
Abstract
Background
Auxin is critical to plant growth and development, as well as stress responses. Small auxin-up RNA (SAUR) is the largest family of early auxin responsive genes in higher plants. However, the function of few SAUR genes is known owing to functional redundancy among the many family members.
Results
In this study, we conducted a phylogenetic analysis using protein sequences of 795 SAURs from Anthoceros angustus, Marchantia polymorpha, Physcomitrella patens, Selaginella moellendorffii, Ginkgo biloba, Gnetum montanum, Amborella trichopoda, Arabidopsis thaliana, Oryza sativa, Zea mays, Glycine max, Medicago truncatula and Setaria italica. The phylogenetic trees showed that the SAUR proteins could be divided into 10 clades and three subfamilies, and that SAUR proteins of three bryophyte species were only located in subfamily III, which suggested that they may be ancestral. From bryophyta to anthophyta, SAUR family have appeared very large expansion. The number of SAUR gene in Fabaceae species was considerably higher than that in other plants, which may be associated with independent whole genome duplication event in the Fabaceae lineages. The phylogenetic trees also showed that SAUR genes had expanded independently monocotyledons and dicotyledons in angiosperms. Conserved motif and protein structure prediction revealed that SAUR proteins were highly conserved among higher plants, and two leucine residues in motif I were observed in almost all SAUR proteins, which suggests the residues plays a critical role in the stability and function of SAUR proteins. Expression analysis of SAUR genes using publicly available RNA-seq data from rice and soybean indicated functional similarity of members in the same clade, which was also further confirmed by qRT-PCR. Summarization of SAUR functions also showed that SAUR functions were usually consistent within a subclade.
Conclusions
This study provides insights into the evolution and function of the SAUR gene family from bryophyta to anthophyta, particularly in Fabaceae plants. Future investigation to understand the functions of SAUR family members should employ a clade as the study unit.
Similar content being viewed by others
Background
As sessile organisms, plants must adapt to environmental variation by integrating developmental and environmental signals. Auxin was the first plant hormone to be identified, and plays important roles in plant growth, development and stress responses [1]. Three gene families are regarded as early and primary auxin-responsive genes: Auxin/Indoleacetic Acid (Aux/IAA), Gretchen Hagen 3 (GH3) and Small Auxin-Up RNA (SAUR) [2, 3]. SAUR genes can be induced originally by exogenous auxin within 2–5 min [4]. Interestingly, the majority of SAUR genes lack an intron [3, 5], and may be regulated at a number of levels; for example, SAUR genes can be regulated post-transcriptionally owing to a highly conserved downstream element (DST) located in the 3′-untranslated region, which leads to mRNA instability in an auxin-independent manner [1, 6,7,8].
The first SAUR gene was identified from elongating hypocotyls of auxin-treated soybean (Glycine max) [9]. Over the past 30 years, members of the SAUR gene family have been identified in many other species of angiosperms and thus comprise a large gene family in plants, such as Arabidopsis thaliana (81 SAUR genes, including two pseudogenes) [2], Oryza sativa (58 including two pseudogenes) [10], Zea mays (79) [3], Gossypium raimondii (145) [11], Citrus sinensis (70) [12], Phyllostachys edulis (44) [13], Boehmeria nivea (71) [14] and Citrullus lanatus (65) [15]. Many members of the SAUR gene family are a result of a high frequency of tandem and segmental duplications, and which have contributed to functional redundancy among the paralogues [6].
Until now, several SAUR genes have been shown to play roles in diverse processes of plant growth, development and stress responses. For example, AtSAUR63 promotes cell elongation, thus resulting in longer hypocotyls, stamen filaments, petals and inflorescence stems [16]. Overexpression of AtSAUR36 and AtSAUR49 promotes leaf senescence [17,18,19]. The atsaur62 and atsaur75 mutants show normal pollen viability but defective pollen tube growth in vitro and in vivo [20]. The SAUR50-like protein is involved in heliotropic movements in the common sunflower (Helianthus annuus) [21]. Many SAUR homologues play crucial roles in circadian floral opening and closure in waterlily [22, 23]. The SAUR41 subfamily is inducible by abscisic acid to modulate cell expansion and salt tolerance in Arabidopsis thaliana seedlings [24]. The thermo-responsive AtSAUR26 subfamily exhibits a high frequency of gene variation associated with adaption to local temperature climate [25].
To improve our understanding of the evolution and functions of the SAUR gene family in plants, in this study we first conducted a phylogenetic analysis using 795 SAUR protein sequences from Anthoceros angustus, Marchantia polymorpha, Physcomitrella patens, Selaginella moellendorffii, Ginkgo biloba, Gnetum montanum, Amborella trichopoda, Arabidopsis thaliana, Oryza sativa, Zea mays, Setaria italica, Glycine max and Medicago truncatula. Conserved motif and protein structure prediction and gene expression profiling of the SAUR family were performed to explore possible functions of SAUR genes. In addition, we summarized functions of all identified SAUR genes.
Results
SAUR protein identification from alga to higher plant species
We identified the SAUR protein family members from the following plant species in BLAST searches of the Phytozome 12 database: Physcomitrella patens (18 members), Arabidopsis thaliana (81, including two pseudogenes), Oryza sativa (58, including two pseudogenes), and maize (79). This was consistent with the results of previous studies [2, 3, 10, 26]. Meanwhile, we firstly identified 3, 5 and 15 SAUR protein sequences in Anthoceros angustus, Marchantia polymorpha and Selaginella moellendorffii, respectively. In gymnosperm, SAUR family members of Ginkgo biloba (42) and Gnetum montanum (37) were firstly identified. For anthophyta, 26 and 58 SAUR protein sequences were firstly identified in Amborella trichopoda and Setaria italica, respectively. Interestingly, up to 141 and 236 SAUR protein sequences were identified in Medicago truncatula and soybean. These results indicated that SAUR family members had expanded largely in the process of evolution. In addition, none SAUR homolog was found in genomes of the seven chlorophyte species (Chlamydomonas reinhardtii, Dunaliella salina, Volvox carteri, Coccomyxa subellipsoidea C-169, Micromonas pusilla CCMP1545, Micromonas sp. RCC299 and Ostreococcus lucimarinus) and four recently annotated streptophyte algae species (Spirogloea muscicola, Mesotaenium endlicherianum, Mesostigma viride and Chlorokybus atmophyticus) using SAUR protein sequences of Arabidopsis thaliana as queries. All identified SAUR protein sequences from different species were used for downstream analysis (Supplementary Table 1).
The encoded SAUR proteins were polypeptides of 55–423 amino acids in length, with a predicted molecular mass range of 6.38–46.81 kDa and the theoretical isoelectric point (pI) ranged from 4.58 to 12. In further analysis, we used the Plant-mPLoc server (http://www.csbio.sjtu.edu.cn/bioinf/plant-multi/) [27] to predict the probable protein localization of identified SAURs. More than 60.8% of the SAUR proteins contained a nucleus-targeting signal and the other proteins were predicted to be localized to the cell membrane, cytoplasm, chloroplasts or mitochondria, etc. (Supplementary Table 2).
