
Abstract
Background
Renal hypouricemia (RHUC) is a hereditary disorder where mutations in SLC22A12 gene and SLC2A9 gene cause RHUC type 1 (RHUC1) and RHUC type 2 (RHUC2), respectively. These genes regulate renal tubular reabsorption of urates while there exist other genes counterbalancing the net excretion of urates including ABCG2 and SLC17A1. Urate metabolism is tightly interconnected with glucose metabolism, and SLC2A9 gene may be involved in insulin secretion from pancreatic β-cells. On the other hand, a myriad of genes are responsible for the impaired insulin secretion independently of urate metabolism.
Case presentation
We describe a 67 year-old Japanese man who manifested severe hypouricemia (0.7 mg/dl (3.8–7.0 mg/dl), 41.6 μmol/l (226–416 μmol/l)) and diabetes with impaired insulin secretion. His high urinary fractional excretion of urate (65.5%) and low urinary C-peptide excretion (25.7 μg/day) were compatible with the diagnosis of RHUC and impaired insulin secretion, respectively. Considering the fact that metabolic pathways regulating urates and glucose are closely interconnected, we attempted to delineate the genetic basis of the hypouricemia and the insulin secretion defect observed in this patient using whole exome sequencing. Intriguingly, we found homozygous Trp258* mutations in SLC22A12 gene causing RHUC1 while concurrent mutations reported to be associated with hyperuricemia were also discovered including ABCG2 (Gln141Lys) and SLC17A1 (Thr269Ile). SLC2A9, that also facilitates glucose transport, has been implicated to enhance insulin secretion, however, the non-synonymous mutations found in SLC2A9 gene of this patient were not dysfunctional variants. Therefore, we embarked on a search for causal mutations for his impaired insulin secretion, resulting in identification of multiple mutations in HNF1A gene (MODY3) as well as other genes that play roles in pancreatic β-cells. Among them, the Leu80fs in the homeobox gene NKX6.1 was an unreported mutation.
Conclusion
We found a case of RHUC1 carrying mutations in SLC22A12 gene accompanied with compensatory mutations associated with hyperuricemia, representing the first report showing coexistence of the mutations with opposed potential to regulate urate concentrations. On the other hand, independent gene mutations may be responsible for his impaired insulin secretion, which contains novel mutations in key genes in the pancreatic β-cell functions that deserve further scrutiny.
Similar content being viewed by others

Background
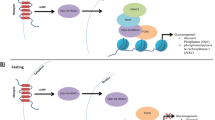
The serum urate concentrations are tightly regulated through multiple complex processes including hepatic production and renal excretion as well as intestinal secretion [1, 2]. The renal tubular transport of urates is regulated bidirectionally [3]: The reabsorption is regulated mainly through two major solute carrier (SLC) transporters, SLC22A12 (also known as URAT1, urate anion transporter 1) [4, 5] or SLC2A9 (also known as GLUT9, glucose transporter 9) [6,7,8] while several transporters for excretion have been identified including ABCG2 (ATP-binding cassette transporter G2) [9, 10], ABCC4 [11], SLC17A1 [12, 13] and OAT (organic anion transporter) family members [11]. The net renal urate excretion is largely determined by the balance of these reabsorption and excretion. The loss of function mutations in SLC22A12 and SLC2A9 causes renal hypouricemia (RHUC) type 1 and type 2, respectively [14]. The SLC22A12 is expressed at the apical membrane of proximal tubules while SLC2A9 isoforms are localized to both the apical and basolateral membrane [11] (Fig. 1).

