
Abstract
Background
The genus Erianthus, which belongs to the “Saccharum complex”, includes C4 warm-season grasses. Erianthus species are widely distributed throughout Southeast Asia, East Asia and South Asia. Erianthus arundinaceus (Retz.) Jeswiet is highly adaptable to the environment, has a high percentage of dry matter, and is highly productive. Recently, this species has attracted attention as a novel bioenergy crop and as a breeding material for sugarcane improvement. Such interest in E. arundinaceus has accelerated the collection and conservation of its genetic resources, mainly in Asian countries, and also evaluation of morphological, agricultural, and cytogenetic features in germplasm collections. In Thailand, genetic resources of E. arundinaceus have been collected over the past 20 years and their phenotypic traits have been evaluated. However, the genetic differences and relatedness of the germplasms are not fully understood.
Results
A set of 41 primer pairs for nuclear simple sequence repeats (SSRs) developed from E. arundinaceus were used to assess the genetic diversity of 121 Erianthus germplasms collected in Thailand; of these primer pairs, 28 detected a total of 316 alleles. A Bayesian clustering approach with these alleles classified the accessions into four main groups, generally corresponding to the previous classification based on phenotypic analysis. The results of principal coordinate analysis and phylogenetic analysis of the 121 accessions on the basis of the SSR markers showed the same trend as Bayesian clustering, whereas sequence variations of three non-coding regions of chloroplast DNA revealed eight haplotypes among the accessions. The analysis of genetic structure and phylogenetic relationships, however, found some accessions whose classification contradicted the results of previous phenotypic classification.
Conclusions
The molecular approach used in this study characterized the genetic diversity and relatedness of Erianthus germplasms collected across Thailand. This knowledge would allow efficient maintenance and conservation of the genetic resources of this grass and would help to use Erianthus species as breeding materials for development of novel bioenergy crops and sugarcane improvement.
Similar content being viewed by others

Background
According to the Fifth Assessment Report (AR5: 2013–2014) of the Intergovernmental Panel on Climate Change (IPCC), since the middle of the twentieth century, the natural environment has been exposed to severe global warming caused by human activities, and urgent mitigation efforts are needed, including energy-saving measures and the introduction of renewable energy sources [1]. Global warming will unavoidably harm food production, ecosystems, and water resources, and advance preparation is necessary to adapt to climate change [1]. Global climate change may affect stable agricultural production by causing droughts, floods, high-temperature injury, and emergence of novel pests [2, 3]. Thus, the establishment of sustainable agricultural production systems resilient to global climate change is one of the critical challenges that humans will face in the near future [1].
Diverse germplasm collections are essential for stable preservation and maintenance of ecotypes that might adapt to climate changes; these collections could provide indispensable materials for the development of novel cultivars with such adaptations. The genus Erianthus, which belongs to the “Saccharum complex”, includes C4 warm-season grasses. The complex includes five interbreeding genera—Saccharum L. (sugarcane), Miscanthus Andersson sect. Diandra Keng, Erianthus sect. Ripidium, Narenga Bor, and Sclerostachya A. Camus—that grow in a wide range of climatic conditions [4,5,6]. Erianthus sect. Ripidium has a basic chromosome number of x = 10 [7] and comprises seven closely related species: E. elephantinus Hook. f. (2n = 20), E. hostii Griseb. (2n = 20), E. ravennae (L.) P. Beauv. (2n = 20), E. procerus (Roxb.) Raizada (2n = 40), E. kanashiroi Ohwi (2n = 60), E. arundinaceus (Retz.) Jeswiet (2n = 30, 40, 60), and E. bengalense (Retz.) Bharadw. (2n = 20, 30, 40, 60) [7]. On the basis of the analysis of repulsion-phase linkage, Chen et al. [8] reported that Erianthus behaves like a true autopolyploid. Of the seven species, E. arundinaceus and E. procerus have recently attracted attention for their use in Erianthus and sugarcane breeding to increase biomass production [9, 10]. Erianthus arundinaceus is highly adaptable to the environment and is distributed in tropical and subtropical areas throughout Southeast Asia, East Asia, and South Asia. Accessions adapted to temperate zones [11] and high altitude [12] have also been found in East Asia. The plant height reaches more than 3 m [13, 14], and the high percentage of dry matter and high productivity of this species have attracted attention to E. arundinaceus as a novel bioenergy crop [11, 15]. Stress tolerance of this species in adverse environments also makes it a promising breeding material for the improvement of sugarcane [6, 10, 16, 17]. Such wide interest in E. arundinaceus has accelerated its collection and conservation, mainly in Asian countries, including India, China, and Thailand, which are rich in Erianthus genetic resources [9, 12, 18]. A number of E. arundinaceus accessions have also been collected in the temperate zone in Japan with the aim of exploring breeding materials for genetic improvement of its overwintering ability as a bioenergy crop candidate [19].
The collected genetic resources can be effectively used in breeding programs after multidimensional evaluation of their characteristics. In China [14] and Japan [11], morphological, agricultural, and cytogenetic features have been evaluated in E. arundinaceus germplasm collections. Genetic diversity assessment using molecular markers has been reported in E. arundinaceus accessions collected from India, China, the Philippines, Vietnam, Indonesia, and Japan for the purpose of bioenergy crop development and sugarcane improvement [20, 21]. Recently, the first cultivar of E. arundinaceus, ‘JES1’, was developed from a Japanese wild accession and was used as raw material for pellet fuel instead of wood pulp [11]. Intergeneric hybrids between Saccharum spp. and E. arundinaceus or between E. procerus and S. officinarum have been used to produce progeny by backcrossing for sugarcane improvement [17, 22, 23]. The genetic resources of Erianthus species from all over Thailand have been explored in the past 20 years, and two species, E. arundinaceus (2n = 40, 60) and E. procerus, have been found there. Thai E. arundinaceus accessions have been classified into Types I–III on the basis of phenotypic traits [9]. Types I and II are hexaploids (2n = 6x = 60). Type I has a hairy leaf sheath and can adapt to various environments such as open hill slopes and streambeds. Type II has a hairless leaf sheath covered with a waxy substance. This type has large buds capable of germinating and prominent root primordia, and is distributed mainly in the south of Thailand. Type III is a tetraploid (2n = 4x = 40) and is similar to Type II, but lacks wax on the leaf sheath. This type is found mainly along rivers and streams in Thailand. Erianthus procerus lacks hair and wax on the leaf sheath. This species has no prominent buds or root primordia, and is found in Thailand mainly on mountain slopes and on forest and field edges. There are no reports of the analysis of these genetic resources with molecular markers, and little is known about the genetic diversity of these Erianthus species in Thailand.
