z, Li F, Shen Z, Qiao Y, Li L, et al. Large-scale analysis reveals the specific clinical and immune features of B7-H3 in glioma. Oncoimmunology. 2018;7(11):e1461304.
Zhong S, Wu B, Dong X, Han Y, Jiang S, Zhang Y, et al. Identification of driver genes and key pathways of Glioblastoma shows JNJ-7706621 as a Novel Antiglioblastoma Drug. World Neurosurg. 2018;109:e329–42.
Du D, Yuan J, Ma W, Ning J, Weinstein JN, Yuan X, et al. Clinical significance of FBXO17 gene expression in high-grade glioma. BMC Cancer. 2018;18(1):773.
Wang HX, Chen YH, Zhou JX, Hu XY, Tan C, Yan Y, et al. Overexpression of G-protein-coupled receptors 65 in glioblastoma predicts poor patient prognosis. Clin Neurol Neurosurg. 2018;164:132–7.
Werner JM, Kuhl S, Ulrich K, Krischek B, Stavrinou P, Goldbrunner R, et al. Expression of CD40 correlates negatively with overall and progression-free survival of low- and high-Grade Gliomas. World Neurosurg. 2019;130:e17–25.
Guo XX, Su J, He XF. A 4-gene panel predicting the survival of patients with glioblastoma. J Cell Biochem. 2019;120(9):16037–43.
Yin W, Tang G, Zhou Q, Cao Y, Li H, Fu X, et al. Expression profile analysis identifies a novel five-gene signature to improve prognosis prediction of glioblastoma. Front Genet. 2019;10:419.
Cao M, Cai J, Yuan Y, Shi Y, Wu H, Liu Q, et al. A four-gene signature-derived risk score for glioblastoma: prospects for prognostic and response predictive analyses. Cancer Biol Med. 2019;16(3):595–605.
Zuo S, Zhang X, Wang L. A RNA sequencing-based six-gene signature for survival prediction in patients with glioblastoma. Sci Rep. 2019;9(1):1–10.
Cheng Q, Huang C, Cao H, Lin J, Gong X, Li J, et al. A Novel Prognostic signature of transcription factors for the prediction in patients with GBM. Front Genet. 2019;10:906.
Hsu JB, Chang TH, Lee GA, Lee TY, Chen CY. Identification of potential biomarkers related to glioma survival by gene expression profile analysis. BMC Med Genomics. 2019;11(Suppl 7):34.
Wang Z, Gao L, Guo X, Feng C, Lian W, Deng K, et al. Development and validation of a nomogram with an autophagy-related gene signature for predicting survival in patients with glioblastoma. Aging. 2019;11(24):12246–69.
Yuan Q, Cai HQ, Zhong Y, Zhang MJ, Cheng ZJ, Hao JJ, et al. Overexpression of IGFBP2 mRNA predicts poor survival in patients with glioblastoma. Biosci Rep. 2019;39(6):BSR20190045.
Masiulionytė B, Valiulytė I, Tamašauskas A, Skiriutė D. Metallothionein genes are highly expressed in Malignant Astrocytomas and Associated with patient survival. Sci Rep. 2019;9(1):5406.
Morelli MB, Amantini C, Tomassoni D, Nabissi M, Arcella A, Santoni G. Transient receptor potential Mucolipin-1 channels in Glioblastoma: Role in Patient’s survival. Cancers (Basel). 2019;11(4):525.
Kruthika BS, Sugur H, Nandaki K, Arimappamagan A, Paturu K, Santosh V. Expression pattern and prognostic significance of myosin light chain 9 (MYL9): a novel biomarker in glioblastoma. J Clin Pathol. 2019;72(10):677–81.
Liu J, Liu D, Yang Z, Yang Z. High LAMC1 expression in glioma is associated with poor prognosis. Onco Targets Ther. 2019;12:4253–60.
Yang Q, Wang R, Wei B, Peng C, Wang L, Hu G, et al. Gene and microRNA signatures are Associated with the Development and Survival of Glioblastoma patients. DNA Cell Biol. 2019;38(7):688–99.
Liu S, Zhang C, Maimela NR, Yang L, Zhang Z, Ping Y, et al. Molecular and clinical characterization of CD163 expression via large-scale analysis in glioma. Oncoimmunology. 2019;8(7):1601478.
Zhou Y, Yang L, Zhang X, Chen R, Chen X, Tang W, et al. Identification of potential biomarkers in Glioblastoma through Bioinformatic Analysis and evaluating their Prognostic Value. Biomed Res Int. 2019;2019:6581576.
Wang J, Matosevic S. NT5E/CD73 as correlative factor of patient survival and natural killer cell infiltration in Glioblastoma. J Clin Med. 2019;8(10):1526.
Fu H, Ge B, Chen D, Wu Y, Luo Q, Li X, et al. Phytanoyl-CoA 2-Hydroxylase-interacting protein-like Gene is a therapeutic target gene for Glioblastoma Multiforme. Med Sci Monit. 2019;25:2583–90.
Hasan T, Caragher SP, Shireman JM, Park CH, Atashi F, Baisiwala S, et al. Interleukin-8/CXCR2 signaling regulates therapy-induced plasticity and enhances tumorigenicity in glioblastoma. Cell Death Dis. 2019;10(4):292.
Ye N, Jiang N, Feng C, Wang F, Zhang H, Bai HX, et al. Combined Therapy Sensitivity Index based on a 13-Gene signature predicts prognosis for IDH Wild-type and MGMT promoter unmethylated glioblastoma patients. J Cancer. 2019;10(22):5536–48.
Zeng F, Wang K, Huang R, Liu Y, Zhang Y, Hu H. RELB: a novel prognostic marker for glioblastoma as identified by population-based analysis. Oncol Lett. 2019;18(1):386–94.