Schematic description of urate transport in renal tubular cells. Arrows indicate the direction of urate transport
As exemplified by the fact that SLC2A9 facilitates the transport of not only urates but also glucose and fructose, urate metabolism is closely interconnected with glucose metabolism. Indeed, both positive and negative correlations between serum urates and blood glucose have been demonstrated. Insulin resistance increases serum urate levels independently of obesity [15]. Based on this positive correlation, serum urate levels were proposed as a potential predictor of type 2 diabetes occurrence [16]. Despite the accumulating observations supporting this positive correlations, negative correlations have long been well appreciated clinically as well [17, 18]. Renal urate excretion has been known to be competed with glycosuria [19], that might be at least in part explained by the dual transporting properties of SLC2A9 for glucose and urates [20]. Moreover, SLC2A9 has been reported to be expressed in pancreatic β-cells where SLC2A9 was proposed to facilitate the glucose uptake to increase glucose induced insulin secretion [21].
Here we report a case with severe hypouricemia accompanied by diabetes with impaired insulin secretion where whole exome sequencing revealed gene mutations responsible for these metabolic disorders.
Case presentation
A 67 year-old Japanese man was referred to our hospital for the treatment of diabetes with the concomitant concern about his severe hypouricemia. He was admitted to our hospital and biochemical data were collected under hospitalized conditions. On admission, his body mass index (BMI) was 25.1 (height: 174.1 cm, weight: 76.2 kg), excluding the possible contributions of obesity-induced alteration of urate metabolism. Although the historical onset and progression of his hypouricemia was unclear, he exhibited severe hypouricemia with hyperuricosuria (serum urate levels: 0.7 mg/dl (3.8–7.0 mg/dl) (41.6 μmol/l (226–416 μmol/l)), fractional excretion of urate (FEUA) was 65.5%) without any signs of kidney dysfunction (serum creatinine levels: 0.7 mg/dl, creatinine clearance: 109.1 ml/min, estimated glomerular filtration rate (eGFR): 85.4 ml/min/1.73m2). His urinary fractional excretion of urates was elevated but relatively modest compared to the reported cases of severe hypouricemia [8, 22]. He did not have any past medical history of either nephrolithiasis or exercise-induced acute renal failure to which hypouricemia sometimes predisposes [23].
We also assessed his glucose metabolism biochemically. On admission, he was treated with 25 units of insulin degludec with 1500 mg of metformin, 20 mg of teneligliptin and 20 mg of tohogliflozin with 8.9% of glycated hemoglobin (HbA1c) levels. Fasting and postprandial serum C-peptide levels-blood glucose levels were 0.09 ng/ml - 5.67 mmol/l, 1.45 ng/ml - 13.0 mmol/l, respectively while his urinary excretion of C-peptide was 25.7 μg/day, indicating impaired insulin secretion. Glutamate decarboxylase (GAD) auto-antibody was negative, and he did not have any medical histories of autoimmune diseases, excluding the possibility of autoimmune diabetes.
Discussion and conclusions
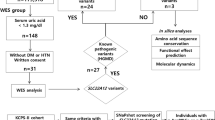
Considering the complex web of interconnections between urate and glucose metabolism, we attempted to delineate the molecular basis behind the coprevalence of diseases observed in this case by taking advantage of whole exome sequencing. We extracted genomic DNA from his peripheral blood mononuclear cells using the QIAamp DNA Blood Maxi Kit (QIAGEN) and the sequencing library was produced by SureSelectXT Reagent Kit/SureSelectXT Human all Exon Kit V6 (Agilent Technologies). The captured DNA was sequenced using the Illumina HiSeq2500 platform with paired-end reads of 100 bp according to the manufacturer’s instructions. Data analysis was performed using the CLC Genomics Workbench (CLC Bio) and non-synonymous single nucleotide variants (SNVs) were identified following the standard workflow (Table 1). He provided written informed consent and this study was approved by the University of Tsukuba Hospital Ethics Committee with the protocol number H30–329.
