
Abstract
Background
Berry size is considered as one of the main selection criteria in table grape breeding programs. However, this is a quantitative and polygenic trait, and its genetic determination is still poorly understood. Considering its economic importance, it is relevant to determine its genetic architecture and elucidate the mechanisms involved in its expression. To approach this issue, an RNA-Seq experiment based on Illumina platform was performed (14 libraries), including seedless segregants with contrasting phenotypes for berry weight at fruit setting (FST) and 6–8 mm berries (B68) phenological stages.
Results
A group of 526 differentially expressed (DE) genes were identified, by comparing seedless segregants with contrasting phenotypes for berry weight: 101 genes from the FST stage and 463 from the B68 stage. Also, we integrated differential expression, principal components analysis (PCA), correlations and network co-expression analyses to characterize the transcriptome profiling observed in segregants with contrasting phenotypes for berry weight. After this, 68 DE genes were selected as candidate genes, and seven candidate genes were validated by real time-PCR, confirming their expression profiles.
Conclusions
We have carried out the first transcriptome analysis focused on table grape seedless segregants with contrasting phenotypes for berry weight. Our findings contributed to the understanding of the mechanisms involved in berry weight determination. Also, this comparative transcriptome profiling revealed candidate genes for berry weight which could be evaluated as selection tools in table grape breeding programs.
Similar content being viewed by others


Background
Grape (Vitis vinifera L.) is the main fruit crop of temperate regions, reaching nearly 77 million tons of fruit produced throughout the world in 2013 [1]. It also exhibits a high level of genetic diversity; the genus Vitis includes more than 50 species [2–4].
Berry weight is considered as one of the main selection criteria in table grape breeding, due to the consumer preferences for large and seedless berries along with organoleptic quality traits such as flavor and aroma [5]. However, berry weight is a quantitative and polygenic trait, probably determined by numerous processes such as cell multiplication, cell wall modification, water and sugar transport. Despite its relatively high heritability which is mostly additive, the genetic determination of berry weight was until recently scarcely documented [6, 7]. Therefore, considering the economic importance of berry weight for table grapes, it is relevant to determine its genetic architecture and elucidate the mechanisms involved in the expression of its driver genes. This information is required for the development of new cultivars involving the combination of desirable traits, which include not just berry size and lack of seeds, but also cluster architecture compatible with a proper berry spatial distribution [8], response to gibberellic acid (GA3) [9], yield [10] and tolerance to fungal diseases [11, 12], among others production traits.
As in other plant species, growth and cell proliferation of grape berries correspond to different processes which together determine the final fruit dimensions [13]. The development and maturation of grapevine berries has been studied as a model because of the uniqueness of this process in plant biology and its molecular regulation [14, 15].
Berry development presents a characteristic double sigmoid curve with three main phases, encompassing a series of physical and biochemical changes such as cell division and elongation, primary and secondary metabolism and resistance/susceptibility to biotic/abiotic stress [16]. Phase I involves events associated with cell division and cell elongation [17], the latter based on the accumulation of organic acids into the vacuole [6, 14, 18]. In this stage the berry is hard, green and grows slowly [14]; malic acid is the predominant metabolite. In Phase II, slower growth is observed and berry softening begins; numerous changes occur associated with gene expression and berry physiology reprogramming. Phase III is when berries reach their mature weight. This stage is characterized by the onset of sugar accumulation, a decrease in organic acid content and concomitantly, accumulation of anthocyanins in colored cultivars and volatile secondary metabolites associated with flavor and aroma [14].
A positive correlation has been described between the final berry weight and seed content [19] in segregating populations [20–24], possibly being the result of growth regulators produced by seeds [6, 25]. Interestingly, in stenospermocarpic varieties pollination occurs normally although the embryo development process aborts early, approximately 2 to 4 weeks after fertilization, while berry development continues normally [5, 24]. However, seedless varieties such as cv. Sultanina exhibit a reduced berry weight at harvest [26, 27], requiring two or three exogenous applications of gibberellic acid along with cluster thinning in order to maximize the potential berry growth; both practices demand high labor force, which increases production costs.