Zhang Z, Wang Z, Huang K, Liu Y, Wei C, Zhou J, et al. PLK4 is a determinant of temozolomide sensitivity through phosphorylation of IKBKE in glioblastoma. Cancer Lett. 2019;443:91–107.
Chen L, Su G. Identification of CTRP1 as a Prognostic Biomarker and Oncogene in Human Glioblastoma. Biomed Res Int. 2019;2019:2582416.
Tang JH, Yang L, Chen JX, Li QR, Zhu LR, Xu QF, et al. Bortezomib inhibits growth and sensitizes glioma to temozolomide (TMZ) via down-regulating the FOXM1-Survivin axis. Cancer Commun (Lond). 2019;39(1):81.
Waters MR, Gupta AS, Mockenhaupt K, Brown LN, Biswas DD, Kordula T. RelB acts as a molecular switch driving chronic inflammation in glioblastoma multiforme. Oncogenesis. 2019;8(6):37.
Yuan F, Yi L, Hai L, Wang Y, Yang Y, Li T, et al. Identification of Key pathways and genes in the Orai2 mediated classical and mesenchymal subtype of Glioblastoma by Bioinformatic Analyses. Dis Markers. 2019;2019:7049294.
Yang P, Kang W, Pan Y, Zhao X, Duan L. Overexpression of HOXC6 promotes cell proliferation and migration via MAPK signaling and predicts a poor prognosis in glioblastoma. Cancer Manag Res. 2019;11:8167–79.
Zeng C, Xing W, Liu Y. Identification of UGP2 as a progression marker that promotes cell growth and motility in human glioma. J Cell Biochem. 2019;120(8):12489–99.
Xu J, Su Z, Ding Q, Shen L, Nie X, Pan X, et al. Inhibition of proliferation by Knockdown of Transmembrane (TMEM) 168 in Glioblastoma Cells via suppression of Wnt/β-Catenin pathway. Oncol Res. 2019;27(7):819–26.
Fernández-García P, Rosselló CA, Rodríguez-Lorca R, Beteta-Göbel R, Fernández-Díaz J, Lladó V, et al. The Opposing Contribution of SMS1 and SMS2 to Glioma Progression and their value in the therapeutic response to 2OHOA. Cancers (Basel). 2019;11(1):88.
Luo X, Xu S, Zhong Y, Tu T, Xu Y, Li X, et al. High gene expression levels of VEGFA and CXCL8 in the peritumoral brain zone are associated with the recurrence of glioblastoma: a bioinformatics analysis. Oncol Lett. 2019;18(6):6171–9.
Ji X, Zhu H, Dai X, Xi Y, Sheng Y, Gao C, et al. Overexpression of GBP1 predicts poor prognosis and promotes tumor growth in human glioblastoma multiforme. Cancer Biomark. 2019;25(3):275–90.
Dong Y, Sun Y, Huang Y, Dwarakanath B, Kong L, Lu JJ. Upregulated necroptosis-pathway-associated genes are unfavorable prognostic markers in low-grade glioma and glioblastoma multiforme. Transl Cancer Res. 2019;8(3):821–7.
Wang H, Guan Q, Nan Y, Ma Q, Zhong Y. Overexpression of human MX2 gene suppresses cell proliferation, migration, and invasion via ERK/P38/NF-κB pathway in glioblastoma cells. J Cell Biochem. 2019;120(11):18762–70.
Wang H, Song X, Huang Q, Xu T, Yun D, Wang Y, et al. LGALS3 promotes Treatment Resistance in Glioblastoma and is Associated with Tumor Risk and Prognosis. Cancer Epidemiol Biomarkers Prev. 2019;28(4):760–9.
Steponaitis G, Kazlauskas A, Skiriute D, Vaitkiene P, Skauminas K, Tamasauskas A. Significance of Amphiregulin (AREG) for the outcome of low and high Grade Astrocytoma patients. J Cancer. 2019;10(6):1479–88.
Wang KY, Huang RY, Tong XZ, Zhang KN, Liu YW, Zeng F, et al. Molecular and clinical characterization of TMEM71 expression at the transcriptional level in glioma. CNS Neurosci Ther. 2019;25(9):965–75.
Zhang K, Wang J, Wang J, Luh F, Liu X, Yang L, et al. LKB1 deficiency promotes proliferation and invasion of glioblastoma through activation of mTOR and focal adhesion kinase signaling pathways. Am J Cancer Res. 2019;9(8):1650–63.
Jovčevska I, Zottel A, Šamec N, Mlakar J, Sorokin M, Nikitin D, et al. High FREM2 gene and protein expression are Associated with favorable prognosis of IDH-WT Glioblastomas. Cancers (Basel). 2019;11(8):1060.
Komaki S, Sugita Y, Furuta T, Yamada K, Moritsubo M, Abe H, et al. Expression of GLUT1 in Pseudopalisaded and Perivascular Tumor cells is an independent prognostic factor for patients with glioblastomas. J Neuropathol Exp Neurol. 2019;78(5):389–97.
Sharma N, Saxena S, Agrawal I, Singh S, Srinivasan V, Arvind S, et al. Differential expression Profile of NLRs and AIM2 in Glioma and implications for NLRP12 in Glioblastoma. Sci Rep. 2019;9(1):8480.
Prasad B, Tian Y, Li X. Large-scale analysis reveals Gene signature for Survival Prediction in primary glioblastoma. Mol Neurobiol. 2020;57(12):5235–46.
Vachher M, Arora K, Burman A, Kumar B. NAMPT, GRN, and SERPINE1 signature as predictor of disease progression and survival in gliomas. J Cell Biochem. 2020;121(4):3010–23.
Nesterova DS, Midya V, Zacharia BE, Proctor EA, Lee SY, Stetson LC, et al. Sexually dimorphic impact of the iron-regulating gene, HFE, on survival in glioblastoma. Neurooncol Adv. 2020;2(1):vdaa001.