Firstly, we found homozygous Trp258* mutations in SLC22A12 gene (rs121907892), that is the most commonly observed dysfunctional mutation in Japanese hypouricemic subjects (Table 2, Fig. 2a) [24, 25]. Interestingly, we additionally found novel heterozygous Glu110Lys mutation in SLC22A12 gene (Fig. 2b). This mutation may be a C to T transition that occurred de novo, which is most frequently encountered in both the CpG and non-CpG context, typically being caused by deamination of methylated cytosines [26]. The Glu110Lys mutation would not influence the urate transport activity of SLC22A12 in this case since the SLC22A12 with Glu110Lys mutation is truncated and inactivated by the Trp258* mutation. However, considering the fact that SLC22A12 is a urate-anion exchanger [4], charged residues would be playing fundamental role in the substrate recognition or maintenance of the structural integrity. The mutation of an acidic amino acid to a basic residue would significantly alter ionic properties of SCL22A12 molecule. Whether Glu110Lys mutation on its own is sufficient to cause functional alteration of SLC22A12 and can be a risk allele for dysregulation of urate metabolism in general population awaits further investigation. On the other hand, we found two non-synonymous mutations in SLC2A9 gene, Gly25Arg (rs2276961) and Arg265His (rs3733591), that are not causative for hypouricemia (Fig. 2c, d). Although the correlation between Arg265His mutation and hyperuricemia remains enigmatic [6, 27,28,29,30,31], this mutation might play a compensatory role in this hypouricemic case toward raising the serum urate levels. We further examined other genes known to be involved in urate metabolism. We found the heterozygous ABCG2 Gln141Lys mutation (rs2231142) [9, 10, 32, 33] that has been firmly established to be associated with hyperuricemia as well as homozygous SLC17A1 Thr269Ile mutation (rs1165196) [12, 13] also reported to be associated with hyperuricemia (Fig. 3a, b). These mutations in two genes may contribute to maintain his serum urate concentrations in the presence of hypouricemia-prone mutations (Fig. 1). Notably, ABCG2 has been reported to control serum urate levels at the level of intestine [34, 35], therefore excretion of urates into urine are increased while that into intestine may be decreased in this case. This study represents the first report showing RHUC1 gene mutations in the presence of hyperuricemia-prone gene mutations. Since the net effect of multiple gene mutations determines the serum urate levels, focused sequencing approaches for limited genes may cause some pitfalls and comprehensive cataloguing of gene mutations linked to urate levels would offer a promise to better understand the biochemical kinetics of urate metabolism in human subjects, which is critically important since urate metabolism in humans is unequivocally different from that in animal models [36]. Although nephrolithiasis and exercise-induced acute kidney failure, two major complications in RHUC, have been reported to be observed mostly in RHUC2 [8, 37], these complications can be seen in RHUC1 with SLC22A12 Trp258* mutations as well [25]. The patient did not have any past history of these complications, which might be in part explained by the presence of the compensatory and adaptive mutations.

Hypouricemia associated genes. SLC22A12 and SLC2A9 mutations observed in this case. aSLC22A12 Trp258* mutation. bSLC22A12 Glu110Lys mutation. cSLC2A9 Gly25Arg mutation. dSLC2A9 Arg265His mutation

Hyperuricemia associated genes. aABCG2 Gln141Lys mutation. bSLC17A1 Thr269Ile mutation
While dysfunction of SLC2A9 has been implicated in both hypouricemia [7, 8] and impaired insulin secretion [21], we could not find either dysfunctional SCL2A9 mutations or other gene mutations causative for both of these two disorders. Therefore, we decided to search for independent gene mutations that could explain his impaired insulin secretion. Among the mutations found in genes associated with diabetes, the most promising was HNF1A gene (Maturity onset diabetes of the young 3, MODY3) [38, 39] where we found as many as four non-synonymous mutations accumulated in this patient: Ile27Leu (rs1169288) [40,41,42], Ser487Asn (rs2464196) [40, 41], Leu551Ser (rs1169304) and Ser581Gly (rs587778398) (Fig. 4a-d). Since it was reported that mutations in exons 8–10 present only in the longest isoform of HNF1A gene are associated with a later onset of MODY [43, 44], the latter two mutations may be of relevance to phenotypic manifestations in this case. We additionally found gene mutations in other genes associated with diabetes (Table 3) among which heterozygous Leu80fs in NKX6.1 was an unreported mutation of potential interest (Fig. 5). The critical role of NKX6.1 in insulin secretion from pancreatic β-cells has been demonstrated [45, 46] and this frame-shift mutation was inserted way upstream of the DNA binding domain of NKX6.1. Functional characterization of the mutant NKX6.1 protein and whether this can be a risk allele for diabetes in a large cohort deserve further investigation. Collectively, accumulation of these gene mutations, rather than monogenic mutations, presumably contributed to his impaired insulin secretion.