Molecular markers provide an objective evaluation of genetic variation in germplasms, which, being not influenced by environmental changes, are thus widely used to assess genetic diversity of plant genetic resources [24]. In the past decade, next generation sequencing (NGS) methodologies, which allow the genome-wide development of molecular markers and genotyping, have been applied to the polyploid species of warm-season grasses such as Paspalum [25], Panicum [26], and Pennisetum [27]. Using NGS data, we have developed simple sequence repeat (SSR or microsatellite) markers from the genomic DNA of hexaploid E. arundinaceus [28]. These SSR markers have been used successfully to estimate genetic diversity in E. arundinaceus collected in Japan and Indonesia [21]. In contrast to nuclear DNA markers, which are biparentally inherited, uniparentally inherited loci such as those in the chloroplast genome might provide information about the evolutionally history of germplasms [29, 30]. Therefore, a comparative analysis of both types of DNA polymorphisms could provide a more complementary and comprehensive insight into genetic diversification.
Genetic improvement depends on the diversity of available genetic resources; thus, it is essential to understand the genetic diversity of E. arundinaceus to facilitate breeding programs in this species. The main objective of the present research is to characterize the genetic variability in the Erianthus species of Thailand that has resulted in inter- and intraspecific variation in morphology. We investigated variability of SSR markers and partial chloroplast genome sequences of the Erianthus genetic resources of Thailand. On the basis of these data, we also analyzed the geographical distribution of the genetic diversity of Erianthus species in Thailand.
Results
SSR analysis
Among 41 SSR primer pairs developed from Japanese E. arundinaceus [28], 28 primer pairs resulted in scorable amplicons of the expected sizes in all accessions tested (E. arundinaceus and E. procerus included), with a total of 316 alleles (Tables S1 and S2). Allele number per locus ranged from 1 (ETR098) to 32 (ETR129), with an average of 11.3 (Table 1). Of the 28 SSRs, 27 (except ETR098) detected polymorphic loci.
To evaluate the informativeness of the 28 SSR primer pairs, we calculated polymorphic information content (PIC), marker index (MI), and resolving power (Rp) for each locus in all accessions tested. These genetic parameters, as well as expected fragment sizes, observed size ranges, numbers of amplified fragments (NA), and percentage of polymorphic fragments (NP) and the numbers of genotypes (NG), are shown in Table 1 and S1. In all 121 accessions, PIC values ranged from 0.00 to 0.36, with an average of 0.21; MI ranged from 0.00 to 4.94, with an average of 2.22; and Rp ranged from 0.00 to 6.48, with an average of 3.15 (Table 1). In the 71 hexaploid E. arundinaceus accessions, the average values were 0.23 (range, 0.00–0.34) for PIC, 2.23 (0.00–5.82) for MI, and 3.19 (0.00–7.69) for Rp (Table S1). In the 16 tetraploid E. arundinaceus accessions, the average values were 0.24 (0.00–0.43) for PIC, 1.22 (0.00–4.77) for MI, and 1.88 (0.00–6.50) for Rp (Table S1). In the 34 E. procerus accessions, the average values were 0.25 (0.00–0.42) for PIC, 1.34 (0.00–2.93) for MI, and 2.09 (0.00–4.06) for Rp (Table S1). In all 121 accessions, the values of these parameters for 7 loci were larger than the average values of all loci, indicating their high discriminatory power (bold loci in Table 1).
Genetic structure
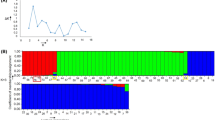
We used SSR genotyping data to determine the genetic structure of 121 Erianthus accessions. In the distribution of ΔK values, we found high values of ΔK at K = 2 and K = 3 (Fig. 1a). We assessed the individual proportion membership (qi) in the groups using the threshold value of 0.8, as used in other grass species; individuals with a value of ≥0.8 were considered to have a strong affinity to a group, and those with < 0.80 as an admixture [31,32,33,34]. With the threshold value of qi ≥0.80, the structure at K = 2 revealed two groups (S1 and S2; Fig. 1b, Table S2). Group S1 consisted of 87 E. arundinaceus accessions (71 hexaploids and 16 tetraploids). Group S2 consisted of 34 E. procerus accessions. At K = 3, three groups of accessions were defined (T1–T3; Fig. 1b, Table S2) with the same cutoff for membership assignment. Group T1 (54 accessions) included E. arundinaceus hexaploids. Group T2 included 24 E. arundinaceus accessions (8 hexaploids and 16 tetraploids). Group T3 included 34 E. procerus accessions. Seven hexaploid E. arundinaceus accessions—ThE01–012 (map No. 21), ThE01–013 (22), ThE02–091 (45), ThE02–086 (64), ThE10–003 (66), ThE10–008 (69), and ThE10–021 (71)—were identified as the admixture group; all accessions were an admixture of T1 and T2.

Genetic assignment of 121 Erianthus accessions using data from genotyping with 28 SSR primer pairs. a Estimation of the most likely number of groups using structure analysis: the mean values of log-likelihood for 10 independent runs for each value of K (left) and ΔK statistics for different K values based on the second-order rate of change in the log-likelihood function (right). b Bar plots of ancestry proportions for the ΔK values at K = 2, K = 3, and K = 4. Accessions identified as the admixture group are marked with asterisks. Ea-TI: E. arundinaceus Type I; Ea-TII: E. arundinaceus Type II; Ea-TIII: E. arundinaceus Type III; Ep: E. procerus
The structure at K = 4 revealed four groups (F1–F4; Fig. 1b, Table S2). Groups F1 (54 accessions) and F2 (8 accessions) included E. arundinaceus hexaploids. Group F3 included 16 tetraploid E. arundinaceus accessions. Group F4 included 34 E. procerus accessions. The remaining seven accessions of hexaploid E. arundinaceus—ThE01–012 (21), ThE01–013 (22), ThE02–091 (45), ThE02–086 (64), ThE02–087 (65), ThE10–003 (66), and ThE10–008 (69)—were identified as the admixture group; Nos. 65 and 69 were admixtures of F2 and F3, and the other accessions were admixtures of F1 and F2. The analysis also found two accessions, ThE10–004 (59) and ThE10–005 (60), with ambiguous phenotypic clustering.
Phylogenetic relationships
To better understand the genetic structure and relationships among Thai Erianthus accessions, we performed a principal coordinate analysis (PCoA) and a phylogenetic analysis based on the genotyping data obtained from 28 SSRs. The results of PCoA revealed four main groups (G1–G4; Fig. 2, Table S2). The admixture group overlapped with groups G1 and G2 (Fig. 2, Table S2). Phylogenetic analysis of the 121 accessions using Nei’s minimum distance on the basis of 316 alleles detected with the 28 SSR loci showed four major groups (C1–C4; Fig. 3), supporting the population structure determined by structure analysis. Groups C1 (59 accessions) and C2 (12 accessions) included hexaploid E. arundinaceus. Group C3 contained 16 tetraploid E. arundinaceus accessions. Group C4 contained 34 E. procerus accessions. The grouping of two accessions—ThE10–004 (59) and ThE10–005 (60)—based on phenotypic variation [9] was inconsistent with that based on genetic variation (Figs. 2 and 3; Table S2). On the basis of phenotypic variations, these accessions have been classed into Type I [9], but our PCoA and phylogenetic analysis classed them into groups G2 and C2, respectively, which consisted mostly of Type II accessions.