Haddad AF, Chen JS, Oh T, Pereira MP, Joshi RS, Aghi MK. Higher cytolytic score correlates with an immunosuppressive tumor microenvironment and reduced survival in glioblastoma. Sci Rep. 2020;10(1):17580.
Sun Z, Qi X, Zhang Y. Bioinformatics Analysis of the expression of ATP Binding Cassette Subfamily C Member 3 (ABCC3) in human glioma. Open Med (Wars). 2020;15:107–13.
Du S, Guan S, Zhu C, Guo Q, Cao J, Guan G, et al. Secretory pathway kinase FAM20C, a marker for Glioma Invasion and Malignancy, predicts poor prognosis of Glioma. Onco Targets Ther. 2020;13:11755–68.
Pan Y, Zhang JH, Zhao L, Guo JC, Wang S, Zhao Y, et al. A robust two-gene signature for glioblastoma survival prediction. J Cell Biochem. 2020;121(7):3593–605.
Valiulyte I, Steponaitis G, Kardonaite D, Tamasauskas A, Kazlauskas A. A SEMA3 signaling pathway-based Multi-biomarker for Prediction of Glioma Patient Survival. Int J Mol Sci. 2020;21(19):7396.
Liu C, Zhang Z, Ping Y, Qin G, Zhang K, Maimela NR, et al. Comprehensive analysis of PD-1 gene expression, Immune characteristics and prognostic significance in 1396 Glioma patients. Cancer Manag Res. 2020;12:4399–410.
Zhu X, Jiang L, Yang H, Chen T, Wu X, Lv K. Analyzing the lncRNA, miRNA, and mRNA-associated ceRNA networks to reveal potential prognostic biomarkers for glioblastoma multiforme. Cancer Cell Int. 2020;20:393.
Faried A, Hermanto Y, Tjahjono FP, Valentino A, Arifin MZ. Identification of Periostin as a potential biomarker in Gliomas by Database Mining. World Neurosurg. 2020;135:e137–63.
Hu C, Fang D, Xu H, Wang Q, Xia H. The androgen receptor expression and association with patient’s survival in different cancers. Genomics. 2020;112(2):1926–40.
Li JL, Chen C, Chen W, Zhao LF, Xu XK, Li Y, et al. Integrative genomic analyses identify WDR12 as a novel oncogene involved in glioblastoma. J Cell Physiol. 2020;235(10):7344–55.
Yang J, Wang L, Xu Z, Wu L, Liu B, Wang J, et al. Integrated Analysis to evaluate the Prognostic Value of signature mRNAs in Glioblastoma Multiforme. Front Genet. 2020;11:253.
Yarmishyn AA, Yang YP, Lu KH, Chen YC, Chien Y, Chou SJ, et al. Musashi-1 promotes cancer stem cell properties of glioblastoma cells via upregulation of YTHDF1. Cancer Cell Int. 2020;20(1):597.
Gan C, Pierscianek D, El Hindy N, Ahmadipour Y, Keyvani K, Sure U, et al. The predominant expression of cancer stem cell marker ALDH1A3 in tumor infiltrative area is associated with shorter overall survival of human glioblastoma. BMC Cancer. 2020;20(1):672.
Peng Q, Li R, Li Y, Xu X, Ni W, Lin H, et al. Prediction of a competing endogenous RNA co-expression network as a prognostic marker in glioblastoma. J Cell Mol Med. 2020;24(22):13346–55.
Song ZB, Yang HP, Xu AQ, Zhan ZM, Song Y, Li ZY. Connective tissue growth factor as an unfavorable prognostic marker promotes the proliferation, migration, and invasion of gliomas. Chin Med J (Engl). 2020;133(6):670–8.
Yang L, Zeng W, Sun H, Huang F, Yang C, Cai X, et al. Bioinformatical Analysis of Gene Expression Omnibus Database Associates TAF7/CCNB1, TAF7/CCNA2, and GTF2E2/CDC20 pathways with Glioblastoma Development and Prognosis. World Neurosurg. 2020;138:e492–514.
Wu X, Hou P, Qiu Y, Wang Q, Lu X. Large-scale analysis reveals the specific clinical and Immune features of DGCR5 in Glioma. Onco Targets Ther. 2020;13:7531–43.
Shi H, Luo K, Huang W. Bone Marrow Stromal Antigen 2 is a potential unfavorable prognostic factor for high-Grade Glioma. Onco Targets Ther. 2020;13:8723–34.
Mao H, Nian J, Wang Z, Li X, Huang C. KDELR2 is an unfavorable prognostic biomarker and regulates CCND1 to promote tumor progression in glioma. Pathol Res Pract. 2020;216(7):152996.
Fuentes-Fayos AC, Vázquez-Borrego MC, Jiménez-Vacas JM, Bejarano L, Pedraza-Arévalo S, L-López F, et al. Splicing machinery dysregulation drives glioblastoma development/aggressiveness: oncogenic role of SRSF3. Brain. 2020;143(11):3273–93.
Miao Y, Wang J, Li Q, Quan W, Wang Y, Li C, et al. Prognostic value and immunological role of PDCD1 gene in pan-cancer. Int Immunopharmacol. 2020;89:107080. Pt B).
Chi Y, Liang Z, Guo Y, Chen D, Lu L, Lin J, et al. WBSCR22 confers cell survival and predicts poor prognosis in glioma. Brain Res Bull. 2020;161:1–12.
Kao Y, Tsai WC, Chen SH, Hsu SY, Huang LC, Chang CJ, et al. Shugosin 2 is a biomarker for pathological grading and survival prediction in patients with gliomas. Sci Rep. 2021;11(1):18541.
Yi L, Fan X, Li J, Yuan F, Zhao J, Nistér M, et al. Enrichment of branched chain amino acid transaminase 1 correlates with multiple biological processes and contributes to poor survival of IDH1 wild-type gliomas. Aging. 2021;13(3):3645–60.