Phylogenetic analysis of SAUR proteins
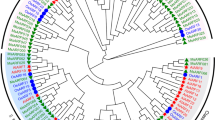
To gain insights into the evolution of SAUR genes in plants, we used SAUR protein sequences, excluding pseudogenes, from Anthoceros angustus (3), Marchantia polymorpha (5), Physcomitrella patens (18), Selaginella moellendorffii (15), Ginkgo biloba (42), Gnetum montanum (37), Amborella trichopoda (26), Arabidopsis thaliana (79), Oryza sativa (56), Zea mays (79), Setaria italica (58), Glycine max (236), and Medicago truncatula (141) to construct two phylogenetic trees for the SAUR protein family via FastTree v2.1 and IQ-TREE v2.0.6 respectively. Based on these two trees, we divided the SAUR proteins into three subfamilies and 10 clades designated groups 1 to 10, although there was a slight difference in several clades between the two trees (Fig. 1, Supplementary Fig. 1, Supplementary Datasheets 1, 2). All SAUR proteins of three bryophyte species were placed in two clades of subfamily III of the two trees, which suggested that these clades may be ancestral. The majority of monocotyledon and eudicotyledon sequences were each grouped together in one clade, which indicated that the SAUR gene family expanded independently in monocotyledons and eudicotyledons. Most SAUR members of Ginkgo biloba, Gnetum montanum had formed independent branches in parallel with ones of anthophyta, which was consistent with their evolutionary status. Most of members of clade 1 in both FastTree and IQ-TREE trees were proteins from soybean and Medicago truncatula (Fig. 1, Supplementary Fig. 1). This may be a result of gene duplication in the clade or independent genome replication among Fabaceae plants. In addition, both FastTree and IQ-TREE trees showed that the Fabaceae species formed a distinct evolutionary lineage (clade 1) and the number of SAUR protein members of eudicotyledons was roughly equal in their other shared branches (Fig. 1, Supplementary Fig. 1). We further reconstructed the phylogenetic tree using SAUR gene family members from Mimosa pudica and Cercis canadensis, which were sister taxa outside of Fabaceae family, together with Arabidopsis thaliana, Glycine max, Medicago truncatula. The phylogenetic tree could be divided ten clades, and SAUR gene family from Medicago truncatula and Glycine max only had extremely large expansion in Clade 1 (Supplementary Fig. 2). Synteny analysis is a useful tool for establishing both orthology relationships and functional linkages between genes. Then we performed synteny analysis of SAUR genes from Arabidopsis thaliana, Glycine max and Medicago truncatula (Supplementary Fig. 3). The results showed that SAUR gene cluster at a certain chromosome of Arabidopsis thaliana had evident synteny relationships with multiple SAUR gene clusters at multiple chromosomes of Medicago truncatula and Glycine max. These results suggested that the occurrence of many SAUR members in Fabaceae resulted from independent genome replication in Fabaceae.

Maximum likelihood phylogenetic tree of small auxin-up RNAs (SAURs) in the thirteen plant species. The tree was constructed from multiple sequences alignment of all SAUR proteins from Anthoceros angustus, Marchantia polymorpha, Physcomitrella patens, Selaginella moellendorffii, Ginkgo biloba, Gnetum montanum, Amborella trichopoda, Arabidopsis thaliana, Oryza sativa, Zea mays, Setaria italica, Glycine max and Medicago truncatula using the maximum likelihood method in FastTree v2.1 with the JTT + CAT model. Then SAUR members were clarified in FigTree v1.4.4. There SAUR family had been divided into three subfamily and 10 clades. Bootstrap support rates were labeled at corresponding branches. SAUR subclades reported and members mentioned in the article were also labeled on it. Branches leading to genes from the different phyla were colored according to the simplified phylogeny of land plants that is shown in the top left corner. Different colorful dots were used to represent SAUR members from different species
Conserved core region and structure specific to SAUR proteins
Multiple sequence alignment of all SAUR proteins from Anthoceros angustus, Marchantia polymorpha, Physcomitrella patens, Selaginella moellendorffii, Ginkgo biloba, Gnetum montanum, Amborella trichopoda, Arabidopsis thaliana, Oryza sativa, Zea mays, Glycine max, Medicago truncatula and Setaria italica revealed that the sequences shared a common conserved core region of ~ 60 amino acid residues, which is regarded as the SAUR domain (Figs. 2, 3), consistent with previous reports [1, 6]. In addition, we observed that the majority of SAUR members in eudicotyledons contained a larger number of conserved amino acid sites compared with monocotyledons generally. Two leucine residues, which formed a hydrophobic core, were observed in almost all SAUR protein sequences, which suggested the residues play a critical role in the folding and basic functions of SAUR proteins (Fig. 2).

The conserved sequence of SAUR proteins from the thirteen plant species. 795 SAUR proteins from Anthoceros angustus, Marchantia polymorpha, Physcomitrella patens, Selaginella moellendorffii, Ginkgo biloba, Gnetum montanum, Amborella trichopoda, Arabidopsis thaliana, Oryza sativa, Zea mays, Setaria italica, Glycine max and Medicago truncatula were used to carry out multiple sequence alignment in Clustal Omega. The amino acid residues at SAUR domain (~ 60 amino acid residues) were labeled in different color. Red arrows indicated highly conserved leucine residues positions

Sequence analysis and structure prediction of SAUR proteins in clade 2. a Multiple sequence alignment of SAUR proteins in clade 2 using Clustal Omega. Some nonconserved insertions of a few SAUR members were removed from the alignment. The dashed grey lines represented vacant sites (no amino acid residues) in multiple sequences alignment. The region of the SAUR domain was labeled in a black box. b The predicted tertiary structure of AtSAUR8 and OsSAUR8 using conserved domain only
We identified five motifs within the conserved regions among the SAUR proteins from Physcomitrella patens, Arabidopsis thaliana, Oryza sativa, Zea mays, Setaria italica, Glycine max and Medicago truncatula (Supplementary Fig. 4). The majority of SAUR members contained motifs I to IV and ~ 20% of the SAUR sequences contained motif V. It is noteworthy that SAUR proteins of Physcomitrella patens only contained motifs I to III, indicating that SAUR proteins in higher plants have evolved novel motifs. However, the functions of these motifs are currently unknown and require further study.
Previous research showed that members of the AtSAUR10 subclade in clade 2 of the two trees induce cell elongation (Fig. 1, Supplementary Fig. 1) [28]. SAUR members that show a close homologous relationship with AtSAUR10 in clade 2 were highly conserved, although the N- and C-terminal regions were less conserved than the core region (Fig. 3a). The majority of SAUR proteins shown in Fig. 3a were predicted to localize in chloroplasts (Supplementary Table 2), which may be associated with the similarity of the N- or C-terminal regions. The conserved sequences of AtSAUR8 and OsSAUR8 displayed similar tertiary structures and similar motifs (Fig. 3b), as predicted using the SWISS-MODEL server [29] and subjected to molecular dynamics simulation using GROMACS2019.1 (http://manual.gromacs.org/2019.1/index.html) [30] for structure optimization: motif I developed a β-sheet, motif II formed a typical α-helix, motif III formed a β-sheet, and motif IV was the linker between motif I and motif II (Fig. 3b), which indicated that the functions of AtSAUR8 and OsSAUR8 may be similar. However, the precise functions of each motif in maintaining protein stability or activities require further study.