Mutations in HNF1A gene observed in this case. a Ile27Leu, b Ser487Asn, c Leu551Ser, d Ser581Gly

The novel mutation observed in NKX6.1 gene
Analysis of genetic inheritability was very limited in this study since he had only a child alive in his family members to whom we were not able to reach. Analysis with larger sample sizes or family-based linkage analysis would help to resolve some questions raised in this study.
In conclusion, we analyzed the genome from a patient showing severe hypouricemia with diabetes with impaired insulin secretion using whole exome sequencing. Interestingly he carried hypouricemic mutations in SLC22A12 gene as well as hyperuricemia-prone mutations. Although preceding studies have been sequencing the mutations in specific genes of interest, it would be helpful to sequence the genome in an unbiased manner to better understand urate metabolism especially from a kinetic point of view. The impaired insulin secretion may be at least in part attributed to the multiple mutations in HNF1A gene. We also identified novel mutations in SLC22A12 and NKX6.1 genes that deserve further scrutiny. This case study has instructive implications about how combined mutations in several genes affecting pathophysiology could present clinical traits in a body.
Availability of data and materials
The datasets generated during the current study are not publicly available because it is possible that individual privacy could be compromised.
Abbreviations
- SLC:
-
Solute carrier family
- RHUC:
-
Renal hypouricemia
- ABC:
-
ATP-binding cassette transporter
- MODY:
-
Maturity onset diabetes of the young
- BMI:
-
Body mass index
- HNF:
-
Hepatocyte nuclear factor
- NKX6.1:
-
NK6 homeobox 1
- URAT1:
-
Urate anion transporter 1
- GLUT9:
-
Glucose transporter 9
- OAT:
-
Organic anion transporter
- FEUA:
-
Fractional excretion of urate
- eGFR:
-
Estimated glomerular filtration rate
- GAD:
-
Glutamate decarboxylase
- SNV:
-
Single nucleotide variant
References
So A, Thorens B. Uric acid transport and disease. J Clin Invest. 2010;120(6):1791–9.
Xu X, Li C, Zhou P, Jiang T. Uric acid transporters hiding in the intestine. Pharm Biol. 2016;54(12):3151–5.
Xu L, Shi Y, Zhuang S, Liu N. Recent advances on uric acid transporters. Oncotarget. 2017;8(59):100852–62.
Enomoto A, Kimura H, Chairoungdua A, Shigeta Y, Jutabha P, Cha SH, Hosoyamada M, Takeda M, Sekine T, Igarashi T, et al. Molecular identification of a renal urate anion exchanger that regulates blood urate levels. Nature. 2002;417(6887):447–52.
Sakiyama M, Matsuo H, Shimizu S, Nakashima H, Nakamura T, Nakayama A, Higashino T, Naito M, Suma S, Hishida A, et al. The effects of URAT1/SLC22A12 nonfunctional variants, R90H and W258X, on serum uric acid levels and gout/hyperuricemia progression. Sci Rep. 2016;6:20148.
Vitart V, Rudan I, Hayward C, Gray NK, Floyd J, Palmer CN, Knott SA, Kolcic I, Polasek O, Graessler J, et al. SLC2A9 is a newly identified urate transporter influencing serum urate concentration, urate excretion and gout. Nat Genet. 2008;40(4):437–42.
Matsuo H, Chiba T, Nagamori S, Nakayama A, Domoto H, Phetdee K, Wiriyasermkul P, Kikuchi Y, Oda T, Nishiyama J, et al. Mutations in glucose transporter 9 gene SLC2A9 cause renal hypouricemia. Am J Hum Genet. 2008;83(6):744–51.