In relation to hormonal regulation, ethylene, auxins, ABA, cytokinins and gibberellins can influence berry development and ripening [28]. The concentration of auxins, cytokinins and gibberellins tends to increase during Phase I, in pre-véraison stages, and later decreasing in véraison, where a peak of abscisic acid has been described [28, 29].
Previous studies have described QTLs associated with berry weight in chromosomes 1 and 12 [23], 5 and 13 [30], 8, 11 and 17 [6], 15 [21] and 18 [22, 24]. In addition, [31] recently reported the VvCEB1 gene, a bHLH transcription factor, as possibly involved in the regulation of cell size in cv. Cabernet Sauvignon. Also, the VvNAC26 gene has been proposed as probably associated with berry weight variation in V. vinifera [32]. However, the genetics and information on the molecular mechanisms behind berry development in table grapes are still scarce and limited.
Diverse transcriptome studies based on microarrays [16, 33–35] as well as high-throughput RNA-Seq sequencing [36, 37] have been developed in grapes, focused on understanding the developmental and maturation process of the berry. However, these studies were directed to improve the understanding of organic acids, resveratrol, anthocyanin and tannin content and metabolism in relation to wine quality [36–40].
Due to the economic importance of berry weight in table grapes, it is relevant to determine the underlying mechanisms controlling this trait, in order to reveal positive and negative genetic factors involved in the expression of this complex trait.
We carried out the first transcriptome analysis with the aim of elucidating the mechanisms involved in berry weight determination. We contrasted seedless table grape segregants with opposite phenotypes for this trait in order to explore its genetic architecture. This comparative transcriptome profiling revealed candidate genes associated with berry weight, which could be evaluated as selection tools in table grape breeding programs.
Results and Discussion
RNA isolation from contrasting segregants for berry weight and library construction
The feasibility of this study was based on the availability of seedless segregants for berry weight (RxS crossing), maintained under the same climatic and agronomic conditions, which offer a unique opportunity to analyze transcriptome changes associated with this complex trait.
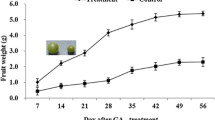
In order to study the underlying differences between large and small berries, six seedless segregants derived from a ‘Ruby Seedless’ x ‘Sultanina’ crossing (RxS; n = 139) with contrasting phenotypes for berry weight were selected and phenotyped during three seasons, 2009–2010 to 2011–2012 (Fig. 1, Additional file 1: Table S1). According to ANOVA, the genotype effect was the most significant (83 %), the season effect corresponding to 8.5 % and the genotype x season interaction was 5.9 %. The linear model explained 97 % of the phenotypic variance (Table 1).

Berry fresh weight at harvest (18°Brix) of six RxS segregants exhibiting contrasting phenotypes, including parents cv. Ruby Seedless and Sultanina. Each value corresponded to phenotypic mean values during the 2009–2010, 2010–2011 and 2011–2012 seasons. Error bars represent one standard error of the mean (SEM)
Thus a transcriptome experiment based on Illumina platform (RNA-Seq) was undertaken focused on early stages of berry development, i.e., fruit setting (FST) and berry 6–8 mm stages (B68) [14]; mRNA samples isolated in both stages were sequenced independently (Fig. 2a). These two stages are part of Phase I of the double sigmoid curve during berry growth, when the final number of cells is being defined, followed by cell expansion associated with water and organic acid accumulation in the vacuole [6, 14], critical processes defining the final fruit size [18, 31]. During the FST stage the berry cell machinery is receptive to exogenous gibberellin (GA) applications, increasing berry weight and reducing seed weight [41]. GA1 and GA4, the two endogenous bioactive GAs synthetized in the berry, have their maximum peaks in the FST and B68 stages, respectively (Ravest et al., in preparation).