Expression analysis of SAUR genes
Expression pattern analysis is a useful tool for understanding gene functions. A comprehensive expression analysis was performed using RNA-seq data to explore expression profiles of SAUR genes in rice and soybean (Fig. 4, Supplementary Fig. 5). The expression level was estimated for all OsSAUR genes detected from 11 tissues and organs, except for eight genes (OsSAUR39, OsSAUR37, OsSAUR4, OsSAUR48, OsSAUR47, OsSAUR46, OsSAUR33 and OsSAUR21) (Fig. 4). The majority of OsSAUR genes in clade 5 of the two trees were highly expressed in the vegetative phase but were weakly expressed in the reproductive phase, which showed a good agreement with their chloroplast localization (Figs. 1, 4, Supplementary Fig. 1, Supplementary Table 2). OsSAUR20 and OsSAUR27 of clade 8 of the two trees were expressed highly in the embryo at 25 days after pollination (DAP) (Figs. 1, 4, Supplementary Fig. 1), which suggested members of this clade are involved in embryo development in rice. Almost all GmSAUR genes in clade 3 of FastTree tree (clade 6 of IQ-TREE tree) showed an identical expression pattern, namely expressed highly in seeds within 25 ~ 42 days after flowering (DAF) and weakly in the pod shell and flower within 14 DAF (Fig. 1, Supplementary Figs. 1, 5). Four GmSAUR genes (Glyma. 3 g259700, Glyma. 7 g051700, Glyma. 16 g020700 and Glyma. 1 g137500) in clade 4 of the two trees were predominantly expressed in the seed (Fig. 1, Supplementary Figs. 1, 3), which implied that members of this clade were involved in seed development. Compared with OsSAUR genes, a greater number of GmSAUR genes showed a similar expression pattern. In addition, based on digital gene expression libraries, we observed that the expression of many OsSAUR genes was induced by abiotic and biotic stresses, especially salt, drought and rice blast (Supplementary Fig. 6), which indicated that many SAUR genes mediate stress responses, although few studies have investigated such functions to date. Then we investigated the expression patterns of OsSAUR genes in clade 6 of FastTree tree (clade 7 of IQ-TREE tree) treated by dark. The results showed that OsSAUR3 and OsSAUR10 were highly expressed in etiolated seedlings (Fig. 5a). Jain et al. (2006) observed that the expression levels of OsSAUR7 in the same clade were also significantly higher in etiolated seedlings than in normal seedlings [10], which indicated they play similar roles in light and hormone responses. These results indicated that the expression patterns of members of the same clade were often similar.

OsSAURs gene expression patterns from 11 different development tissues or organ systems by RNA-seq. S: Shoot; FSS: Four-leaf Stage Seedling; TL: Twenty-day Leaf; PoI: Post-emergence Inflorescence; PrI: Pre-emergence Inflorescence; A: Anther; P: Pistil; FDS: Five DAP Seed; TDEm: Twenty-five DAP Embryo; TDS: Ten DAP Seed; TDEn: Twenty-five DAP Endosperm; DAP: Days After Pollination

Expression analysis of small auxin-up RNA (SAUR) genes in rice. a Relative expression level of OsSAUR3 and OsSAUR10 in clade 6 of FastTree tree (clade 7 of IQ-TREE tree) from leaf blade of normal seedings and etiolated seedings under different days (ndL, n-day-old light-grown seedings, n = 3, 6, 9, 12; 3dD, 3-day-old etiolated seedings, etc.). b Relative expression level of the seven OsSAUR genes in rice seedlings treated by 10 μM IAA. The expression of OsUBQ5 was used as an internal control. Data were mean ± SE from three biological replicates. * indicate statistically significant differences by student t-test: P < 0.05. ** P < 0.01
To clarify the response of SAUR genes to auxin, we also investigated the expression patterns of OsSAURs genes selected randomly from different clades in the leaf blade of rice seedlings treated with 10 μM IAA. The results revealed extremely diverse auxin-responsive expression patterns (Fig. 5b). Three genes (OsSAUR33, OsSAUR11 and OsSAUR5) were induced rapidly at 5 min after auxin treatment, whereas three genes (OsSAUR17, OsSAUR36 and OsSAUR38) were strongly inhibited within 5 min by exogenous auxin. Strangely, OsSAUR57 was induced rapidly at 5 min and then was strongly suppressed at 30 min after 10 μM IAA treatment. These results showed that expression of SAUR genes was closely associated with exogenous auxin application time and indicated that SAUR genes play an extremely complex role in the auxin-mediated signal pathway.
Functions of identified SAUR genes
To explore SAUR functions, we summarized the functions of all currently identified SAUR genes (Fig. 6, Supplementary Table. 3). Although SAUR proteins influence diverse aspects of plant growth and development, the molecular mechanisms could be summarized in relatively few processes. The AtSAUR19-subclade and AtSAUR26-subclade in clade 1 of the two trees inhibit PP2C-D phosphatases and then activate plasma-membrane H+-ATPase activity to promote cell expansion (Figs. 1, 6, Supplementary Fig. 1) [25, 31]. The AtSAUR76-subclade in clade 9 of the two trees mediates ethylene signaling via interaction with Ethylene Insensitive 4 (EIN4) and Ethylene Receptor 2 (ETR2) to promote plant growth (Figs. 1, 6, Supplementary Fig. 1) [32]. AtSAUR49 activates Senescence-Associated Receptor-Like Kinase (SARK)-mediated leaf senescence signaling by suppression of Senescence Suppressed Protein Phosphatase (SSPP) [19]. AtSAUR70 binds to calmodulin in a calcium-dependent manner in vivo [42], which is indicative of a link between auxin signaling and the second messenger Ca2+/calmodulin. OsSAUR39 overexpression negatively regulates auxin biosynthesis and transport [43]. In addition, AbSAUR1 overexpression significantly improves the overall yield of tropane alkaloids, which are regarded as anticholinergic drugs in the clinic, attaining a yield 3.55 times that of the control [44]. The researchers also observed that the genes in the same evolutionary lineage showed similar functions, and thus investigated the SAUR genes group as one unit, for example, the AtSAUR19-subclade and AtSAUR26-subclade in clade 1, AtSAUR10-subclade in clade 2, AtSAUR63-subclade in clade 5, AtSAUR41-subclade in clade 4, and AtSAUR76-subclade in clade 9 of the two trees (Figs. 1, 6, Supplementary Fig. 1).