Principal coordinate analysis of 121 Erianthus accessions based on the Bruvo distance between individuals calculated in Polysat software. Colors indicate four groups corresponding to those in Fig. 1 at K = 4. The admixture group is indicated by grey circles. Map numbers of seven accessions assigned to the admixture group are shown next to the circles. Two accessions with ambiguous phenotypic clustering are marked with asterisks next to the map numbers. Dashed circles indicate grouping (G1–G4). Numbers on each axis indicate the proportion of variance explained by each principal coordinate

Unrooted neighbor-joining tree of 121 Erianthus accessions based on Nei’s minimum distance. The color of each accession corresponds to that in Fig. 1 at K = 4. Two accessions with ambiguous phenotypic clustering are marked with asterisks
Genetic diversity and differentiation among groups
To evaluate genetic diversity and differentiation in groups based on polymorphic SSRs, we calculated the genetic diversity parameters for the groups defined at K = 4, which could explain the genetic characteristics of Thai Erianthus species in more detail. In this analysis (Table 2), we excluded the seven accessions identified as the admixture group. NA ranged from 5.54 in group F3 to 10.14 in F1, with an average of 7.01. The effective number of alleles (NAe) ranged from 3.97 (F3) to 5.13 (F1), with an average of 4.51. Allelic richness (AR) ranged from 4.47 (F3) to 5.76 (F1), with an average of 5.04. Gene diversity (He) ranged from 0.72 (F3) to 0.78 (F1), with an average of 0.75. The values of the inbreeding coefficient (Fi) were negative in all groups and ranged from − 0.27 (F1) to − 0.39 (F3), with an average of − 0.34. Group F1 had the highest values of all parameters except Fi.
To estimate the degree of relative genetic differentiation among the groups, we calculated population differentiation (Fst) among all groups (Table 3). Fst ranged from 0.03 to 0.05 (average 0.04). The effective migration rate (Nm) ranged from 4.88 between groups F3 and F4 to 8.22 between F1 and F4 (Table 3), with an average of 6.58.
Geographical distribution of genetic diversity
To evaluate the relationships between genetic diversity and geographical distribution, we investigated the correlation between genetic distance and geographic distance among accessions using Mantel’s test. The correlation was significant for the 71 hexaploid E. arundinaceus accessions (r = 0.369, p = 0.0001), but not for the 16 tetraploid E. arundinaceus accessions (r = 0.135, p = 0.1112) or the 34 E. procerus accessions (r = 0.154, p = 0.0118), when p < 0.001 was considered statistically significant. In addition, we determined the correlations between genetic diversity parameters (AR, He, and Fi) and geographic distribution (Fig. 4). AR, He, and Fi for hexaploid E. arundinaceus were positively correlated with latitude (r = 0.89*, 0.98*** and 0.79*, respectively). In tetraploid accessions (E. arundinaceus and E. procerus), no significant correlations were found. These values were not significantly correlated with longitude in either tetraploid or hexaploid Erianthus (data not shown).

Correlations between genetic diversity parameters (AR, He, and Fi) and latitude. The analysis was performed for hexaploid E. arundinaceus (orange), tetraploid E. arundinaceus (blue), and E. procerus (yellow). Latitudes were rounded to integers. Significant correlations are indicated (*p < 0.01 and ***p < 0.0001)
Chloroplast haplotype variation
We also compared sequences of three non-coding regions (rps16–trnQ, atpA–rps14, and rpl16–rps3) of chloroplast DNA (cpDNA) among the 121 Erianthus accessions. The lengths of these regions were 747, 810–839, and 529–544 bp, respectively, and the lengths of concatenated sequences were 2093–2126 bp (Table S3). The aligned sequences of these regions were 747, 846 and 544 bp, respectively, and the concatenated alignment of the three regions was 2126 bp (Tables S3, S4). We identified 11 variations in the sequence, of which 10 were parsimony-informative sites. On the basis of the concatenated sequences, haplotypes H1–H8 were identified (Table S4). Haplotype diversity, nucleotide diversity, and neutrality were estimated in the absence of indels for each group identified in the structure analysis (K = 4) and are summarized in Table 4. Overall haplotype diversity (h) was 0.78. Haplotypes H1–H7 were identified in group F1, which had the highest value of haplotype diversity (h = 0.78). The overall values of nucleotide diversity parameters were 1.23 for Pi, 1.68 for θ, and 0.81 for π. The highest values of Pi (1.36), θ (1.74), and π (0.084) were found in group F1. The neutrality test statistics (DFu and Li, FFu and Li, FFu, and DTajima) were not significant in any group and did not reveal a deviation from neutrality in the regions examined in all 121 accessions.
A parsimonious network among haplotypes of the 121 accessions was generated from the sequence dataset of the three chloroplast non-coding regions (Fig. 5). The haplotypes in the network were clearly split into the upper clade (H1, H4, H7, and H8) and lower clade (H2, H3, H5, and H6), and the connection among lineages extended to three steps. Haplotype H1 was predominant (56 of the 121 accessions, 46.3%). In the upper clade, H7 and H8 were generated from H1 by one mutational step, and H4 was generated from H7 by one additional mutational step. In the lower clade, H6, connected to the upper clade, was generated from H1 via three mutational steps. H3 and H5 were separated from H6 by one mutational step, and H2 was generated from H5 by one additional mutational step.

Median-joining network of eight haplotypes (H1–H8) based on sequence variations in three non-coding regions of E. arundinaceus cpDNA. a Locations of the three regions (rps16–trnQ, atpA–rps14, and rpl16–rps3). The position (bp) of each region in full-length cpDNA of E. arundinaceus accession ‘JW630’ is given in parentheses. LSC, large single-copy region; IR, inverted repeat; SSC, small single-copy region. b Circle sizes are proportional to haplotype frequency, and their colors correspond to those in Fig. 1 at K = 4. Positions of mutational steps in concatenated sequences between haplotypes are shown next to the branches. Positions of mutational steps in each cpDNA region are shown in parentheses. Haplotype of each accession is indicated in Table S2
Discussion
The development of a new cultivar is influenced by the distribution of diversity in available genetic resources. There are some reports of genetic diversity assessment of the Erianthus genetic resources. Here, we characterized previously unknown genetic properties of Erianthus species in Thailand at the molecular level.
Two Erianthus species, E. arundinaceus and E. procerus, with different phenotypic characteristics and ploidy, have been identified in Thailand [9]. We used 41 SSR primer pairs developed from the nuclear genome of a Japanese E. arundinaceus accession [28] to genotype the Thai Erianthus collection. Of these primer pairs, 28 amplified products of the expected sizes in all 121 E. arundinaceus and E. procerus accessions, suggesting the usefulness of these primer pairs in genetic diversity analysis of Erianthus species in Thailand.