Geraldo LH, Xu Y, Jacob L, Pibouin-Fragner L, Rao R, Maissa N, et al. SLIT2/ROBO signaling in tumor-associated microglia and macrophages drives glioblastoma immunosuppression and vascular dysmorphia. J Clin Invest. 2021;131(16):e141083.
Fang R, Chen X, Zhang S, Shi H, Ye Y, Shi H, et al. EGFR/SRC/ERK-stabilized YTHDF2 promotes cholesterol dysregulation and invasive growth of glioblastoma. Nat Commun. 2021;12(1):177.
Jiang Z, Shi Y, Zhao W, Zhang Y, Xie Y, Zhang B, et al. Development of an Immune-related Prognostic Index Associated with Glioblastoma. Front Neurol. 2021;12:610797.
Wang H, Wang X, Xu L, Lin Y, Zhang J, Cao H. Low expression of CDHR1 is an independent unfavorable prognostic factor in glioma. J Cancer. 2021;12(17):5193–205.
Maimaiti A, Wang X, Hao Y, Jiang L, Shi X, Pei Y, et al. Integrated Gene expression and methylation analyses identify DLL3 as a biomarker for prognosis of malignant glioma. J Mol Neurosci. 2021;71(8):1622–35.
Wang Y, Cai R, Wang P, Huang C, Zhang C, Liu Z. FAM46A expression is elevated in glioblastoma and predicts poor prognosis of patients. Clin Neurol Neurosurg. 2021;201:106421.
Zhao H, Zhou X, Yuan G, Hou Z, Sun H, Zhai N, et al. CDC6 is up-regulated and a poor prognostic signature in glioblastoma multiforme. Clin Transl Oncol. 2021;23(3):565–71.
Morrison C, Weterings E, Mahadevan D, Sanan A, Weinand M, Stea B. Expression levels of RAD51 inversely correlate with survival of Glioblastoma patients. Cancers (Basel). 2021;13(21):5358.
Xu B. Prediction and analysis of hub genes between glioblastoma and low-grade glioma using bioinformatics analysis. Med (Baltim). 2021;100(3):e23513.
Jeong HY, Kim HJ, Kim CE, Lee S, Choi MC, Kim SH. High expression of RFX4 is associated with tumor progression and poor prognosis in patients with glioblastoma. Int J Neurosci. 2021;131(1):7–14.
Cao F, Fan Y, Yu Y, Yang G, Zhong H. Dissecting prognosis modules and biomarkers in Glioblastoma based on Weighted Gene Co-expression Network Analysis. Cancer Manag Res. 2021;13:5477–89.
Cheng L, Yuan M, Li S, Lian Z, Chen J, Lin W, et al. Identification of an IFN-β-associated gene signature for the prediction of overall survival among glioblastoma patients. Ann Transl Med. 2021;9(11):925.
Hu N, Hu WH, Zhou SL, Yang Z, Liang WL, Yang RY, et al. SLC7A11 negatively associates with mismatch repair gene expression and endows glioblastoma cells sensitive to radiation under low glucose conditions. Neoplasma. 2021;68(6):1147–56.
Phan TL, Kim HJ, Lee SJ, Choi MC, Kim SH. Elevated RGMA expression predicts poor prognosis in patients with Glioblastoma. Onco Targets Ther. 2021;14:4867–78.
Hu L, Han Z, Cheng X, Wang S, Feng Y, et al. Expression Profile Analysis identifies a Novel Seven Immune-related gene signature to improve prognosis prediction of Glioblastoma. Front Genet. 2021;12:638458.
Lan Y, Zhao E, Zhang X, Zhu X, Wan L, Ping AS. Prognostic impact of a lymphocyte activation-associated gene signature in GBM based on transcriptome analysis. PeerJ. 2021;9:e12070.
Łysiak M, Smits A, Roodakker KR, Sandberg E, Dimberg A, Mudaisi M, et al. Deletions on chromosome Y and downregulation of the SRY Gene in Tumor Tissue Are Associated with worse survival of Glioblastoma patients. Cancers (Basel). 2021;13(7):1619.
Chen SH, Lin HH, Li YF, Tsai WC, Hueng DY. Clinical significance and systematic expression analysis of the thyroid receptor interacting protein 13 (TRIP13) as human gliomas biomarker. Cancers (Basel). 2021;13(10):2338.
Hsieh YH, Tsai JP, Yu CL, Lee CC, Hsu JC, Chen JC. Overexpression of Lipocalin-2 inhibits proliferation and invasiveness of human Glioblastoma Multiforme Cells by activating ERK Targeting cathepsin D expression. Biology (Basel). 2021;10(5):390.
Shi Y, Sun Y, Cheng H, Wang C. EFNB1 acts as a novel prognosis marker in Glioblastoma through Bioinformatics methods and experimental validation. J Oncol. 2021;2021:4701680.
Yang J, Yang Q. Identification of Core genes and screening of potential targets in Glioblastoma Multiforme by Integrated Bioinformatic Analysis. Front Oncol. 2021;10:615976.
Zhang Y, Yang X, Zhu XL, Wang ZZ, Bai H, Zhang JJ, et al. A Novel Immune-related Prognostic Biomarker and Target Associated with Malignant Progression of Glioma. Front Oncol. 2021;11:643159.
Urbantat RM, Blank A, Kremenetskaia I, Vajkoczy P, Acker G, Brandenburg S. The CXCL2/IL8/CXCR2 pathway is relevant for brain tumor malignancy and endothelial cell function. Int J Mol Sci. 2021;22(5):2634.
Krassnig S, Wohlrab C, Golob-Schwarzl N, Raicht A, Schatz C, Birkl-Toeglhofer AM, et al. A Profound Basic characterization of eIFs in Gliomas: identifying eIF3I and 4H as potential novel target candidates in Glioma Therapy. Cancers (Basel). 2021;13(6):1482.