The simplified schema and functions of SAUR genes in plant evolution. During the process of plant evolution, SAUR gene family from bryophyta to anthophyta expended largely, and every clade experienced expansion at different degrees (left panel). Right panel of Fig. 6 showed the function of SAUR genes in response to hormonal and environmental signals: based on acid growth mediated by plasma membrane H+-ATPase, auxin-responsive SAUR proteins (AtSAUR 19- and AtSAUR 26-subclades in clade 1) activate plasma membrane H+-ATPase by inhibiting PP2C-D phosphatases to promote cell expansion [31]. The AtSAUR76-subclade in clade 9 mediates ethylene signaling via interaction with Ethylene Insensitive 4 (EIN4) and Ethylene Receptor 2 (ETR2) to promote plant growth [32]. The SAUR41-subclade is inducible by abscisic acid to modulate cell expansion and salt tolerance, and maintain calcium homeostasis [24]. Auxin Response Factor 6 (ARF6), Brassinazole Resistant1 (BZR1) and Phytochrome Interacting Factor 4 (PIF4) may directly regulate transcription of AtSAUR19-subclade and AtSAUR26-subclade independently or as a complex [33] sensing hormonal and environmental signals including auxin, brassinosteroids (BR), gibberellin (GA), jasmonic acid (JA), light, heat and shade [25, 28, 33,34,35,36,37,38,39,40]. Arabidopsis zinc-finger protein 1 (AZF1) and AZF2 in the ABA-response pathway can repress the expression of AtSAUR63 subclade [41]
Discussion
In the process of adaptation to diverse terrestrial environments, plants have evolved a suite of hormones to respond to developmental and environmental signals [45]. Auxin signaling occurs later in evolution compared with cytokinin and ethylene, and was initially discovered in streptophyte algae [46, 47]. But none SAUR homolog was found in streptophyte algae and other alga, speculating that SAUR proteins appeared relatively later. SAUR genes of bryophyte were first identified in Physcomitrella patens [26]. In the present study we first identified SAUR genes from the reference genome sequences for Anthoceros angustus (3), Marchantia polymorpha (5), Selaginella moellendorffii (15), Ginkgo biloba (42), Gnetum montanum (37), Setaria italica (58), Mimosa pudica (62), Cercis canadensis (48) Medicago truncatula (141), and Glycine max (236). So large expansion of SAUR family from bryophyta to anthophyta should contribute to greatly balance developmental and environmental signals, which is critical for blossom of terrestrial plant (Fig. 6). Compared with the number of SAUR genes in other dicotyledons, a greater number are present in the genomes of soybean and Medicago truncatula (Figs. 1, 6, Supplementary Fig. 1). Combined with SAUR gene synteny analysis between Fabaceae and Arabidopsis thaliana (Supplementary Fig. 3), which suggested that SAUR family expansion might be related to whole genome duplications in Fabaceae plants [48]. We observed less expansion in monocotyledons than in eudicotyledons in general, which was also verified by previous studies on SAUR gene number in plant species such as cotton (145) [11], citrus (70) [12], bamboo (44) [13], ramie (71) [14] and watermelon (65) [15] (Supplementary Fig. 7).
Understanding protein structure is vital to determining the function of a protein and its interaction with other proteins. Prediction of protein structure has profound theoretical and practical influences on biological research [49]. In the present study, we identified five conserved motifs among SAUR proteins and observed that the majority of SAURs contain motifs I to IV and ~ 20% of SAUR family members contain motif V (Supplementary Fig. 4). This result is similar to predictions for cotton [11]. The tertiary structures of AtSAUR8 and OsSAUR8 showed that motifs I to IV generated a similar structure (Fig. 3b), however, the fine functions of each motif in maintaining protein stability and activities require further study. In addition, we observed that two leucine residues in motif I located at the hydrophobic core positions were present in almost all SAUR proteins (Fig. 2), and might be involved in protein interactions, such as with PP2C.D [31]. However, their critical role in the biochemical function of SAUR proteins needs to be confirmed by conducting point-mutation genetic experiments in the future.
The functions of SAUR proteins remain mysterious despite previous notable breakthroughs (Fig. 6, Supplementary Table 3). We examined the expression level of seven randomly chosen OsSAUR genes in response to IAA treatment. The expression level of four genes was upregulated, whereas three genes were downregulated, at 5 min after IAA treatment (Fig. 5b). Digital gene expression analysis also suggested that many OsSAUR genes are responsive to salt, drought and rice blast (Supplementary Fig. 4). The function of SAUR genes in resistance to rice blast is worth further study. A recent study reported that SlSAUR69 enhances the sensitivity of tomato fruit to ethylene through repression of polar auxin transport to influence the unripening-to-ripening transition, which revealed that SlSAUR69 mediates crosstalk between auxin and ethylene [50]. The AtSAUR26 subfamily, which was identified as a QTL for growth thermo-responsiveness, shows temperature-related natural polymorphisms that influence the thermo-responsiveness of plant architecture for adaptation to the local temperature [25]. These results indicated that SAUR genes from different clades participate in a multitude of processes involved in plant growth and stress response.
The present expression analysis of SAUR genes showed that the expression pattern was similar among members of the same clade. For example, OsSAUR3, OsSAUR10 and OsSAUR7 of clade 6 of FastTree tree (clade 7 of IQ-TREE tree) were highly expressed in etiolated seedlings (Fig. 5a) [10]. The majority of OsSAUR genes in clade 5 were highly expressed in the vegetative phase and localized in chloroplasts (Fig. 4, Supplementary Table 2). Four GmSAUR genes (Glyma.3 g259700, Glyma.7 g051700, Glyma.16 g020700 and Glyma.1 g137500) in clade 4 were predominantly expressed in the seed (Fig. 1, Supplementary Fig. 1, Supplementary Fig. 5). These results indicated that genes in the same evolutionary lineage show functional similarity, which is confirmed by functional experiments. For example, the AtSAUR19-subclade and AtSAUR26-subclade in clade 1 are involved in so-called “acid growth” by invoking plasma membrane H+-ATPase activation [51], and the AtSAUR41-subclade in clade 4 maintains calcium homeostasis and modulates drought and salt tolerance (Figs. 1, 6, Supplementary Fig. 1).
Conclusions
In this study, we present a phylogenetic framework for the SAUR gene family from thirteen plant species. The SAUR gene family shows notable expansion from bryophyta to anthophyta, particularly in Fabaceae plants, and can be divided into three subfamilies and 10 clades (Fig. 1, Supplementary Fig. 1, Supplementary Datasheets 1, 2). Multiple sequence alignment and motif and protein structure prediction indicate that SAUR proteins are highly conserved (Figs. 2, 3). SAUR proteins participate in a multitude of processes involved in plant growth and development. Although SAUR genes from different clades show diverse expression patterns, members of the same clade show functional similarity. Future elucidation of the functions of SAUR family members will require a concerted effort by adoption of diverse approaches, including molecular genetic analysis based on an evolutionary lineage as the study unit.