Genetic structure of Thai Erianthus
Structure analysis based on genotyping data from the 28 SSR primers indicated high values of ΔK at K = 2 and K = 3. The structure at K = 2 revealed two groups (S1 and S2; Fig. 1, Table S2), corresponding to the two Erianthus species. At K = 3, three groups of accessions were defined (T1–T3; Fig. 1, Table S2). Of the three structure groups, one included mainly hexaploid E. arundinaceus, and the other two included tetraploid species (E. arundinaceus and E. procerus). In this analysis, seven accessions showed admixed ancestry between tetraploid and hexaploid Erianthus (Table S2). However, no traces of mating between accessions with different ploidy were identified from the fragment patterns. Using cytogenetic analysis, Tagane et al. [9] have reported that the E. arundinaceus accessions identified as admixed in this study were hexaploids. Another population structure was indicated by ΔK at K = 4. This structure could explain genetic characteristics of Thai Erianthus species in more detail. Tetraploid and hexaploid E. arundinaceus accessions, which were identified as the same group (T2) at K = 3, were defined as different groups (F2 and F3) at K = 4, except ThE02–087 (65). At K = 4, eight hexaploid accessions (Nos. 59, 60, 62, 63, 67, 68, 70, and 71) were defined as a group distinct from the 16 tetraploid E. arundinaceus accessions, although they belonged to the same group as tetraploid E. arundinaceus or to the admixture group at K = 3. The results of PCoA and phylogenetic analysis of the 121 accessions showed the same trend as the structure analysis at K = 4. On the basis of phenotypic analysis, Tagane et al. [9] divided Thai E. arundinaceus into Types I and II (2n = 6x = 60), and Type III (2n = 4x = 40). Our results of grouping at K = 4 are generally consistent with those results, except for seven accessions (Nos. 21, 22, 59, 60, 65, 66, and 69). We used the grouping at K = 4 in subsequent analysis.
Genetic differentiation among groups
Genetic differentiation among all pairs of groups from the structure analysis (K = 4) was estimated using Fst. At Fst > 0.15, genetic differentiation between populations is considered to be significant [35]. The genetic differentiation among the groups was low, as the Fst values were < 0.15 (0.030–0.051).
The analysis of genetic structure and phylogenetic relationships found some accessions that were genetically admixed (Nos. 21, 22, 45, 64, 65, 66, and 69) and some with ambiguous phenotypic clustering (Nos. 59 and 60). These results suggest natural hybridization events. Tagane et al. [9] plotted ThE02–091 (No. 45 in this study) and ThE02–086 (No. 64) in the intermediate regions in principal component analysis and canonical discriminant analysis based on phenotypic variations, and assumed these accessions to be hybrids between hexaploid (Type I) and tetraploid E. arundinaceus (Type III). Because flowering time differs among the three types of Thai E. arundinaceus [9], intercrossing among them seems to be difficult. This is one of the factors causing genetic differentiation in populations, and it could prevent genetic diversification based on hybridization events. In relation to gene flow, Nm between groups (4.88–8.22) indicated that the possibility of random mating between the populations was high (Nm > 4 [36, 37];), suggesting that gene flow has occurred among some accessions and thus may have led to a low genetic differentiation between groups. The flowering periods of the hexaploid E. arundinaceus Types I and II overlap slightly [9]. Because some accessions with contradictory phenotypic and genetic groupings were collected at locations geographically close to each other, the possibility that they have arisen by clonal propagation or genetic exchange between the types should not be completely dismissed. Further analysis with a greater number of DNA markers could clarify the origin of hybridity in these accessions.
Genetic diversity and distribution
The SSR markers used in this study indicated a high degree of genetic diversity in group F1, which consisted of hexaploid E. arundinaceus. The basic chromosome number in E. arundinaceus is x = 10, and individuals with 2n = 30, 40, or 60 have been found [7, 38]. Type I E. arundinaceus is hexaploid and, among all regions, is found mainly in Thailand. Altered ploidy is one of the factors affecting genetic diversity [39, 40]. Among tetraploids, the mean values of genetic variation as indicated by Nei’s similarity index were 0.69 (range 0.07–0.86) in E. arundinaceus and 0.75 (range 0.50–0.96) in E. procerus, and were lower than in the hexaploid accessions (mean 0.86, range 0.04–0.96; data not shown). Zhang et al. [41] suggested that physical isolation by geographical barriers such as oceans, mountains, and rivers might decrease the level of genetic variation in Erianthus germplasms collected across China, including in an island area, because limited gene flow from outside reduces genetic diversity. Such effect of isolation on genetic diversity can also be inferred from the significant correlation between genetic distance and physical distance among the hexaploid E. arundinaceus accessions in this study. The diversity of accessions from Indonesia is lower than that in other Asian countries [20], as suggested by amplified fragment length polymorphism (AFLP) analysis. Correlation between genetic diversity and geographical distribution in this study showed that the degree of genetic diversity in hexaploid E. arundinaceus tended to increase with latitude. From phenotypic differences, it was suggested that genetic divergence of E. arundinaceus in Thailand could have occurred through adaptive evolution to different natural habitats [9]. These ideas together with our results suggest that not only isolation by geographical barriers but also adaptive radiation and parallel or convergent evolution accompanying changes in ecological conditions could affect genetic diversity in Thai Erianthus germplasms. The assessment of genetic diversity among germplasms collected over a wider area could improve our understanding of the geographical distribution of genetic diversity in Erianthus species and provide novel insights into the genetic basis of environmental adaptability of these species.
Information on the evolutionary history would also help us to understand the establishment of the current genetic diversity of Erianthus in Thailand. Type I hexaploid E. arundinaceus and tetraploid E. procerus were clustered into different groups in the structure and phylogenetic analyses. However, the degree of genetic divergence between these two groups was not high in comparison with that among other groups (Table 3). Because the chloroplast genome is maternally inherited in most angiosperm species, its diversity provides insight into the maternal evolutionary history and relationships among species with different chromosome numbers. Fu and Allaby [42] have reported that Linum species with different chromosome numbers share sequence variations in their chloroplast genomes. In this study, the network analysis based on variations in partial cpDNA sequences detected 8 haplotypes (Table 5), of which 5 (except H2, H3, and H8) were detected across the phenotypic types (Types I–III) and species (E. arundinaceus and E. procerus), even if the accessions had different chromosome numbers. For example, the tetraploid E. procerus tended to share sequence variants with hexaploid E. arundinaceus of Type I, suggesting that the current hexaploid E. arundinaceus and E. procerus could have diverged from a common matrilineal ancestor relatively recently. Therefore, the low degree of genetic divergence between groups detected in this study is not surprising. Similar results have been reported by Tagane et al. [9], who showed that these species are phenotypically similar and overlap in principal component analysis of phenotypic traits. A more detailed analysis of cpDNA polymorphisms could provide information useful for understanding the evolutionary relationships between and within Erianthus species.