Experimental design, gene differential expression and hierarchical clustering of differentially expressed genes. a Phenological stages considered for the transcriptomic study. RNA samples were obtained from large (LB) and small (SB) berry genotypes, at phenological stages of fruit-setting (FST) and berry 6–8 mm stages (B68), modified from [15]. b Differentially expressed genes after comparison between RxS segregants with contrasting phenotypes for berry weight in both phenological stages. c Hierarchical clustering of a group of 526 differentially expressed genes among LB and SB segregants in the FST and B68 stages. Pearson correlation was used as distance and five clusters were identified
Illumina GAII mRNA sequencing
A total of 14 libraries were analyzed; 155,060,882 reads of 50 bp were obtained (Additional file 2: Table S2). After quality trimming 152,897,297 reads were kept, representing a loss of about 2 % of the reads for each library (Additional file 2: Table S2). Of this total, 91 % of the reads were mapped as unique and multiple alignments (Additional file 3: Table S3). The total of mapped reads corresponded to 147.8 million reads, of which 63 to 69 % mapped in exons, 15 to 19 % in UTR regions, 8 to 9 % within intron regions, and 6 to 9 % in intergenic regions; the percentage of usable reads (UTR and exons) varied from 80 to 85 % (Additional file 4: Table S4). A total of 8.5 million reads obtained from the 14 libraries were not mapped to the reference genome PN40024. They were used to construct 2,625 de novo contigs, with an average length of 673 bp. Of them, 457 contigs were mapped to the reference genome and reanalyzed (Additional file 5: Table S5).
Global analysis of gene expression changes from fruit set (FST) to berry 6–8 mm (B68) stages
To determine which genes are changing their expression profiles and at what stage, comparisons between individuals with contrasting phenotypes for berry weight were performed (Fig. 2b, c). A group of 526 differentially expressed genes (DE) was identified comparing large (LB) versus small berry (SB) segregants in the two phenological stages (cuffdiff2 p < 0.01, FDR < 0.05) (Fig. 2b). In particular, 101 genes were identified from FST (39 up-regulated/62 down-regulated) (Additional file 6: Table S6) and 463 genes from B68 (172 up-regulated/291 down-regulated) (Additional file 7: Table S7). Interestingly, 37 of these were differentially expressed in both stages, with 34 coincidentally raising or decreasing their expression level, including transcripts coding for stilbene synthases (STS) (14) (Additional file 8: Table S8); this is equivalent to what has been observed in previous transcriptome studies during berry development in cv. Corvina [36] and cv. Cabernet Sauvignon [39].
A hierarchical clustering was performed using gene expression measured as fpkm observed in the group of 526 DE genes (Fig. 2c), and using Pearson correlation as distance in the transcriptional dendrogram. According to the expression profiles, five groups of DE genes were identified containing 60, 58, 101, 169 and 138 DE genes (Fig. 2c). In addition, a functional enrichment analysis (Gene Ontology) was developed to assess main processes over-represented in each cluster of transcripts using the agriGO platform [42] (Additional file 9: Figure S1). No over-represented category was identified in the case of cluster 2. Concomitantly, GO analysis of the groups of 101 DE genes identified in the FST and 463 in the B68 stage were performed and the results agreed with the global analysis.
Functional analysis of DE genes comparing large and small berry segregants at fruit set (FST) and berry 6–8 mm (B68) development stages
Selection of a subset of candidate genes able to explain the difference in berry size
In order to identify the genes involved in berry size determination, a principal components analysis was performed considering the 526 DE genes. The results showed that two components explained 87 % of the phenotypic variance (Fig. 3). The first component explained 55 % of the variation and clearly discriminates between contrasting phenotypes. The second component explained 31.7 % of the observed variation and discriminated between phenological stages (Fig. 3). Subsequently, correlation analyses were performed and significant correlations (p < 0.05) between DE genes and the two components were performed in order to select candidate genes, defined as transcripts whose expression level discriminates between individual classes [40].

Principal components analysis (PCA) using normalized expression data (fpkm). Analysis included the group of 526 DE genes derived from comparison between LB (in blue) and SB segregants (in red) in the FST and B68 stages
A group of 68 DE genes were significantly correlated with component 1 and 16 with component 2 (Table 2). Interestingly, both subsets of DE genes were identified in the B68 stage (Additional file 7: Table S7).