Methods
Identification and sequence analysis of SAUR family members
SAUR protein sequences of Arabidopsis thaliana were used as queries in a BLAST search (score > 50, E-value < 0.01) of the Phytozome 12 database (https://phytozome.jgi.doe.gov). Sequences for SAUR members from Arabidopsis thaliana, Glycine max, Medicago truncatula, Marchantia polymorpha, Physcomitrella patens, Selaginella moellendorffii and Amborella trichopoda were downloaded. Sequences were obtained for SAUR members in Oryza sativa, Zea mays and Setaria italica with the same approach but using SAUR protein sequences of Oryza sativa as queries. We used SAUR protein sequences of Arabidopsis thaliana as queries to search SAUR members identification of Anthoceros angustus, Gnetum montanum, Ginkgo biloba, Mimosa pudica and Cercis canadensis via local blastp (score > 50, E-value < 0.01) of ncbi-blast-2.10.1+ (ftp://ftp.ncbi.nlm.nih.gov/blast/executables/blast+/LATEST/). Protein sequences of these five species were downloaded from the DRYAD website (https://datadryad.org/stash/dataset/doi:10.5061/dryad.msbcc2ftv, https://datadryad.org/stash/dataset/doi:10.5061/dryad.0vm37) and the GigaScience GigaDB repository (https://db.cngb.org/search/literature/27871309, http://gigadb.org/dataset/101044, http://gigadb.org/dataset/101049) respectively. SAUR members identification of chlorophyte species (Chlamydomonas reinhardtii, Dunaliella salina, Volvox carteri, Coccomyxa subellipsoidea C-169, Micromonas pusilla CCMP1545, Micromonas sp. RCC299 and Ostreococcus lucimarinus) and streptophyte algae species (Spirogloea muscicola, Mesotaenium endlicherianum, Mesostigma viride and Chlorokybus atmophyticus) was carried out via a BLAST search using SAUR protein sequences of Arabidopsis thaliana as queries in Phytozome 12 database (https://phytozome.jgi.doe.gov) and NCBI database (https://www.ncbi.nlm.nih.gov) respectively. Seven hundred ninety-five SAUR protein sequences from Anthoceros angustus, Marchantia polymorpha, Physcomitrella patens, Selaginella moellendorffii, Ginkgo biloba, Gnetum montanum, Amborella trichopoda, Arabidopsis thaliana, Oryza sativa, Zea mays, Glycine max, Medicago truncatula and Setaria italica were aligned with clustalx 2.1 [52] and inspected manually with Jalview [53]. Conserved regions of SAUR gene family (~ 60 amino acids) were greatly aligned and there was local homology in non-conserved regions. Some extremely gapped positions were manually removed, and nearly all non-conserved regions with local homology were retained. Finally obtained sequence alignment was used to constructed FastTree and IQ-TREE tree. Physicochemical parameters of SAUR proteins were analyzed using ProtParam (http://web.expasy.org/protparam) [54]. The Plant-mPLoc online tool (http://www.csbio.sjtu.edu.cn/bioinf/plant-multi/) [27] was used to predict the subcellular localization of SAUR proteins.
Phylogenetic analysis of SAUR family
Phylogenetic analysis of the multiple sequence alignment of SAUR proteins was conducted. Phylogenetic trees were constructed using FastTree v2.1 [55] with the JTT + CAT model and IQ-TREE v2.0.6 [56] with the JTT + R9 model respectively. Support for each node was assessed by performing a bootstrap analysis with 1000 replicates.
Multiple sequence alignment and motif prediction of SAUR proteins
Protein sequences of SAUR family members were used to generate a multiple sequence alignment and visualization analyses using Clustal Omega (https://www.ebi.ac.uk/Tools/msa/clustalo) [57] and Jalview [53], respectively. Only the conserved portion of each sequence was retained, which was then applied for motif prediction using MEME (http://meme-suite.org).
Plant materials and growth conditions
Asian cultivated rice (Oryza sativa subsp. japonica cv. Nipponbare, of which was completed the genome sequencing in 2004) was used in this study. Collection of this rice variety was complied with the institutional and national guidelines in China, and seeds were stored in our lab. Rice seeds were treated and grown as described previously [58]. For auxin treatment, the 2-week-old seedlings were incubated in Yoshida nutrition solution [59] containing 10 μM IAA and then sampled at 5, 10, 15, 30 and 60 min, respectively. For dark treatment, the seedlings were grown in the plant incubator without light all the time and sampled at 3, 6, 9 and 12 day, respectively. Control (CK) plants were all grown in the plant incubator with normal condition.
Quantitative real-time PCR analysis
Total RNA was extracted with TRIzol Reagent from leaf blade of rice seedlings grown under the normal condition and different treatments. For plants, growing parts (such as root and leaf) often can produce a large amount of auxin to maintain the needs of plant growth. The genes OsSAUR3 and OsSAUR10 were chosen to examined to respond to light. So, RNA was used from leaf blade to run qRT-PCR. The cDNA was synthesized using the PrimeScript RT Reagent Kit with gDNA Eraser (Takara, Kyoto, Japan). PCR amplifications were performed using the TransStart Tip Green qPCR SuperMix (TransGen Biotech, Beijing, China) on the CFX96™ Real-Time PCR Detection System (Bio-Rad, Hercules, CA, USA). Gene-specific primers used in the experiments are listed in Supplementary Table 4. Three biological replicates were performed for each reaction. Relative gene expression levels were calculated from the qRT-PCR data using the 2−△△Ct method [60].
Expression analysis based on RNA-seq and digital gene expression data
The expression data were derived from the Rice Genome Annotation Project (http://rice.plantbiology.msu.edu/expression.shtml) and Soybase Database (https://www.soybase.org/soyseq/) [61]. These data were gene-wise normalized within different plant tissues and hierarchically clustered on the basis of Pearson correlation coefficients using the weighted pair group method with averaging linkage with Genesis (v1.7.6) software [62]. Digital gene expression data was visualized in Genesis using white and black color to represent “unexpressed” and “expressed” of genes.
Protein tertiary structure prediction and optimization
Prediction of the tertiary structure prediction of SAUR proteins was performed using SWISS-MODEL (https://swissmodel.expasy.org/) [29]. Sequence identities of AtSAUR8 and OsSAUR8 with templates were 23.40 and 21.15%, respectively. The predicted structures were subjected to MD simulation using GROMACS 2019.1 software package (http://manual.gromacs.org/2019.1/index.html) with the Charmm 27 force field and TIPS3P water model in a box large enough to enclose the whole protein, and then subjected to energy minimization with 50,000 steps of steepest descent. The minimized structure was equilibrated with NVT and NPT simulation in turn. Finally, we carried out 1 ns long NVT MD simulation and structural parameters like RMSD converged after 800 ps. Analysis of RMSD and Ramachandran plot for structure optimization were shown in Supplementary Fig. 8. Visualization of protein structures was conducted with NOC 3.01 (http://noch.sourceforge.net/).