Dinour D, Gray NK, Campbell S, Shu X, Sawyer L, Richardson W, Rechavi G, Amariglio N, Ganon L, Sela BA, et al. Homozygous SLC2A9 mutations cause severe renal hypouricemia. J Am Soc Nephrol. 2010;21(1):64–72.
Matsuo H, Takada T, Ichida K, Nakamura T, Nakayama A, Ikebuchi Y, Ito K, Kusanagi Y, Chiba T, Tadokoro S, et al. Common defects of ABCG2, a high-capacity urate exporter, cause gout: a function-based genetic analysis in a Japanese population. Sci Transl Med. 2009;1(5):5ra11.
Nakayama A, Matsuo H, Takada T, Ichida K, Nakamura T, Ikebuchi Y, Ito K, Hosoya T, Kanai Y, Suzuki H, et al. ABCG2 is a high-capacity urate transporter and its genetic impairment increases serum uric acid levels in humans. Nucleosides Nucleotides Nucleic Acids. 2011;30(12):1091–7.
Dalbeth N, Stamp LK, Merriman TR. The genetics of gout: towards personalised medicine? BMC Med. 2017;15(1):108.
Iharada M, Miyaji T, Fujimoto T, Hiasa M, Anzai N, Omote H, Moriyama Y. Type 1 sodium-dependent phosphate transporter (SLC17A1 protein) is a Cl(−)-dependent urate exporter. J Biol Chem. 2010;285(34):26107–13.
Chiba T, Matsuo H, Kawamura Y, Nagamori S, Nishiyama T, Wei L, Nakayama A, Nakamura T, Sakiyama M, Takada T, et al. NPT1/SLC17A1 is a renal urate exporter in humans and its common gain-of-function variant decreases the risk of renal underexcretion gout. Arthritis Rheum. 2015;67(1):281–7.
Nakayama A, Matsuo H, Ohtahara A, Ogino K, Hakoda M, Hamada T, Hosoyamada M, Yamaguchi S, Hisatome I, Ichida K, et al. Clinical practice guideline for renal hypouricemia (1st edition). Hum Cell. 2019;32(2):83–7.
Facchini F, Chen YD, Hollenbeck CB, Reaven GM. Relationship between resistance to insulin-mediated glucose uptake, urinary uric acid clearance, and plasma uric acid concentration. Jama. 1991;266(21):3008–11.
Kramer CK, von Muhlen D, Jassal SK, Barrett-Connor E. Serum uric acid levels improve prediction of incident type 2 diabetes in individuals with impaired fasting glucose: the Rancho Bernardo study. Diabetes Care. 2009;32(7):1272–3.
Padova J, Patchefsky A, Onesti G, Faludi G, Bendersky G. The effect of glucose loads on renal uric acid excretion in diabetic patients. Metab Clin Exp. 1964;13:507–12.
Herman JB, Goldbourt U. Uric acid and diabetes: observations in a population study. Lancet. 1982;2(8292):240–3.
Skeith MD, Healey LA, Cutler RE. Urate excretion during mannitol and glucose diuresis. J Lab Clin Med. 1967;70(2):213–20.
Cheeseman C. Solute carrier family 2, member 9 and uric acid homeostasis. Curr Opin Nephrol Hypertens. 2009;18(5):428–32.
Evans SA, Doblado M, Chi MM, Corbett JA, Moley KH. Facilitative glucose transporter 9 expression affects glucose sensing in pancreatic beta-cells. Endocrinology. 2009;150(12):5302–10.
Shen H, Feng C, Jin X, Mao J, Fu H, Gu W, Liu A, Shu Q, Du L. Recurrent exercise-induced acute kidney injury by idiopathic renal hypouricemia with a novel mutation in the SLC2A9 gene and literature review. BMC Pediatr. 2014;14:73.
Kuwabara M, Niwa K, Ohtahara A, Hamada T, Miyazaki S, Mizuta E, Ogino K, Hisatome I. Prevalence and complications of hypouricemia in a general population: a large-scale cross-sectional study in Japan. PLoS One. 2017;12(4):e0176055.