Flor S, Oliva CR, Ali MY, Coleman KL, Greenlee JD, Jones KA, et al. Catalase overexpression drives an aggressive phenotype in Glioblastoma. Antioxid (Basel). 2021;10(12):1988.
Cocola C, Magnaghi V, Abeni E, Pelucchi P, Martino V, Vilardo L, et al. Transmembrane protein TMEM230, a target of Glioblastoma Therapy. Front Cell Neurosci. 2021;15:703431.
Cheng S, Wang LY, Wang CH, Wang FK, Zhu B, Zhang P, et al. Transmembrane protein DCBLD2 is correlated with poor prognosis and affects phenotype by regulating epithelial-mesenchymal transition in human glioblastoma cells. NeuroReport. 2021;32(6):507–17.
Liu J, Gao L, Ji B, Geng R, Chen J, Tao X, et al. BCL7A as a novel prognostic biomarker for glioma patients. J Transl Med. 2021;19(1):335.
Li X, Meng Y. SUMOylation Regulator-Related molecules can be used as prognostic biomarkers for Glioblastoma. Front Cell Dev Biol. 2021;9:658856.
Ye T, Cheng Y, Li C. Adaptor Protein Complex 1 Sigma 3 is highly expressed in Glioma and could enhance its progression. Comput Math Methods Med. 2021;2021:5086236.
Zhou K, Jiang T, Liu Y, Zhao Z, Huang L, Li G. FXYD2 mRNA expression represents a new independent factor that affects survival of glioma patients and predicts chemosensitivity of patients to temozolomide. BMC Neurol. 2021;21(1):438.
Tu Z, Shu L, Li J, Wu L, Tao C, Ye M, et al. A Novel signature constructed by RNA-Binding protein coding genes to improve overall survival prediction of Glioma patients. Front Cell Dev Biol. 2021;8:588368.
Zhou D, Wang M, Zhang Y, Wang K, Zhao M, Wang Y, et al. Screening and identification of LMNB1 and DLGAP5, two key biomarkers in gliomas. Biosci Rep. 2021;41(5):BSR20210231.
Moriconi C, Civita P, Neto C, Pilkington GJ, Gumbleton M. Caveolin-1, a key Mediator Across multiple pathways in Glioblastoma and an independent negative biomarker of patient survival. Front Oncol. 2021;11:701933.
Stanke KM, Wilson C, Kidambi S. High expression of glycolytic genes in clinical glioblastoma patients correlates with Lower Survival. Front Mol Biosci. 2021;8:752404.
Redekar SS, Varma SL, Bhattacharjee A. Identification of key genes associated with survival of glioblastoma multiforme using integrated analysis of TCGA datasets. Comput Methods Programs Biomed Updat. 2022;2:100051.
Liao Y, Luo Z, Lin Y, Chen H, Chen T, Xu L, et al. PRMT3 drives glioblastoma progression by enhancing HIF1A and glycolytic metabolism. Cell Death Dis. 2022;13(11):943.
Zhao S, Ji W, Shen Y, Fan Y, Huang H, Huang J, et al. Expression of hub genes of endothelial cells in glioblastoma-A prognostic model for GBM patients integrating single-cell RNA sequencing and bulk RNA sequencing. BMC Cancer. 2022;22(1):1274.
Nimbalkar VP, Kruthika BS, Sravya P, Rao S, Sugur HS, Chickabasaviah YT, et al. Chitinase 3-Like 2. Am J Clin Pathol. 2022;158(4):521–9.
Wu Y, Huang Y, Zhou C, Wang H, Wang Z, Wu J, et al. A Novel necroptosis-related prognostic signature of Glioblastoma Based on Transcriptomics Analysis and single cell sequencing analysis. Brain Sci. 2022;12(8):988.
Zhang S, Zhang W, Wu B, Xia L, Li L, Jin K, et al. Hub gene target of glioblastoma: LOX, SERPINH1 and TGFBI. Med (Baltim). 2022;101(45):e31418.
Pan C, Liang L, Wang Z, Zhang B, Li Q, Tian Y, et al. Expression and significance of SOX B1 genes in glioblastoma multiforme patients. J Cell Mol Med. 2022;26(3):789–99.
Yuan Q, Zuo FX, Cai HQ, Qian HP, Wan JH. Identifying Differential expression genes and prognostic signature based on Subventricular Zone involved Glioblastoma. Front Genet. 2022;13:912227.
Jin Y, Wang Z, Xiang K, Zhu Y, Cheng Y, Cao K, et al. Comprehensive development and validation of gene signature for predicting survival in patients with glioblastoma. Front Genet. 2022;13:900911.
Li ZH, Ma Y, Zhou Y, Huang ZH. Expression profiles of HOXC6 predict the survival of Glioblastoma patients and correlate with cell cycle. J Oncol. 2022;2022:8656865.
Santoni G, Maggi F, Amantini C, Arcella A, Marinelli O, Nabissi M, et al. Coexpression of TRPML1 and TRPML2 mucolipin channels affects the survival of Glioblastoma patients. Int J Mol Sci. 2022;23(14):7741.
Dubois N, Berendsen S, Tan K, Schoysmans L, Spliet W, Seute T, et al. STAT5b is a marker of poor prognosis, rather than a therapeutic target in glioblastomas. Int J Oncol. 2022;61(4):124.
Trivedi T, Panchal K, Bhalala N, Trivedi P. Prognostic significance of STAT3 gene expression in patients with glioblastoma tumors: a study from western India. J Egypt Natl Canc Inst. 2022;34(1):30.
Xiong Z, Wu S, Li FJ, Luo C, Jin QY, Connolly ID, et al. Elevated ETV6 expression in Glioma promotes an aggressive in Vitro Phenotype Associated with shorter patient survival. Genes (Basel). 2022;13(10):1882.