Gene synteny analysis between Fabaceae plants and Arabidopsis thaliana
Genome assembly sequence and gene annotation of Arabidopsis thaliana, Glycine max and Medicago truncatula were download from Ensembl Plants database (http://plants.ensembl.org/index.html). The genes synteny analysis was built with MCScanX [63] program in TBtools [64]. Synteny relationship of SAUR genes was highlighted with blue lines.
Availability of data and materials
The datasets supporting the conclusions of this article are included within the article and its supplementary information files. The data of the multiple sequence alignment used for FastTree and IQ-TREE trees was available in TreeBASE Web (http://purl.org/phylo/treebase/phylows/study/TB2:S26122).
Abbreviations
- SAUR :
-
Small auxin-up RNA
- GH3 :
-
Gretchen hagen 3
- Aux/IAA :
-
Auxin/indoleacetic acid
- qRT-PCR:
-
Quantitative real-time PCR
- DGE:
-
Digital gene expression
- ML:
-
Maximum likelihood
- PP2C:
-
Type 2C protein phosphatase
- EIN:
-
Ethylene insensitive
- ETR:
-
Ethylene receptor
- SARK:
-
Senescence-associated receptor-like kinase
- SSPP:
-
Senescence suppressed protein phosphatase
- DAP:
-
Days after pollination
- DAF:
-
Days after flowering
- CK:
-
Cytokinin
- QTL:
-
Quantitative trait locus
- PM H+-ATPases:
-
Plasma membrane H+-ATPases
- ABA:
-
Abscisic acid
- BR:
-
Brassinosteroids
- GA:
-
Gibberellin
- JA:
-
Jasmonic acid
- AZF:
-
Arabidopsis zinc-finger protein
- ARF:
-
Auxin response factor
- BZR:
-
Brassinazole resistant
- PIF:
-
Phytochrome interacting factor
- S:
-
Shoot
- FSS:
-
Four-leaf stage seedling
- TL:
-
Twenty-day leaf
- PoI:
-
Post-emergence inflorescence
- PrI:
-
Pre-emergence inflorescence
- A:
-
Anther
- P:
-
Pistil
- FDS:
-
Five DAP seed
- TDEm:
-
Twenty-five DAP Embryo
- TDS:
-
Ten DAP seed
- TDEn:
-
Twenty-five DAP endosperm
- WGD:
-
Whole genome duplication
References
Ren H, Gray WM. SAUR proteins as effectors of hormonal and environmental signals in plant growth. Mol Plant. 2015;8(8):1153–64.
Hagen G, Guilfoyle T. Auxin-responsive gene expression: genes, promoters and regulatory factors. Plant Mol Biol. 2002;49(3–4):373–85.
Chen Y, Hao X, Cao J. Small auxin upregulated RNA (SAUR) gene family in maize: identification, evolution, and its phylogenetic comparison with Arabidopsis, rice, and sorghum. J Integr Plant Biol. 2014;56(2):133–50.
Franco AR, Gee MA, Guilfoyle TJ. Induction and superinduction of auxin-responsive mRNAs with auxin and protein synthesis inhibitors. J Biol Chem. 1990;265(26):15845–9.
Wu J, Liu SY, He YJ, Guan XY, Zhu XF, Cheng L, Wang J, Lu G. Genome-wide analysis of SAUR gene family in Solanaceae species. Gene. 2012;509(1):38–50.
Stortenbeker N, Bemer M. The SAUR gene family: the plant's toolbox for adaptation of growth and development. J Exp Bot. 2019;70(1):17–27.
Sullivan ML, Green PJ. Mutational analysis of the DST element in tobacco cells and transgenic plants: identification of residues critical for mRNA instability. RNA. 1996;2(4):308–15.
Newman TC, Ohme-Takagi M, Taylor CB, Green PJ. DST sequences, highly conserved among plant SAUR genes, target reporter transcripts for rapid decay in tobacco. Plant Cell. 1993;5(6):701–14.
McClure BA, Guilfoyle T. Characterization of a class of small auxin-inducible soybean polyadenylated RNAs. Plant Mol Biol. 1987;9(6):611–23.
Jain M, Tyagi AK, Khurana JP. Genome-wide analysis, evolutionary expansion, and expression of early auxin-responsive SAUR gene family in rice (Oryza sativa). Genomics. 2006;88(3):360–71.
Li X, Liu G, Geng Y, Wu M, Pei W, Zhai H, Zang X, Li X, Zhang J, Yu S, et al. A genome-wide analysis of the small auxin-up RNA (SAUR) gene family in cotton. BMC Genomics. 2017;18(1):815.
Xie R, Dong C, Ma Y, Deng L, He S, Yi S, Lv Q, Zheng Y. Comprehensive analysis of SAUR gene family in citrus and its transcriptional correlation with fruitlet drop from abscission zone a. Funct Integr Genomics. 2015;15(6):729–40.
Bai Q, Hou D, Li L, Cheng Z, Ge W, Liu J, Li X, Mu S, Gao J. Genome-wide analysis and expression characteristics of small auxin-up RNA (SAUR) genes in moso bamboo (Phyllostachys edulis). Genome. 2017;60(4):325–36.
Huang X, Bao YN, Wang B, Liu LJ, Chen J, Dai LJ, Baloch SU, Peng DX. Identification of small auxin-up RNA (SAUR) genes in Urticales plants: mulberry (Morus notabilis), hemp (Cannabis sativa) and ramie (Boehmeria nivea). J Genet. 2016;95(1):119–29.
Zhang N, Huang X, Bao Y, Wang B, Zeng H, Cheng W, Tang M, Li Y, Ren J, Sun Y. Genome-wide identification of SAUR genes in watermelon (Citrullus lanatus). Physiol Mol Biol Plants. 2017;23(3):619–28.
Chae K, Isaacs CG, Reeves PH, Maloney GS, Muday GK, Nagpal P, Reed JW. Arabidopsis SMALL AUXIN UP RNA63 promotes hypocotyl and stamen filament elongation. Plant J. 2012;71(4):684–97.
Stamm P, Kumar PP. Auxin and gibberellin responsive Arabidopsis SMALL AUXIN UP RNA36 regulates hypocotyl elongation in the light. Plant Cell Rep. 2013;32(6):759–69.
Hou K, Wu W, Gan SS. SAUR36, a small auxin up RNA gene, is involved in the promotion of leaf senescence in Arabidopsis. Plant Physiol. 2013;161(2):1002–9.
Wen Z, Mei Y, Zhou J, Cui Y, Wang D, Wang NN. SAUR49 can positively regulate leaf senescence by suppressing SSPP in Arabidopsis. Plant Cell Physiol. 2020;61(3):644–58.
He SL, Hsieh HL, Jauh GY. SMALL AUXIN UP RNA62/75 are required for the translation of transcripts essential for pollen tube growth. Plant Physiol. 2018;178(2):626–40.
Atamian HS, Creux NM, Brown EA, Garner AG, Blackman BK, Harmer SL. Circadian regulation of sunflower heliotropism, floral orientation, and pollinator visits. Science. 2016;353(6299):587–90.