Iwai N, Mino Y, Hosoyamada M, Tago N, Kokubo Y, Endou H. A high prevalence of renal hypouricemia caused by inactive SLC22A12 in Japanese. Kidney Int. 2004;66(3):935–44.
Komoda F, Sekine T, Inatomi J, Enomoto A, Endou H, Ota T, Matsuyama T, Ogata T, Ikeda M, Awazu M, et al. The W258X mutation in SLC22A12 is the predominant cause of Japanese renal hypouricemia. Pediatr Nephrol. 2004;19(7):728–33.
Smith TCA, Arndt PF, Eyre-Walker A. Large scale variation in the rate of germ-line de novo mutation, base composition, divergence and diversity in humans. PLoS Genet. 2018;14(3):e1007254.
Tu HP, Chen CJ, Tovosia S, Ko AM, Lee CH, Ou TT, Lin GT, Chang SJ, Chiang SL, Chiang HC, et al. Associations of a non-synonymous variant in SLC2A9 with gouty arthritis and uric acid levels in Han Chinese subjects and Solomon Islanders. Ann Rheum Dis. 2010;69(5):887–90.
Urano W, Taniguchi A, Anzai N, Inoue E, Sekita C, Endou H, Kamatani N, Yamanaka H. Association between GLUT9 and gout in Japanese men. Ann Rheum Dis. 2010;69(5):932–3.
McArdle PF, Parsa A, Chang YP, Weir MR, O'Connell JR, Mitchell BD, Shuldiner AR. Association of a common nonsynonymous variant in GLUT9 with serum uric acid levels in old order amish. Arthritis Rheum. 2008;58(9):2874–81.
Hollis-Moffatt JE, Gow PJ, Harrison AA, Highton J, Jones PB, Stamp LK, Dalbeth N, Merriman TR. The SLC2A9 nonsynonymous Arg265His variant and gout: evidence for a population-specific effect on severity. Arthritis Res Ther. 2011;13(3):R85.
Hurba O, Mancikova A, Krylov V, Pavlikova M, Pavelka K, Stiburkova B. Complex analysis of urate transporters SLC2A9, SLC22A12 and functional characterization of non-synonymous allelic variants of GLUT9 in the Czech population: no evidence of effect on hyperuricemia and gout. PLoS One. 2014;9(9):e107902.
Stiburkova B, Pavelcova K, Zavada J, Petru L, Simek P, Cepek P, Pavlikova M, Matsuo H, Merriman TR, Pavelka K. Functional non-synonymous variants of ABCG2 and gout risk. Rheumatology. 2017;56(11):1982–92.
Stiburkova B, Pavelcova K, Pavlikova M, Jesina P, Pavelka K. The impact of dysfunctional variants of ABCG2 on hyperuricemia and gout in pediatric-onset patients. Arthritis Res Ther. 2019;21(1):77.
Hosomi A, Nakanishi T, Fujita T, Tamai I. Extra-renal elimination of uric acid via intestinal efflux transporter BCRP/ABCG2. PLoS One. 2012;7(2):e30456.
Ichida K, Matsuo H, Takada T, Nakayama A, Murakami K, Shimizu T, Yamanashi Y, Kasuga H, Nakashima H, Nakamura T, et al. Decreased extra-renal urate excretion is a common cause of hyperuricemia. Nat Commun. 2012;3:764.
Alvarez-Lario B, Macarron-Vicente J. Uric acid and evolution. Rheumatology. 2010;49(11):2010–5.
Jeannin G, Chiarelli N, Gaggiotti M, Ritelli M, Maiorca P, Quinzani S, Verzeletti F, Possenti S, Colombi M, Cancarini G. Recurrent exercise-induced acute renal failure in a young Pakistani man with severe renal hypouricemia and SLC2A9 compound heterozygosity. BMC Med Genet. 2014;15:3.