Zhang M, Zhou Z, Liu Z, Liu F, Zhao C. Exploring the potential biomarkers for prognosis of glioblastoma via weighted gene co-expression network analysis. PeerJ. 2022;10:e12768.
Wang ZL, Huang RY, Han B, Wu F, Sun ZY, et al. Identification of tumor-associated antigens and immune subtypes of lower-grade glioma and glioblastoma for mRNA vaccine development. Chin Neurosurg J. 2022;8(1):34.
Park SY, Jeong HY, Batara DC, Lee SJ, Cho JY, Kim SH. Sprouty 1 is associated with stemness and cancer progression in glioblastoma. IBRO Neurosci Rep. 2022;13:120–6.
Moreno DA, da Silva LS, Gomes I, Leal LF, Berardinelli GN, Gonçalves GM, et al. Cancer immune profiling unveils biomarkers, immunological pathways, and cell type score associated with glioblastoma patients’ survival. Ther Adv Med Oncol. 2022;14:17588359221127678.
Akçay S, Güven E, Afzal M, Kazmi I. Non-negative matrix factorization and differential expression analyses identify hub genes linked to progression and prognosis of glioblastoma multiforme. Gene. 2022;824:146395.
Liu D, Wang X, Liu Y, Li C, Zhang Z, Lv P. Actin-binding LIM 1 (ABLIM1) inhibits Glioblastoma Progression and serves as a Novel Prognostic Biomarker. Dis Markers. 2022;2022:9516808.
Lin D, Li W, Zhang N, Cai M. Identification of TNFAIP6 as a hub gene associated with the progression of glioblastoma by weighted gene co-expression network analysis. IET Syst Biol. 2022;16(5):145–56.
Güven E, Afzal M, Kazmi I. Screening the Significant Hub Genes by Comparing Tumor Cells, Normoxic and Hypoxic Glioblastoma Stem-like Cell Lines Using Co-Expression Analysis in Glioblastoma. Genes (Basel). 2022 15;13(3):518.
Zheng ZQ, Yuan GQ, Kang NL, Nie QQ, Zhang GG, Wang Z. Chromobox 7/8 serve as independent indicators for glioblastoma via promoting proliferation and invasion of glioma cells. Front Neurol. 2022;13:912039.
Liu Y, Xie M, Zhou Y, Che L, Zhang B. Interleukin-17 receptor D is a favorable biomarker of glioblastoma. J Neurosurg Sci. 2022. Epub ahead of print.
Lin Z, Zhang Z, Zheng H, Xu H, Wang Y, Chen C, et al. Molecular mechanism by which CDCP1 promotes proneural-mesenchymal transformation in primary glioblastoma. Cancer Cell Int. 2022;22(1):151.
Cui G, Xiao Y. Identification of SAA1 as a prognostic biomarker associated with immune infiltration in glioblastoma. Autoimmunity. 2022;55(6):418–27.
Gao W, Li Y, Zhang T, Lu J, Pan J, Qi Q, et al. Systematic analysis of chemokines reveals CCL18 is a prognostic biomarker in Glioblastoma. J Inflamm Res. 2022;15:2731–43.
Han MH, Min KW, Noh YK, Kim JM, Cheong JH, Ryu JI, et al. Identification of genes from ten oncogenic pathways associated with mortality and disease progression in glioblastoma. Front Oncol. 2022;12:965638.
Wei L, Zou C, Chen L, Lin Y, Liang L, Hu B, et al. Molecular insights and Prognosis Associated with RBM8A in Glioblastoma. Front Mol Biosci. 2022;9:876603.
Vedunova M, Turubanova V, Vershinina O, Savyuk M, Efimova I, Mishchenko T, et al. DC vaccines loaded with glioma cells killed by photodynamic therapy induce Th17 anti-tumor immunity and provide a four-gene signature for glioma prognosis. Cell Death Dis. 2022;13(12):1062.
Zhang S, Xiao X, Wang Y, Song T, Li C, Bao H, et al. Developing an Immune-Related signature for Predicting Survival Rate and the response to Immune checkpoint inhibitors in patients with glioma. Front Genet. 2022;13:899125.
Zhu J, Lin Q, Zheng H, Rao Y, Ji T. The pro-invasive factor COL6A2 serves as a novel prognostic marker of glioma. Front Oncol. 2022;12:897042.
Chen L, Xiong Z, Zhao H, Teng C, Liu H, Huang Q, et al. Identification of the novel prognostic biomarker, MLLT11, reveals its relationship with immune checkpoint markers in glioma. Front Oncol. 2022;12:889351.
Xie Y, Ding W, Xiang Y, Wang X, Yang J. Calponin 3 acts as a potential diagnostic and prognostic marker and promotes Glioma Cell Proliferation, Migration, and Invasion. World Neurosurg. 2022;165:e721–31.
Fuentes-Fayos AC, Pérez-Gómez JM, G-García ME, Jiménez-Vacas JM, Blanco-Acevedo C, et al. SF3B1 inhibition disrupts malignancy and prolongs survival in glioblastoma patients through BCL2L1 splicing and mTOR/ß-catenin pathways imbalances. J Exp Clin Cancer Res. 2022;41(1):39.
Guda MR, Tsung AJ, Asuthkar S, Velpula KK. Galectin-1 activates carbonic anhydrase IX and modulates glioma metabolism. Cell Death Dis. 2022;13(6):574.
Du Z, Wang Y, Liang J, Gao S, Cai X, Yu Y, et al. Association of glioma CD44 expression with glial dynamics in the tumour microenvironment and patient prognosis. Comput Struct Biotechnol J. 2022;20:5203–17.