Genetic resources of Erianthus species are distributed extensively in Southeast Asia, East Asia, and South Asia. Accessions adapted to temperate zones and high altitudes have also been explored and collected, mainly in China and Japan [11, 12]. A previous study in Erianthus species suggested that the degree of genetic diversity or similarity among populations and individuals could be influenced by genetic isolation due to geographical barriers [41]. We did not address the degree of genetic diversity between genetic resources from Thailand and other countries, and further investigations will be needed. In particular, a comprehensive analysis with common DNA markers targeting Erianthus accessions collected in Asian countries, where the genetic resources are plentiful, would not only clarify the position of the Thai germplasms, but also provide information on genetic characteristics of the available resources of Erianthus species. This knowledge would allow efficient maintenance and conservation of the genetic resources of these species of large grasses and would facilitate the use of Erianthus species as breeding materials for the development of novel bioenergy crops and the improvement of sugarcane, which is now in progress in some countries [11, 15, 43, 44]. Such studies might be an important step toward mitigation of and adaptation to global warming.
Conclusions
In this study, we characterized the genetic diversity of Erianthus germplasms collected across Thailand by using SSR markers developed from genomic DNA of Japanese E. arundinaceus. These markers are highly polymorphic in Thai Erianthus accessions and could be useful for evaluation of genetic resources in Erianthus species. Thai Erianthus accessions were classified into four groups, generally corresponding to the previous classification based on phenotypic analysis. Some E. arundinaceus accessions were regarded as intraspecific hybrids. These results reveal the genetic basis for the Thai Erianthus germplasm collection diversity and provide useful resources for genetic study and breeding in Erianthus species.
Methods
Plant materials and DNA extraction
We used 71 hexaploid and 16 tetraploid E. arundinaceus accessions and 34 tetraploid E. procerus accessions (121 accessions in total; Table 5 and S2). All accessions were wild Erianthus collected across Thailand since 1997 in the framework of the collaborative research project between DOA (Development of Agriculture, Ministry of Agriculture and Cooperatives, Thailand) and JIRCAS (Japan International Research Center for Agricultural Sciences, Japan). The accessions were classified on the basis of morphological characters [13, 45]; they are maintained in the Tha Phra field at the Khon Kaen Field Crop Research Center, DOA, Khon Kaen, Thailand. On the basis of the coordinate data, the geographic distribution of these accessions was visualized in DIVA-GIS v. 7.5 software [46] (Fig. S1).
Genomic DNA was extracted from approximately 100 mg of freshly harvested leaves of each accession using a DNeasy Plant Mini Kit (Qiagen, Hilden, Germany). Its quality was assessed by agarose gel electrophoresis, and its concentration was determined with a NanoDrop 1000 spectrophotometer (Thermo Fisher Scientific Inc., Waltham, MA, USA) and adjusted to 10 ng/μL.
Amplification of SSRs
The 41 SSR primer pairs developed from the wild E. arundinaceus accession ‘JW630’, collected in Japan [28], were used for PCR amplification of Thai Erianthus accessions. Of these primer pairs, 28 showed high stability and high reproducibility and were used to genotype the accessions (Table S2). PCR based on the M13-tailed primer method [47] was performed as described [28]. PCR products amplified with universal M13 primers labeled with each fluorophore (FAM, HEX, or NED) were diluted 1:5 and aliquots were combined with 12 μL deionized formamide (Amresco, Solon, OH, USA) and 0.25 μL ROX500 size standard (Life Technologies, Carlsbad, CA, USA). This mixture was heated at 98 °C for 2 min and then chilled on ice to denature the DNA before loading. Alleles were separated in an ABI3100 Genetic Analyzer (Life Technologies), and the allele sizes were determined automatically using GeneMapper v. 5.0 software (Life Technologies). The data included peaks that may be caused by artefacts often encountered in microsatellite analysis, such as stutter, peak broadening and shift. To minimize genotyping error, the allele sizes were checked and edited manually, and some samples were re-analyzed to confirm ambiguous genotypes.
Each fragment was considered as a dominant marker and was scored as 1 (presence) or 0 (absence), because the E. arundinaceus accessions used in this study are polyploid. The binary genotypic data were used to assess the discriminatory power and utility of each locus on the basis of PIC [48], MI [49, 50], Rp [51], expected fragment sizes, observed size range, NA, NP, and NG.
Amplification of chloroplast non-coding regions
Three non-coding intergenic spacer regions of cpDNA—rps16–trnQ, atpA–rps14, and rpl16–rps3—were sequenced in all Erianthus accessions by using Sanger sequencing of each PCR product. Sequence variations in these regions were identified between E. arundinaceus accessions from Japan and Indonesia [52]. Primer pairs to amplify each region (Table S5) were designed from the chloroplast genome sequence of ‘JW630’ (GenBank accession No. LC160130). PCR was performed in a 15-μL mixture containing genomic DNA (20 ng), 5× PrimeSTAR buffer (TaKaRa, Shiga, Japan), 0.4 mM each dNTP (TaKaRa), 5 pmol of each specific forward and reverse primer, and 0.5 units PrimeSTAR HS-DNA polymerase (TaKaRa) in a GeneAmp PCR System 9700 thermal cycler (Life Technologies) as follows: 98 °C for 1 min, followed by 30 cycles of 98 °C for 15 s, 56 °C for 15 s, and 72 °C for 2.5 min. Amplification products were purified with a QuickStep2 PCR Purification Kit (Edge Biosystems, Gaithersburg, MD, USA) and were used as templates for sequencing. Cycle-sequencing was performed with a BigDye Terminator Cycle Sequence Kit v. 3.1 (Life Technologies) using specific primers (Table S5) in the same thermal cycler. Sequencing products were purified on a Sephadex G-50 column (GE Healthcare, Uppsala, Sweden) and sequenced in an ABI3500 genetic analyzer (Life Technologies).
Data analysis
SSR profiles of 121 accessions were used to investigate the population genetic structure through model-based Bayesian clustering analysis in STRUCTURE v. 2.3.4 software [53, 54]. The data for tetraploids were treated as for hexaploids (i.e., the last two of six rows per individual in tetraploids were coded as missing data) to enable simultaneous analysis of the mixed-ploidy data. Recessive alleles were considered to be present (recessive alleles = 1), as described in the recessive allele approach for polyploid species [55]. The population number (K = 1–10) was tested in an admixture ancestry model with correlated allele frequencies. Each run was performed in 10 replicates for each K value, with a burn-in period of 100,000 steps followed by 100,000 Markov Chain Monte Carlo (MCMC) iterations. The optimal K values were determined using the ad hoc statistic ΔK, which was estimated as the rate of change in the log probability of data between successive K values [56] in the online application STRUCTURE HARVESTER [57]. The optimal alignment for 10 replicate runs was determined with the full search algorithm in CLUMPP 1.1.2 software [58], and then the inferred clusters were visualized as color bar plots in DISTRUCT 1.1 software [59].
Structural features were assessed by PCoA and phylogenetic tree analysis using SSR genotyping data. PCoA based on Bruvo distances [60] among individuals was performed in Polysat v. 1.4 in the R statistical software package. We also calculated pairwise Nei’s minimum distance among all accessions and constructed a phylogenetic tree of the 121 accessions by using the neighbor-joining method [61] based on Nei’s similarity index. The analysis was conducted in Populations 1.2.32 [62] and the tree was visualized in Figtree v. 1.4.2 software [63].