Ellard S. Hepatocyte nuclear factor 1 alpha (HNF-1 alpha) mutations in maturity-onset diabetes of the young. Hum Mutat. 2000;16(5):377–85.
Wang H, Hagenfeldt-Johansson K, Otten LA, Gauthier BR, Herrera PL, Wollheim CB. Experimental models of transcription factor-associated maturity-onset diabetes of the young. Diabetes. 2002;51(Suppl 3):S333–42.
Yamagata K, Oda N, Kaisaki PJ, Menzel S, Furuta H, Vaxillaire M, Southam L, Cox RD, Lathrop GM, Boriraj VV, et al. Mutations in the hepatocyte nuclear factor-1alpha gene in maturity-onset diabetes of the young (MODY3). Nature. 1996;384(6608):455–8.
Ben Khelifa S, Martinez R, Dandana A, Khochtali I, Ferchichi S, Castano L. Maturity onset diabetes of the young (MODY) in Tunisia: low frequencies of GCK and HNF1A mutations. Gene. 2018;651:44–8.
Beysel S, Eyerci N, Pinarli FA, Kizilgul M, Ozcelik O, Caliskan M, Cakal E. HNF1A gene p.I27L is associated with early-onset, maturity-onset diabetes of the young-like diabetes in Turkey. BMC Endocr Disord. 2019;19(1):51.
Harries LW, Ellard S, Stride A, Morgan NG, Hattersley AT. Isomers of the TCF1 gene encoding hepatocyte nuclear factor-1 alpha show differential expression in the pancreas and define the relationship between mutation position and clinical phenotype in monogenic diabetes. Hum Mol Genet. 2006;15(14):2216–24.
Bellanne-Chantelot C, Carette C, Riveline JP, Valero R, Gautier JF, Larger E, Reznik Y, Ducluzeau PH, Sola A, Hartemann-Heurtier A, et al. The type and the position of HNF1A mutation modulate age at diagnosis of diabetes in patients with maturity-onset diabetes of the young (MODY)-3. Diabetes. 2008;57(2):503–8.
Schisler JC, Jensen PB, Taylor DG, Becker TC, Knop FK, Takekawa S, German M, Weir GC, Lu D, Mirmira RG, et al. The Nkx6.1 homeodomain transcription factor suppresses glucagon expression and regulates glucose-stimulated insulin secretion in islet beta cells. Proc Natl Acad Sci U S A. 2005;102(20):7297–302.
Tessem JS, Moss LG, Chao LC, Arlotto M, Lu D, Jensen MV, Stephens SB, Tontonoz P, Hohmeier HE, Newgard CB. Nkx6.1 regulates islet beta-cell proliferation via Nr4a1 and Nr4a3 nuclear receptors. Proc Natl Acad Sci U S A. 2014;111(14):5242–7.
Acknowledgements
We thank Dr. Masafumi Muratani (University of Tsukuba) for bioinformatics analysis and valuable discussion.
Funding
No funding was obtained for this study.
Author information
Authors and Affiliations
Contributions
MS and TM was in charge of the medical care for the patient and extracted genomic DNA from his peripheral blood. MS performed the data mining and analysis. TM, YY, YF, MO, YM, YS, YO, HI, NY, SY, HS1 (corresponding to Hiroaki Suzuki) and HS2 (corresponding to Hitoshi Shimano) supported the medical care and manuscript preparation. MS wrote the manuscript. HS2 helped writing the manuscript and supervised this project. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The patient provided a written informed consent and this study was approved by the University of Tsukuba Hospital Ethics Committee (protocol no. H30–329).
Consent for publication
Written consent for publication of clinical data and results of the whole exome analysis was obtained from the patient.
Competing interests
Authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Sekiya, M., Matsuda, T., Yamamoto, Y. et al. Deciphering genetic signatures by whole exome sequencing in a case of co-prevalence of severe renal hypouricemia and diabetes with impaired insulin secretion. BMC Med Genet 21, 91 (2020). https://doi.org/10.1186/s12881-020-01031-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12881-020-01031-z