Phillips E, Balss J, Bethke F, Pusch S, Christen S, Hielscher T, et al. PFKFB4 interacts with FBXO28 to promote HIF-1α signaling in glioblastoma. Oncogenesis. 2022;11(1):57.
Li JW, Huang QR, Mo LG. CNPY4 is a potential promising prognostic-related biomarker and correlated with immune infiltrates in gliomas. Med (Baltim). 2022;101(33):e30044.
Li Z, Fu WJ, Chen XQ, Wang S, Deng RS, Tang XP, et al. Autophagy-based unconventional secretion of HMGB1 in glioblastoma promotes chemosensitivity to temozolomide through macrophage M1-like polarization. J Exp Clin Cancer Res. 2022;41(1):74.
Guan Z, Luo L, Liu S, Guan Z, Zhang Q, Wu Z, et al. The role of TGR5 as an onco-immunological biomarker in tumor staging and prognosis by encompassing the tumor microenvironment. Front Oncol. 2022;12:953091.
Guo YZ, Chen G, Huang M, Wang Y, Liu YY, Jiang Q, et al. TIMM44 is a potential therapeutic target of human glioma. Theranostics. 2022;12(17):7586–602.
Wang X, Han M, Wang S, Sun Y, Zhao W, Xue Z, et al. Targeting the splicing factor NONO inhibits GBM progression through GPX1 intron retention. Theranostics. 2022;12(12):5451–69.
Wan Z, Zuo X, Wang S, Zhou L, Wen X, Yao Y, et al. Identification of angiogenesis-related genes signature for predicting survival and its regulatory network in glioblastoma. Cancer Med. 2023;12(16):17445–67.
Gao D, Wang P, Zhi L, Sun S, Qiu X, Liu Y. Expression of TMEM59L associated with radiosensitive in glioblastoma. J Radiat Res. 2023;64(5):833–41.
Liu Q, Wang R, Li Z, Peng Y, Qin W, Peng XM, et al. Prognostic significance of carboxypeptidase Q and its methylation in glioblastoma. Transl Cancer Res. 2023;12(5):1073–87.
Chen J, Wang H, Deng C, Fei M. SLC12A5 as a novel potential biomarker of glioblastoma multiforme. Mol Biol Rep. 2023;50(5):4285–99.
Zheng ZQ, Yuan GQ, Zhang GG, Nie QQ, Wang Z. Development and validation of a predictive model in diagnosis and prognosis of primary glioblastoma patients based on Homeobox A family. Discov Oncol. 2023;14(1):108.
Yoon HG, Cheong JH, Ryu JI, Won YD, Min KW, Han MH. The genes significantly associated with an improved prognosis and long-term survival of glioblastoma. PLoS ONE. 2023;18(11):e0295061.
Dang HH, Ta HDK, Nguyen TTT, Wang CY, Lee KH, Le NQK. Identification of a Novel eight-gene risk model for Predicting Survival in Glioblastoma: a Comprehensive Bioinformatic Analysis. Cancers (Basel). 2023;15(15):3899.
Singh K, Han C, Fleming JL, Becker AP, McElroy J, Cui T, et al. TRIB1 confers therapeutic resistance in GBM cells by activating the ERK and akt pathways. Sci Rep. 2023;13(1):12424.
Li S, Li S, Li Q, Liu F, Liao W, Yu L, et al. Increased neuromedin B is Associated with a favorable prognosis in Glioblastoma. Front Biosci (Landmark Ed). 2023;28(3):54.
Ali MY, Griguer CE, Flor S, Oliva CR. Mitoferrin-1 promotes proliferation and abrogates protein oxidation via the glutathione pathway in Glioblastoma. Antioxid (Basel). 2023;12(2):349.
Jia F, Zhang L, Jiang Z, Tan G, Wang Z. FZD1/KLF10-hsa-miR-4762-5p/miR-224-3p-circular RNAs axis as prognostic biomarkers and therapeutic targets for glioblastoma: a comprehensive report. BMC Med Genomics. 2023;16(1):21.
Ren D, Zhuang X, Lv Y, Zhang Y, Xu J, Gao F, et al. Identification of family with sequence similarity 110 Member C (FAM110C) as a candidate Diagnostic and Prognostic Biomarker for Glioma. Iran J Public Health. 2023;52(10):2117–27.
Zheng Y, Lang Y, Qi B, Wang Y, Gao W, Li T. TSPAN4 is a prognostic and immune target in Glioblastoma multiforme. Front Mol Biosci. 2023;9:1030057.
Ma K, Guo Q, Zhang X, Li Y. High expression of triggering receptor expressed on myeloid cells 1 predicts poor prognosis in Glioblastoma. Onco Targets Ther. 2023;16:331–45.
Gao L, Tong S, Liu J, Cai J, Ye Z, Zhou L, Song P, Li Z, Lei P, Wei H, Hua Q, Tian D, Cai Q. TMEM2 induces epithelial-mesenchymal transition and promotes resistance to temozolomide in GBM cells. Heliyon. 2023;9(6):e16559.
Chen Y, Pan Y, Gao H, Yi Y, Qin S, Ma F, Zhou X, Guan M. Mechanistic insights into super-enhancer-driven genes as prognostic signatures in patients with glioblastoma. J Cancer Res Clin Oncol. 2023;149(13):12315–32.
Ye Y, Zhong W, Qian J, Zhang J, Xu T, Han R, Han J, Wang C, Song L, Zeng X, Wang H. Comprehensive analysis of the prognosis and immune infiltrates for the BET protein family reveals the significance of BRD4 in glioblastoma multiforme. Front Cell Dev Biol. 2023;11:1042490.
Zhu W, Luo N, Li Q, Chen X, Li X, Fu M, Yang F, Chen Z, Zhang Y, Zhang Y, Peng X, Hu G. Development and validation of an inflammatory response-related prognostic model and immune infiltration analysis in glioblastoma. Ann Transl Med. 2023;11(2):69.