One of the most relevant functional categories associated with this group of genes was stress/defense response (26 %), encompassing HSP and chaperonins up-regulated in LB segregants (Additional file 10: Figure S2). In addition, protein kinase modifications and transcription categories were also relevant, possibly associated with the reprogramming of genes controlling transcription and translation rate in order to remodel the set of cell proteins. Four genes coding for receptor kinase-like (RLK) were up-regulated in SB segregants (Table 2). RLKs play a pivotal role in sensing external stimuli, activating downstream signaling pathways and regulating cell behavior involved in response to pathogens [43] growth and development processes in plants as well as biotic and abiotic stresses, suggesting a possible participation in the defense response in plants [43, 44]. This evidence suggests that a transcriptome reprogramming process is taking place during berry maturation, involving changes in synthesis and activation of proteins, processes that have been previously described in cv. Corvina, as well as a possible compensatory adaptation [16]. Indeed, increments in HSPs and chaperonin expression towards véraison have been reported, with a peak at véraison and subsequent reduction during berry maturation, associated with massive changes in metabolism at this phenological stage which demand the synthesis of new proteins [38, 45, 46].
Considering the observed evidence from other genetic backgrounds such as cv. Corvina, the higher expression level of HSP and chaperonins in LB segregants may be reflecting the adaptation of the berry to environmental stresses such as higher temperatures in the field.
Furthermore, evidence of a strong transcriptional control was found, with seven genes associated with the transcription category, two of them up-regulated in LB segregants, the heat stress transcription factor A-2b and a GCN5-related N-acetyltransferase (GNAT) family protein. Interestingly, the former corresponds to a transcriptional regulator whose orthologue in rice is the protein OsHsfA2e, induced by heat stress and specifically bound to the promotor of heat shock elements and possibly responsible for tolerance to high temperatures. Considering this, its introgression could be considered useful, in order to improve crop tolerance to climate change-associated stresses [47, 48]. The latter gene, a histone acetyltransferase (HAT), is responsible for lysine residue acetylation in histones H2B, H3 and H4, and also acts as a transcriptional activator, implicated in chromatin assembly and DNA replication [49].
In addition, a gene coding for a NAC domain-containing protein 78 was found up-regulated in SB segregants, which are plant-specific transcription factors (TFs). Members of this gene family have been related to plant development [50]. In particular in Vitis vinifera, VvNAC26 gene has been associated with the early development of grape flowers and berries [51], possibly contributing to berry size variation [32].
In the transport category six DE genes were found, two up-regulated in LB segregants, the sugar carrier protein A and the putative mitochondrial 2-oxoglutarate/malate carrier protein, probably associated with the transport of malate to the vacuole and cell turgor; both could be key for cell expansion. Malate is the main organic acid stored in the vacuole of grape berry cells, from FST to véraison [46].
Associated with cell wall metabolism, we found DE genes coding for a probable xyloglucan endotransglucosylase/hydrolase proteins, a lichenase and a probable galacturonosyltransferase 13, up-regulated in SB segregants, and an expansin-A15, up-regulated in LB segregants (Additional file 7: Table S7). This result is concordant with the top over-represented category ‘xyloglucan:xyloglucosyl transferase’ associated with cluster 4 (Fig. 2c, Additional file 9: Figure S1C).
This evidence could be related to cell expansion events described in the B68 stage, which initially requires cell wall softening and later the incorporation of recently synthetized material [18, 31]. Cell wall softening occurs as a result of disruption of chemical bonds between structural cell wall components, by acidification and hydrolase enzymes, modifications which require an accurate and coordinated transcriptional regulation of genes involved in biosynthesis and cell wall adaptations [18, 31]. These enzymes modify hemicelluloses during cell expansion and fruit softening, suggesting a direct influence on growth. Furthermore, cell expansion involves changes in composition as well as the accumulation of different compounds which maintain osmotic pressure and water flux in cells in expansion [31, 52]. Evidence obtained in this study agreed with these events where a strong induction of genes associated with cell expansion was observed, which probably results in larger berry weights.
Our results suggest a relevant role of expansins in the LB phenotype during the B68 stage. In the case of SB segregants, genes with xyloglucan:xyloglucosyl transferase activity were found up-regulated in the same stage (Additional file 7: Table S7). This evidence suggests a differentiation in cell wall modifications, considering that expansins have been proposed as cell wall activator agents without hydrolytic activity. Likewise, up-regulated endoglucanases were identified in LB segregants, which are also associated with cell wall dynamics. Concomitantly, in the B68 stage genes related to auxin metabolism were also identified, up-regulated in the LB phenotype, in line with the putative role of auxins in cell expansion, involved in acid growth mediated by expansins [31, 53] (Additional file 7: Table S7).