Ke M, Gao Z, Chen J, Qiu Y, Zhang L, Chen X. Auxin controls circadian flower opening and closure in the waterlily. BMC Plant Biol. 2018;18(1):143.
Zhang L, Chen F, Zhang X, Li Z, Zhao Y, Lohaus R, Chang X, Dong W, Ho SYW, Liu X, et al. The water lily genome and the early evolution of flowering plants. Nature. 2020;577(7788):79–84.
Qiu T, Qi M, Ding X, Zheng Y, Zhou T, Chen Y, Han N, Zhu M, Bian H, Wang J. The SAUR41 subfamily of SMALL AUXIN UP RNA genes is abscisic acid-inducible to modulate cell expansion and salt tolerance in Arabidopsis thaliana seedlings. Ann Bot. 2020;125(5):805–19. https://doi.org/10.1093/aob/mcz160.
Wang Z, Yang L, Liu Z, Lu M, Wang M, Sun Q, Lan Y, Shi T, Wu D, Hua J. Natural variations of growth thermo-responsiveness determined by SAUR26/27/28 proteins in Arabidopsis thaliana. New Phytol. 2019;224(1):291–305.
Rensing SA, Lang D, Zimmer AD, Terry A, Salamov A, Shapiro H, Nishiyama T, Perroud P-F, Lindquist EA, Kamisugi Y, et al. The Physcomitrella genome reveals evolutionary insights into the conquest of land by plants. Science. 2008;319(5859):64–9.
Chou KC, Shen HB. Plant-mPLoc: a top-down strategy to augment the power for predicting plant protein subcellular localization. PLoS One. 2010;5(6):e11335.
van Mourik H, van Dijk ADJ, Stortenbeker N, Angenent GC, Bemer M. Divergent regulation of Arabidopsis SAUR genes: a focus on the SAUR10-clade. BMC Plant Biol. 2017;17(1):245.
Waterhouse A, Bertoni M, Bienert S, Studer G, Tauriello G, Gumienny R, Heer FT, de Beer TAP, Rempfer C, Bordoli L, et al. SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res. 2018;46(W1):W296–303.
Abraham MJ, van der Spoel D, Lindahl E, Hess B, and the GROMACS development team. GROMACS User Manual version 2019.1.
Spartz AK, Ren H, Park MY, Grandt KN, Lee SH, Murphy AS, Sussman MR, Overvoorde PJ, Gray WM. SAUR inhibition of PP2C-D phosphatases activates plasma membrane H+-ATPases to promote cell expansion in Arabidopsis. Plant Cell. 2014;26(5):2129–42.
Li ZG, Chen HW, Li QT, Tao JJ, Bian XH, Ma B, Zhang WK, Chen SY, Zhang JS. Three SAUR proteins SAUR76, SAUR77 and SAUR78 promote plant growth in Arabidopsis. Sci Rep. 2015;5:12477.
Oh E, Zhu JY, Bai MY, Arenhart RA, Sun Y, Wang ZY. Cell elongation is regulated through a central circuit of interacting transcription factors in the Arabidopsis hypocotyl. eLife. 2014;3:47.
Wiesel L, Davis JL, Milne L, Redondo Fernandez V, Herold MB, Middlefell Williams J, Morris J, Hedley PE, Harrower B, Newton AC, et al. A transcriptional reference map of defence hormone responses in potato. Sci Rep. 2015;5:15229.
Nemhauser JL, Hong F, Chory J. Different plant hormones regulate similar processes through largely nonoverlapping transcriptional responses. Cell. 2006;126(3):467–75.
OuYang F, Mao JF, Wang J, Zhang S, Li Y. Transcriptome analysis reveals that red and blue light regulate growth and phytohormone metabolism in Norway Spruce [Picea abies (L.) Karst]. PLoS One. 2015;10(8):e0127896.
Sun N, Wang J, Gao Z, Dong J, He H, Terzaghi W, Wei N, Deng XW, Chen H. Arabidopsis SAURs are critical for differential light regulation of the development of various organs. Proc Natl Acad Sci U S A. 2016;113(21):6071–6.
Oh E, Zhu JY, Wang ZY. Interaction between BZR1 and PIF4 integrates brassinosteroid and environmental responses. Nat Cell Biol. 2012;14(8):802–9.
Hao Y, Oh E, Choi G, Liang Z, Wang ZY. Interactions between HLH and bHLH factors modulate light-regulated plant development. Mol Plant. 2012;5(3):688–97.
Franklin KA, Lee SH, Patel D, Kumar SV, Spartz AK, Gu C, Ye S, Yu P, Breen G, Cohen JD, et al. Phytochrome-interacting factor 4 (PIF4) regulates auxin biosynthesis at high temperature. Proc Natl Acad Sci U S A. 2011;108(50):20231–5.
Kodaira K-S, Qin F, Tran L-SP, Maruyama K, Kidokoro S, Fujita Y, Shinozaki K, Yamaguchi-Shinozaki K. Arabidopsis Cys2/His2 zinc-finger proteins AZF1 and AZF2 negatively regulate abscisic acid-repressive and auxin-inducible genes under abiotic stress conditions. Plant Physiol. 2011;157(2):742–56.
Popescu SC, Popescu GV, Bachan S, Zhang ZM, Seay M, Gerstein M, Snyder M, Dinesh-Kumar SP. Differential binding of calmodulin-related proteins to their targets revealed through high-density Arabidopsis protein microarrays. Proc Natl Acad Sci U S A. 2007;104(11):4730–5.
Kant S, Bi YM, Zhu T, Rothstein SJ. SAUR39, a small auxin-up RNA gene, acts as a negative regulator of auxin synthesis and transport in rice. Plant Physiol. 2009;151(2):691–701.
Bai F, Li S, Yang C, Zhao T, Zhang T, Lan X, Chen M, Liao Z. Overexpression of the AbSAUR1 gene enhanced biomass production and alkaloid yield in Atropa belladonna. Ind Crop Prod. 2019;140.
One Thousand Plant Transcriptomes I. One thousand plant transcriptomes and the phylogenomics of green plants. Nature. 2019;574(7780):679–85.
Wang S, Li L, Li H, Sahu SK, Wang H, Xu Y, Xian W, Song B, Liang H, Cheng S, et al. Genomes of early-diverging streptophyte algae shed light on plant terrestrialization. Nat Plants. 2020;6(2):95–106.
Cheng S, Xian W, Fu Y, Marin B, Keller J, Wu T, Sun W, Li X, Xu Y, Zhang Y, et al. Genomes of subaerial zygnematophyceae provide insights into land plant evolution. Cell. 2019;179(5):1057–67 e1014.
Wang J, Sun P, Li Y, Liu Y, Yu J, Ma X, Sun S, Yang N, Xia R, Lei T, et al. Hierarchically aligning 10 legume genomes establishes a family-level genomics platform. Plant Physiol. 2017;174(1):284–300.