Li L, Gao R, Huangfu W, Zhang F, Wang R. Clinical significance of NKD inhibitor of WNT signaling pathway 1 (NKD1) in Glioblastoma. Genet Res (Camb). 2023;2023:1184101.
Lin L, Li X, Zhu S, Long Q, Hu Y, Zhang L, et al. Ferroptosis-related NFE2L2 and NOX4 genes are potential risk prognostic biomarkers and correlated with immunogenic features in Glioma. Cell Biochem Biophys. 2023;81(1):7–17.
Hu B, Chen D, Li Y, Yu S, Kuang L, Ma X, et al. Expression of TXLNA in brain gliomas and its clinical significance: a bioinformatics analysis. Chin Neurosurg J. 2023;9(1):27.
Ding J, Ji X, Liu L, Chen DZ, Luo N, Yu XT, et al. A prognostic and immunological analysis of 7B-containing Kelch structural domain (KLHDC7B) in pan-cancer: a potential target for immunotherapy and survival. J Cancer Res Clin Oncol. 2023;149(10):7857–76.
Le MK, Vuong HG, Nguyen TTT, Kondo T. NAA10 overexpression dictates distinct epigenetic, genetic, and clinicopathological characteristics in adult gliomas. J Neuropathol Exp Neurol. 2023;82(7):650–8.
Wang J, Shan A, Shi F, Zheng Q. Molecular and clinical characterization of ANG expression in gliomas and its association with tumor-related immune response. Front Med (Lausanne). 2023;10:1044402.
Wu J, Yu X, Zhu H, Chen P, Liu T, Yin R, et al. RelB is a potential molecular biomarker for immunotherapy in human pan-cancer. Front Mol Biosci. 2023;10:1178446.
Zhu RK, Zhang W, Zhang YX, Hui Z, Wang XW. A pan-cancer analysis to determine the prognostic analysis and immune infiltration of HSPA5. Curr Cancer Drug Targets. 2023. Epub ahead of print.
Azimi P, Karimpour M, Yazdanian T, Totonchi M, Ahmadiani A, Khoshbin S et al. Identification of prognostic values defined by mRNA expression of B3GNT7, CSTA, FCGR2C, PITX1, and SAA1 in glioblastoma patients. J Neurosurg Sci. Epub ahead of print.
Page MJ, McKenzie JE, Bossuyt PM, Boutron I, Hoffmann TC, Mulrow CD, et al. The PRISMA 2020 statement: an updated guideline for reporting systematic reviews. BMJ. 2021;372:n71.
Wells G, Shea B, O’Connell D, Peterson J, Welch V, Losos M et al. The Newcastle-Ottawa Scale (NOS) for assessing the quality of nonrandomized studies in meta-analyses. (2020). Available online at: http://www.ohri.ca/programs/clinical_epidemiology/nosgen.pdf.
Zhang J, Fu M, Zhang M, Zhang J, Du Z, Zhang H, et al. DDX60 is Associated with Glioma Malignancy and serves as a potential immunotherapy biomarker. Front Oncol. 2021;11:665360.
Fu M, Zhang J, Li W, He S, Zhang J, Tennant D, et al. Gene clusters based on OLIG2 and CD276 could distinguish molecular profiling in glioblastoma. J Transl Med. 2021;19(1):404.
Rahiminejad S, Maurya MR, Subramaniam S. Topological and functional comparison of community detection algorithms in biological networks. BMC Bioinformatics. 2019;20(1):212.
Lamano JB, Lamano JB, Li YD, DiDomenico JD, Choy W, Veliceasa D, et al. Glioblastoma-derived IL6 induces immunosuppressive peripheral myeloid cell PD-L1 and promotes Tumor Growth. Clin Cancer Res. 2019;25(12):3643–57.
Duan R, Han L, Wang Q, Wei J, Chen L, Zhang J, et al. HOXA13 is a potential GBM diagnostic marker and promotes glioma invasion by activating the wnt and TGF-β pathways. Oncotarget. 2015;6(29):27778–93.
Zaman N, Dass SS, DU Parcq P, Macmahon S, Gallagher L, Thompson L, et al. The KDR (VEGFR-2) genetic polymorphism Q472H and c-KIT polymorphism M541L are Associated with more aggressive Behaviour in Astrocytic Gliomas. Cancer Genomics Proteom. 2020;17(6):715–27.
Azimi P, Karimpour M, Totonchi M, Ahmadiani A. NOTCH1 mutation as prognostic indicator in glioblastoma patients. Conference: International Congress of Cancer Genomics (CGC2023), https://www.kongreuzmani.com/1-international-congress-of-cancer-genomics-cgc-2023.html, 3–5 May 2023, Tehran, Iran.
Kimberley D, Brosofske RE, Froese MJ, Falkowski, et al. A review of methods for Mapping and Prediction of Inventory attributes for operational Forest Management. For Sci. 2014;60(4):733–56.
Fury W, Batliwalla F, Gregersen PK, Li W. Overlapping probabilities of top ranking gene lists, hypergeometric distribution, and stringency of gene selection criterion. Engineering in Medicine and Biology Society, 2006. EMBS’06. 28th Annual International Conference of the IEEE. 2006, IEEE, 5531–5534.
Acknowledgements
We would like to thank the National Institute for Medical Research Development (NIMAD) for their support throughout the research process.
Funding
Not applicable.
Author information
Authors and Affiliations
Contributions
P.A and T.Y co-designed the study, conducted the searches and performed the bioinformatic analysis. A.A co-designed the study, assisted in the interpretation of the data, and edited the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Azimi, P., Yazdanian, T. & Ahmadiani, A. mRNA markers for survival prediction in glioblastoma multiforme patients: a systematic review with bioinformatic analyses. BMC Cancer 24, 612 (2024). https://doi.org/10.1186/s12885-024-12345-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12885-024-12345-z