On the basis of SSR genotyping data, genetic diversity and allele frequencies in each group were estimated through the NA, NAe, AR, He, and Fi statistics. The coefficients of genetic differentiation between each group assigned by the structure analysis were estimated with the parameter Fst from the analysis of molecular variance (AMOVA); Fst / (1 − Fst) was used. Statistical analyses were implemented in SPAGeDi 1.5 software [64], which was designed to analyze data from polyploid species. Nm between groups was estimated from Fst, taking into account sample size and the number of loci [65, 66], as Nm = (1 − Fst) / 4Fst [67].
A matrix of linear distances (km) among all genotypes was constructed on the basis of the geographic coordinates of the accessions in Geographic Distance Matrix Generator v. 1.2.3 software [68]. This matrix was compared to the genetic distance matrix (Nei’s minimum distance based on SSR genotyping) using Mantel’s correlation test [69] based on 1000 random permutations in GenoDive v. 2.0b27 software [70]. The correlation coefficients between genetic diversity parameters (AR, He, and Fi) and latitude and longitude were calculated in JMP 8.0 software (SAS Institute, Cary, NC, USA).
Sequences of the three chloroplast non-coding regions were concatenated and aligned in BioEdit 7.2.5 software [71]. After manual editing, haplotype and nucleotide diversities were estimated in each group using h [72], Pi [72], Watterson’s θ [73], and Tajima’s π [74] statistics. Tests of neutral evolution were performed as described [75,76,77]. All parameters were calculated in DnaSP v. 5.10.01 software [78]. Indels in the alignment were treated as missing data. The phylogenetic network of the inferred haplotypes was also constructed using concatenated alignments and a maximum-parsimony method based on a median-joining algorithm [79] in Network 4.6 software (http://www.fluxus-engineering.com/).
Availability of data and materials
All data generated and analyzed during this study, including primer sequences and grouping of the accessions, are included in this published article and its supplementary information files. All cpDNA sequences generated in this study are available in the NCBI database (https://www.ncbi.nlm.nih.gov/) with accession numbers LC636829–LC637191 (Table S3).
Abbreviations
- AFLP:
-
Amplified fragment length polymorphism
- AMOVA:
-
Analysis of molecular variance
- A R :
-
Allelic richness
- cpDNA:
-
Chloroplast DNA
- F i :
-
inbreeding coefficient
- F st :
-
population differentiation
- H e :
-
gene diversity
- MI:
-
Marker index
- N A :
-
Number of amplified fragments
- N Ae :
-
effective number of alleles
- NGS:
-
Next generation sequencing
- N m :
-
Effective migration rate
- N G :
-
Number of genotypes
- N P :
-
Percentage of polymorphic fragments
- PCoA:
-
Principal coordinate analysis
- PCR:
-
Polymerase chain reaction
- PIC:
-
Polymorphic information content
- R p :
-
Resolving power
- SSR:
-
Simple sequence repeat
References
IPCC: Climate Change 2014: Synthesis Report. In: Core Writing Team, Pachauri RK, Meyer LA, editors. Contribution of Working Groups I, II and III to the Fifth Assessment Report of the Intergovernmental Panel on Climate Change. Geneva, Switzerland: IPCC; 2014.
FAO, IFAD, UNICEF, WFP and WHO. The state of food security and nutrition in the world 2019. Safeguarding against economic slowdowns and downturns. Rome, France: FAO; 2019.
Gomez-Zavaglia A, Mejuto JC, Simal-Gandara J. Mitigation of emerging implications of climate change on food production systems. Food Res Int. 2020. https://doi.org/10.1016/j.foodres.2020.109256.
Mukherjee SK. Origin and distribution of Saccharum. Bot Gaz. 1957;119:55–61.
Nair NV, Praneetha M. Cyto-morphological studies on three Erianthus arundinaceus (Retz.) Jeswiet accessions from Andaman-Nicobar islands, India. Cytologia. 2006;71:107–9.
Jackson P, Henry RJ. Erianthus. In: Kole C, editor. Wild crop relatives: genomic and breeding resources. Berlin: Springer; 2011. p. 97–109.
Amalraj VA, Balasundaram N. On the taxonomy of the members of ‘Saccharum Complex’. Genet Resour Crop Evol. 2006;53:35–41.
Chen JW, Lao FY, Chen XW, Deng HH, Liu R, He HY, et al. DNA marker transmission and linkage analysis in population derived from a sugarcane (Saccharum spp.) × Erianthus arundinaceus hybrid. PLoS One. 2015;10:e0128865.
Tagane S, Ponragdee W, Sansayawichai T, Sugimoto A, Terajima Y. Characterization and taxonomical note about Thai Erianthus germplasm collection: the morphology, flowering phenology and biogeography among E. procerus and three types of E. arundinaceus. Genet Resour Crop Evol. 2012;59:769–81.
Mohanraj K, Manjunatha T, Mahadevaiah C, Suganya A, Adhini SP, Geetha P. Genetic variability for nitrogen use efficiency in interspecific and intergeneric hybrids of sugarcane. Int J Curr Microbiol App Sci. 2020;9:19–30.
Matsunami H, Kobayashi M, Tsuruta S, Terajima Y, Sato H, Ebina M, et al. Overwintering ability and high-yield biomass production of Erianthus arundinaceus in a temperate zone in Japan. BioEnergy Res. 2018;11:467–79.
Zhang J, Yan J, Zhang Y, Xiao M, Bai S, Wu Y, et al. Molecular insights of genetic variation in Erianthus arundinaceus populations native to China. PLoS One. 2013;8:e008038.
Tagane S, Tagane Yasuda M, Ponragdee W, Sansayawichai T, Sugimoto A. Cytological study of Erianthus procerus and E. arundinaceus (Gramineae) in Thailand. Cytologia. 2011;76:171–5.
Yan J, Zhang J, Sun K, Chang D, Bai S, Shen Y, et al. Ploidy level and DNA content of Erianthus arundinaceus as determined by flow cytometry and the association with biological characteristics. PLoS One. 2016;11:e0151948.
Wang W, Li R, Wang H, Qi B, Jiang X, Zhu Q, et al. Sweetcane (Erianthus arundinaceus) as a native bioenergy crop with environmental remediation potential in southern China: a review. GCB Bioenergy. 2019;11:1012–25.
Berding N, Roach BT. Germplasm collection maintenance, and use. In: Heinz DJ, editor. Sugarcane improvement through breeding. Amsterdam: Elsevier; 1987. p. 143–210.
Fukuhara S, Terajima Y, Irei S, Sakaigaichi T, Ujihara K, Sugimoto A, et al. Identification and characterization of intergeneric hybrid of commercial sugarcane (Saccharum spp. hybrid) and Erianthus arundinaceus (Retz.). Euphytica. 2013;189:321–7.
Nair NV, Sekharan S. Saccharum germplasm collection in Mizoram. India Sugar Tech. 2009;11:288–91.