Evidence obtained from the transcriptome analysis suggested that major differences among LB and SB seedless segregants are triggered at the B68 stage, which may be responsible for the final berry weight observed at harvest. In this stage berry diameter increases by cell expansion [14].
Other functional categories were associated with secondary metabolism, transport of inorganic ions and metals, proteosome-protein degradation, hormone metabolism and signaling, development and chlorophyll biosynthesis (Additional file 10: Figure S2).
Regarding the group of 16 genes significantly correlated with component 2 (Table 2), two genes were identified coding for aspartic proteinase nepenthesin-1, possibly associated with aspartic-type endopeptidase activity [54], and senescence process (development); as well as a serine carboxypeptidase-like 18 and endoglucanase 1, both related to cell wall metabolism (Additional file 11: Figure S3). Furthermore, three genes were found associated with hormonal metabolism and signaling, coding for auxin-induced protein AUX22 and ARG2, and pirin-like protein, related to calcium signaling.
Co-expression network analysis
Network analyses were performed to identify co-expression genes associated with the separation between LB and SB segregants. Subsequently, correlation analyses results lead to identify a total of 4,950 partial correlations, 431 of them significant (p < 0.05). Correlograms were plotted with the total observed correlations (Additional file 12: Figure S4), and correlations of over 90 % were considered as significant (Additional file 13: Figure S5). Furthermore, 15 % of the significant correlations were negative and more variable (CV = 5 %). Positive significant correlations represented 85 % and were less variable (CV = 2.6 %). Five interconnected clusters of nodes were identified (Fig. 4) (Additional file 14: Table S9).

Nodes of co-expressed genes among LB and SB segregants identified using a network analysis. Main components of each node are N1: HSPs, chaperonins; N2: STBs, thaumatins; N3: monooxygenases; N4: cell wall modifications; N5: vacuolar transporters. Lines in red and green represent negative and positive correlations, respectively
These results were concordant with those obtained from hierarchical clustering and PCA; the seven DE genes selected as candidate markers for berry weight from PCA analysis were also present in the network analysis (Additional file 15: Figure S6). In addition, according to the cluster connectivity our results agreed with previous studies which described that highly connected genes were usually involved in the same biological pathways [55].
Cluster one was conformed mostly of genes coding for HSPs and chaperonin proteins, including also a gene coding for GDSL esterase/lipase and expansin-A8 (Fig. 4), all of them up-regulated in LB segregants. This result is concordant with identification of the category ‘Protein folding’ over-represented in cluster 3 (Fig. 2c, Additional file 9: Figure S1B), a process mediated by HSP [56]. As these genes have been associated with heat stress during berry development [56] and the response to microclimate changes in bunches [16, 57], this evidence suggests that LB segregants could respond more efficiently to heat stress.
Negative correlations were among genes coding for major allergen Pru av 1, associated with defense responses [58, 59], and expansin-A8; genes coding for chaperonins or HSPs were also found (Fig. 4).
Cluster two was composed mainly of genes coding for PALs and STS, a gene co-expression previously reported in cv. Syrah [37]; they were up-regulated in SB segregants in this study, in both phenological stages (Fig. 4; Additional files 6 and 7). These results are concordant with the identification of the over-represented categories ‘L-phenylalanine catabolic process’ and ‘Response to biotic stimulus’ found in cluster 5 (Fig. 2c, Additional file 9: Figure S1D, E).
STS expression has been considered as a response to stress factors such as fungal diseases, wounding and UV light [16, 60, 61], and a shift in phenylpropanoid pathway metabolites is highly sensitive to temperature changes [56]. The differential expression of those genes during berry development and maturation have been described in cv. Corvina [16, 40, 62], Norton [33] and Moscatel de Hamburgo [35]. Hence these results suggest that SB segregants presented a higher stress level during berry development than LB segregants, possibly environmental due to high temperatures.
However, positive correlation was observed between genes coding for expansin-A15 (code 80) and F-box/LRR-repeat protein 3 (code 35), both negatively correlated with genes coding for stilbene synthases in the cluster. F-box proteins act as regulators of the ubiquitin kinase dependent pathway associated with protein degradation, an important post-translational mechanism. Thus the removal of unfolded or non-functional proteins facilitates the adaptation of organisms to environmental changes, through rapid intracellular signaling [63].