Pan Y: Protein structure prediction and its understanding based on machine learning methods. IEEE 7th International Symposium on BioInformatics and BioEngineering 2007:7–7.
Shin JH, Mila I, Liu M, Rodrigues MA, Vernoux T, Pirrello J, Bouzayen M. The RIN-regulated small Auxin-up RNA SAUR69 is involved in the unripe-to-ripe phase transition of tomato fruit via enhancement of the sensitivity to ethylene. New Phytol. 2019;222(2):820–36.
Du M, Spalding EP, Gray WM. Rapid auxin-mediated cell expansion. Annu Rev Plant Biol. 2020;71(1):379–402.
Larkin M, Gordon B, Nigel C, Ramu M, Paul M, Hamish V, Franck W, Iain W, Andreas L, Rodrigo T, Julie G, Toby H, Desmond. Clustal W and Clustal X version 2.0. Bioinformatics. 2007;23:2947–48.
Waterhouse AM, Procter JB, Martin DM, Clamp M, Barton GJ. Jalview version 2--a multiple sequence alignment editor and analysis workbench. Bioinformatics. 2009;25(9):1189–91.
Wilkins MR, Gasteiger E, Bairoch A, Sanchez JC, Williams KL, Appel RD, Hochstrasser DF. Protein identification and analysis tools in the ExPASy server. Methods Mol Biol. 1999;112:531–52.
Price MN, Dehal PS, Arkin AP. FastTree: computing large minimum evolution trees with profiles instead of a distance matrix. Mol Biol Evol. 2009;26(7):1641–50.
Nguyen L-T, Schmidt HA, von Haeseler A, Minh BQ. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol. 2014;32(1):268–74.
Madeira F, Park YM, Lee J, Buso N, Gur T, Madhusoodanan N, Basutkar P, Tivey ARN, Potter SC, Finn RD, et al. The EMBL-EBI search and sequence analysis tools APIs in 2019. Nucleic Acids Res. 2019;47(W1):W636–41.
Jin Y, Pan W, Zheng X, Cheng X, Liu M, Ma H, Ge X. OsERF101, an ERF family transcription factor, regulates drought stress response in reproductive tissues. Plant Mol Biol. 2018;98(1–2):51–65.
Yoshida S FD, Cock JH, Gomez KA: Laboratory manual for physiological studies of rice. International Rice Research Institute, Manila, Philippines 1976:61–66.
Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) method. Methods. 2001;25(4):402–8.
Severin AJ, Woody JL, Bolon YT, Joseph B, Diers BW, Farmer AD, Muehlbauer GJ, Nelson RT, Grant D, Specht JE, et al. RNA-Seq atlas of Glycine max: a guide to the soybean transcriptome. BMC Plant Biol. 2010;10:160.
Sturn A, Quackenbush J, Trajanoski Z. Genesis: cluster analysis of microarray data. Bioinformatics (Oxford). 2002;18(1):207–8.
Wang YP, Tang HB, DeBarry JD, Tan X, Li JP, Wang XY, Lee TH, Jin HZ, Marler B, Guo H, et al. MCScanX: a toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 2012;40(7):14.
Chen CJ, Chen H, Zhang Y, Thomas HR, Frank MH, He YH, Xia R. TBtools: an integrative toolkit developed for interactive analyses of big biological data. Mol Plant. 2020;13(8):1194–202.
Acknowledgements
The authors wish to thank Yaqiong Wang (Children’s Hospital of Fudan University) for suggestions of this manuscript. We also thank Robert McKenzie, PhD, from Liwen Bianji, Edanz Group China (www.liwenbianji.cn/ac), for editing the English text of a draft of this manuscript.
Funding
This study was financially supported by National Nature Science Foundation for Young Scientists of China (No. 31700208 to YJ). The funding body had no role in the design of this study and collection, analysis, and interpretation of data and in writing the manuscript.
Author information
Authors and Affiliations
Contributions
YJ and HZ conceived and designed the experiments. HZ performed the phylogenetic analysis, grew plants and carried out qRT-PCR. ZY and XY summarized SAUR functions. JC, XC and HZ collected the leaves samples in the culture room. HZ and YJ wrote the manuscript. YL and FM contributed to the discussion of the manuscript. YJ revised the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests in this paper.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Supplementary Datasheet S1
. Maximum likelihood phylogenetic tree constructed by FastTree v2.1 of small auxin-up RNAs (SAURs) containing detailed names from the thirteen plant species.
Additional file 2: Supplementary Datasheet S2
. Maximum likelihood phylogenetic tree constructed by IQ-TREE v2.0.6 of small auxin-up RNAs (SAURs) containing detailed names from the thirteen plant species.
Additional file 3: Supplementary Table 1
. Identified SAUR protein sequences from Anthoceros angustus, Marchantia polymorpha, Physcomitrella patens, Selaginella moellendorffii, Ginkgo biloba, Gnetum montanum, Amborella trichopoda, Arabidopsis thaliana, Oryza sativa, Zea mays, Setaria italica, Mimosa pudica, Cercis canadensis, Glycine max, and Medicago truncatula.
Additional file 4: Supplementary Table 2
. The length, isoelectric point and subcellular location of small auxin-up RNA (SAUR) proteins from the thirteen plant species.
Additional file 5: Supplementary Table 3
. The functions of all identified SAUR genes.
Additional file 6: Supplementary Table 4
. Gene-specific primers used in this study.
Additional file 7: Supplementary Fig. 1
. Maximum likelihood phylogenetic tree constructed by IQ-TREE of the SAUR gene family from the thirteen plant species.
Additional file 8: Supplementary Fig. 2
. Maximum likelihood phylogenetic tree constructed by FastTree of the SAUR gene family in Arabidopsis thaliana, Mimosa pudica, Cercis canadensis, Medicago truncatula and Glycine max.
Additional file 9: Supplementary Fig. 3
. Gene synteny analysis between Fabaceae plants and Arabidopsis thaliana.
Additional file 10: Supplementary Fig. 4
. The five conserved motifs of SAUR proteins from seven plant species containing Arabidopsis thaliana, Oryza sativa, Zea mays, Glycine max, Medicago truncatula, Setaria italica, Physcomitrella patens.
Additional file 11: Supplementary Fig. 5
. Expression profiles of small auxin-up RNA (SAUR) genes in soybean (Glycine max).
Additional file 12: Supplementary Fig. 6
. Digital gene expression of OsSAURs from different developmental stage of rice varieties under diverse stress conditions.
Additional file 13: Supplementary Fig. 7
. The number of identified SAUR genes from genomes annotated monocotyledons and dicotyledons.
Additional file 14: Supplementary Fig. 8
. Analysis of structural parameter RMSD and Ramachandran plot for structure optimization.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Zhang, H., Yu, Z., Yao, X. et al. Genome-wide identification and characterization of small auxin-up RNA (SAUR) gene family in plants: evolution and expression profiles during normal growth and stress response. BMC Plant Biol 21, 4 (2021). https://doi.org/10.1186/s12870-020-02781-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12870-020-02781-x