Kobayashi M, Tsuruta S, Ebina M. Exploration and collection of Erianthus arundinaceus clones in Shizuoka, Aichi and Ibaraki Prefectures. Ann Rep Explor Introd Plant Genet Resour. 2018;34:79–93 (Japanese with English summary).
Cai Q, Aitken KS, Fan YH, Piperidis G, Liu XL, McIntyre CL, et al. Assessment of the genetic diversity in a collection of Erianthus arundinaceus. Genet Resour Crop Evol. 2012;59:1483–91.
Tsuruta S, Ebina M, Terajima Y, Kobayashi M, Takahashi W. Genetic variability in Erianthus arundinaceus accessions native to Japan based on nuclear DNA content and simple sequence repeat markers. Acta Physiol Plant. 2017. https://doi.org/10.1007/s11738-017-2519-1.
Cai Q, Aitken K, Deng HH, Chen XW, Fu C, Jackson PA, et al. Verification of the introgression of Erianthus arundinaceus germplasm into sugarcane using molecular markers. Plant Breed. 2005;124:322–8.
Nair NV, Mohanraj K, Sunadaravelpandian K, Suganya A, Selvi A, Appunu C. Characterization of an intergeneric hybrid of Erianthus procerus × Saccharum officinarum and its backcross progenies. Euphytica. 2017;213:267.
Nadeem MA, Nawaz MA, Shahid MQ, Doğan Y, Comertpay G, Tildiz M, et al. DNA molecular markers in plant breeding: current status and recent advancements in genomic selection and genome editing. Biotechnol Biotec Eq. 2018;32:261–85.
Giordano A, Cogan NOI, Kaur S, Drayton M, Mouradov A, Panter S, et al. Gene discovery and molecular marker development, based on high-throughput transcript sequencing of Paspalum dilatatum Poir. PLoS One. 2014;9:e85050.
Jiang Y, Li H, Zhang J, Xiang J, Cheng R, Liu G. Whole genomic EST-SSR development based on high-throughput transcript sequencing in proso millet (Panicum miliaceum). Int J Agric Biol. 2018;20:617–20.
Muktar MS, Teshome A, Hanson J, Negawo AT, Habte E, Entfellner J-BD, et al. Genotyping by sequencing provides new insights into the diversity of Napier grass (Cenchrus purpureus) and reveals variation in genome-wide LD patterns between collections. Sci Rep. 2019;9:6936.
Tsuruta S, Ebina M, Kobayashi M, Takahashi W, Terajima Y. Development and validation of genomic simple sequence repeat markers in Erianthus arundinaceus. Mol Breed. 2017. https://doi.org/10.1007/s11032-017-0675-z.
Jose CC, Roberto A, Victoria I, Javier T, Manuel T, Joaquin D. A phylogenetic analysis of 34 chloroplast genomes elucidates the relationships between wild and domestic species within the genus Citrus. Mol Biol Evol. 2015;32:2015–35.
Li W, Liu Y, Yang Y, Xie X, Lu Y, Yang Z, et al. Interspecific chloroplast genome sequence diversity and genomic resources in Diospyros. BMC Plant Biol. 2018;18:210.
Menchari Y, Délye C, Crre VL. Genetic variation and population structure in black-grass (Alopecurus myosuroides Huds.), a successful, herbicide-resistant, annual grass weed of winter cereal fields. Mol Ecol. 2007;16:3161–72.
Brazauskas G, Lenk I, Pedersen MG, Studer B, Lubberstedt T. Genetic variation, population structure, and linkage disequilibrium in European elite germplasm of perennial ryegrass. Plant Sci. 2011;181:412–20.
Wu WD, Liu WH, Sun M, Zhou JQ, Liu W, Zhang CL, et al. Genetic diversity and structure of Elymus tangutorum accessions from western China as unraveled by AFLP markers. Hereditas. 2019;156:8.
Xiong Y, Xiong Y, Yu Q, Zhao J, Lei X, Dong Z, et al. Genetic variability and structure of an important wild steppe grass Psathyrostachys juncea (Triticeae: Poaceae) germplasm collection from north and Central Asia. Peer J. 2020;8:e9033.
Wright S. Evolution and the genetics of population, Vol. 4. In: Variability within and among natural populations. Chicago: University of Chicago Press; 1978. p. 97–159.
Slatkin M. Gene flow in natural populations. Annu Rev Ecol Syst. 1985;16:393–430.
Groeneveld L, Lenstra J, Eding H, Toro M, Scherf B, Pilling D, et al. Genetic diversity in farm animals – a review. Anim Genet. 2010;41:6–31.
Daniels J, Roach BT. Taxonomy and evolution. In: Heinz DJ, editor. Sugarcane improvement through breeding. Amsterdam: Elsevier; 1987. p. 7–84.
Soltis PS, Soltis DE. The role of hybridization in plant speciation. Annu Rev Plant Biol. 2009;60:561–88.
Robertson A, Rich TCG, Allen AM, Houston L, Roberts C, Bridle JR, et al. Hybridization and polyploidy as drivers of continuing evolution and speciation in Sorbus. Mol Ecol. 2010;19:1675–90.
Zhang J, Yan J, Shen X, Chang D, Bai S, Zhang Y, et al. How genetic variation is affected by geographic environments and ploidy level in Erianthus arundinaceus? PLoS One. 2017;12:e0178451.
Fu YB, Allaby RG. Phylogenetic network of Linum species as revealed by non-coding chloroplast DNA sequences. Genet Resour Crop Evol. 2010;57:667–77.
Yang S, Zeng K, Chen K, Wu J, Wang Q, Li X, et al. Chromosome transmission in BC4 progenies of intergeneric hybrids between Saccharum spp. and Erianthus arundinaceus (Retz.) Jeswiet. Sci Rep. 2019;9:2528.
Pachakkil B, Terajima Y, Ohmido N, Ebina M, Irei S, Hayashi H, et al. Cytogenetic and agronomic characterization of intergeneric hybrids between Saccharum spp hybrid and Erianthus arundinaceus. Sci Rep. 2019;9:1748.
Sugimoto A, Ponragdee W, Sansayawichai T, Kawashima T, Thippayarugs S, Suriyaphan P, et al. Collecting and evaluating of wild relatives of sugarcane as breeding materials of new type sugarcane cultivars of cattle feed in Northeast Thailand. JIRCAS Working Rep. 2002;30:55–60.
Hijmans RJ, Guarino L, Bussink C, Mathur P, Cruz M, Barrentes I, et al. DIVA-GIS 7.5. A geographic information system for the analysis of species distribution data. 2012. http://www.diva-gis.org. Accessed 21 Aug 2019.
Schuelke M. An economic method for the fluorescent labeling of PCR fragments. Nature Biotech. 2000;18:233–4.
Roldan-Ruiz I, Dendauw JE, Van Bockstaele E, Depicker A, Loose M. AFLP markers reveal high polymorphic rates in ryegrasses (Lolium spp). Mol Breed. 2000;6:125–6.
Powell W, Margenta M, Andre C, Hanfrey M, Vogel J, Tingey S, et al. The comparison of RFLP, RAPD, AFLP and SSR (microsatellite) markers for germplasm analysis. Mol Breed. 1996;2:225–38.
de Jesus ON, Silva SO, Amorim EP, Ferreira CF, de Campos JMS, de Gaspari SG, et al. Genetic diversity and population structure of Musa accessions in ex situ conservation. BMC Plant Biol. 2013. https://doi.org/10.1186/1471-2229-13-41.
Prevost A, Wilkinson MJ. A new system of comparing PCR primers applied to ISSR fingerprinting of potato cultivars. Theor Appl Genet. 1999;98:107–12.
Tsuruta S, Ebina M, Kobayashi M, Takahashi W. Complete chloroplast genomes of Erianthus arundinaceus and Miscanthus sinensis: comparative genomics and evolution of the Saccharum complex. PLoS One. 2017;12:e0169992.
Pritchard JK, Stephens M, Donnelly P. Inference of population structure using multilocus genotype data. Genetics. 2000;155:945–59.
Falush D, Stephens M, Pritchard JK. Inference of population structure: extensions to linked loci and correlated allele frequencies. Genetics. 2003;164:1567–87.
Falush D, Stephens M, Pritchard JK. Inference of population structure using multilocus genotype data: dominant markers and null alleles. Mol Ecol Notes. 2007;7:574–8.
Evanno G, Regnaut S, Goudet J. Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol. 2005;14:2611–20.
Earl DA, von Holdt BM. STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conser Genet Res. 2012;4:359–61.
Jakobsson M, Rosenberg NA. CLUMPP: a cluster matching and permutation program for dealing with label switching and multimodality in analysis of population structure. Bioinformatics. 2007;23:1801–6.
Rosenberg NA. DISTRUCT: a program for the graphical display of population structure. Mol Ecol Notes. 2004;4:137–8.
Bruvo R, Michiels NK, D’Souza TG, Schulenburg H. A simple method for the calculation of microsatellite genotype distances irrespective of ploidy level. Mol Ecol. 2004;13:2101–6.
Saito N, Nei M. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol. 1987;6:514–25.
Langella, O. Populations 1.2.32. 1999. http://bioinformatics.org/~tryphon/populations/. Accessed 31 May 2017.
Rambaut A. FigTree ver.1.4.2. 2014. http://tree.bio.ed.ac.uk/software/figtree/. Accessed 19 Jan 2017.
Hardy OJ, Vekemans X. SPAGeDi: a versatile computer program to analyse spatial genetic structure at the individual or population levels. Mol Ecol Not. 2002;2:618–20.
Slatkin M. A measure of population subdivision based on microsatellite allele frequencies. Genet. 1995;139:457–62.
Gaggiotti OE, Lange O, Rassmann K, Gliddon C. A comparison of two indirect methods for estimating average levels of gene flow using microsatellite data. Mol Ecol. 1999;8:1513–20.
Wright S. Isolation by distance. Genetic. 1943;28:114–38.
Ersts PJ. Geographic Distance Matrix Generator (version1.2.3). American Museum of Natural History, Center for Biodiversity and Conservation. 2013. http://biodiversityinformatics.amnh.org/open_source/gdmg. Accessed 21 Aug 2019.
Mantel NA. The detection of disease clustering and a generalized regression approach. Cancer Res. 1967;27:209–20.
Meirmans PG, van Tienderen PH. GENOTYPE and GENODIVE: two programs for the analysis of genetic diversity of asexual organisms. Mol Ecol Not. 2004;4:792–4.
Hall TA. BioEdit 7.2.5. Biological sequence alignment editor and analysis program for Windows 95/98/NT. 2013. http://www.mbio.ncsu.edu/bioedit/bioedit.html. Accessed 22 Aug 2017.
Nei M. Molecular evolutionary genetics. New York: Columbia Univ. Press; 1987.
Watterson GA. On the number of segregation sites in genetical models without recombination. Theor Popul Biol. 1975;7:256–76.
Tajima F. The amount of DNA polymorphism maintained in a finite population when the neutral mutation rate varies among sites. Genetics. 1996;143:1457–65.
Fu YX, Li WH. Statistical tests of neutrality of mutations. Genetics. 1993;133:693–709.
Fu YX. Statistical tests of neutrality of mutations against population growth, hitchhiking and background selection. Genetics. 1997;147:915–25.
Tajima F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics. 1989;123:585–95.
Librado P, Rozas J. DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics. 2009;25:1451–2.
Bandelt HJ, Forster P, Röhl A. Median-joining networks for inferring intraspecific phylogenies. Mol Biol Evol. 1999;16:37–48.
Acknowledgements
We thank Tida Chernram and Sangdaum Chanachai of the KKFCRC for their great help in the maintaining and sampling of Erianthus accessions and DNA extraction. We also thank the KKFCRC staff for technical assistance with the experiments. Miho Innami at the NARO and Maki Hoshi at JIRCAS made valuable contributions to the sequencing and genotyping data analysis.
Funding
This work was supported by a research grant from the Japan International Research Center for Agricultural Sciences (Project No. B0000a1B3). The funder had no role in the design of the study, data collection and analysis, interpretation of data, or in writing the manuscript.
Author information
Authors and Affiliations
Author notes
Makoto Kobayashi is deceased.
- Makoto Kobayashi
Contributions
ST, ME, MK, and SSa contributed to the overall project conception and design. AT, WP, and YT managed Erianthus germplasms. SSr, YT, and ST collected leaf samples for DNA extraction. SSr and ST extracted DNA and conducted genotyping and sequencing. ME and SSa coordinated analysis of sequencing and genotyping data. ST analyzed and interpreted the data and drafted the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Table S1
. Characteristics of 28 SSR loci in 121 Thai Erianthus accessions.
Additional file 2: Table S2
. Sampling locations of 121 Erianthus accessions collected in Thailand, their grouping by each analysis, and size of amplified fragment in each accession.
Additional file 3: Table S3
. GenBank accession numbers of three non-coding regions of chloroplast DNA and their concatenated sequences in 121 Thai Erianthus accessions.
Additional file 4: Table S4
. Statistical analysis of haplotype diversity, nucleotide diversity, and neutrality in each chloroplast DNA region in 121 Thai Erianthus accessions.
Additional file 5: Table S5
. Sequences of primers used for amplification and sequencing of chloroplast DNA regions in Thai Erianthus accessions.
Additional file 6: Figure S1
. Geographic locations of 121 Erianthus accessions collected in Thailand, and a pie chart of the populations and a bar chart of ancestry proportion in the 7 admixtures. Colors correspond to those in Fig. 1 at K = 4. Admixture group is indicated in gray. Accession numbers are listed as map No. in Table S2.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Tsuruta, Si., Srithawong, S., Sakuanrungsirikul, S. et al. Erianthus germplasm collection in Thailand: genetic structure and phylogenetic aspects of tetraploid and hexaploid accessions. BMC Plant Biol 22, 45 (2022). https://doi.org/10.1186/s12870-021-03418-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12870-021-03418-3