In particular, expansin-A15 also showed negative correlation with genes coding for thaumatins, proteosome subunits, inorganic transporters and proteins related to pathogenesis (Additional file 14: Table S9), identified up-regulated in SB segregants (Additional files 6 and 7).
Cluster three was composed mostly of genes with monooxygenase and oxide-reductase activities, including cytochrome P450, PR6 protease inhibitor and eugenol synthase (Fig. 4). Genes belonging to the cytochrome P450 family were found up-regulated in SB segregants (Additional files 6 and 7), associated with phenylpropanoids, flavonoids, brassinosteroids and lignin synthesis. Interestingly, it has been reported that cytochrome P450-78A partially controls fruit size in tomato and possibly has a role in the domestication of this species [64].
Biosynthetic enzymes, redox regulators and HSP have been described as effector genes related to abiotic stress responses [65]. However, genes coding for chloroplast beta-amylase 3, gibberellin receptor GID1 and protein WAX2, up-regulated in SB segregants, were negatively correlated (Fig. 4).
WAX2 protein plays a role in the conversion or secretion of common precursors for cutins and wax metabolic pathways; it is also related to cuticle formation and stomata, both involved in transpiration control and drought tolerance as well [66].
Cluster 4 included a cohort of candidate enzymes related to cell wall modification, with xyloglucan endotransglucosylase/hydrolase protein 23 (XTH) and glucan endo-13-beta-glucosidase activities, positively correlated (Fig. 4).
Interestingly, cluster 5 presented no edges with the remaining clusters. Two branches were observed, the first composed of genes coding for 60S ribosomal protein L7 and abscisic acid 8'-hydroxylase 3, all of them positively correlated. The ribosomal protein modulation suggests that the transcriptome reprogramming that occurs during berry maturation involves changes in protein synthesis [16] (Fig. 4). A second branch included genes coding for cysteine-rich receptor-like protein kinase 10; vacuolar amino acid transporter 1, up-regulated in LB segregants (Additional file 6: Table S6), possibly associated with amino acid compartmentalization in the vacuole [67]; cytokinin dehydrogenase 3, as well as galactinol-sucrose galactosyltransferase; glutathione S-transferase, associated with the cellular response induced by heat shock stress and auxins, and metals such as cadmium, silver and copper [68]; and isoflavone-7-O-methyltransferase 9, related with flavonoid/isoflavonoid metabolism and biotic stress responses [69], which were positively regulated (Fig. 4).
Expression analysis of a group of candidate genes associated with berry weight using qPCR
The expression profiles of seven DE genes were experimentally validated by real-time qPCR experiments, in the phenological stages of anthesis (FL), fruit-setting (FST) and berry 6–8 mm (B68) (Fig. 5), in order to select candidate genes as putative factors associated with berry weight determination.

Validation of differentially expressed (DE) genes among LB and SB segregants by real-time PCR. LB, large berries, in black; SB, small berries, in grey. Phenological stages were anthesis (FL), fruit setting (FST) and berry 6–8 mm (B68). Genes are a GDSL esterase/lipase; b cytokinin dehydrogenase 3; c stilbene synthase 6; d gene coding for 17.9 kDa class II HSP; e TF-bHLH60; f TF-bHLH93; g TF-bHLH96; different letters on top of bars indicate significant differences (p < 0.05) according to one-way ANOVA and Tukey’s multiple comparison test among phenotypic category/phenological stage; values are the results of 27 observations categorized by phenotype. The TCPb gene (possible T-complex protein subunit beta, GSVIVG01008708001) was used as reference gene and gene expression was expressed as relative expression
The results of the network and PCA were considered in the selection of candidate genes. Genes coding for GDSL esterase/lipase, cytokinin dehydrogenase 3 and stilbene synthase 6 were selected from the network analysis. In addition, the gene coding for HSP 17.9 kDa class II was significantly correlated with PCA component 1.
In the case of the gene coding for GDSL esterase/lipase, experimental results confirmed its up-regulated expression in LB segregants in the B68 stage (p < 0.05), suggesting an increase in its expression in this stage in both LB and SB segregants (Fig. 5a). In addition, the gene coding for cytokinin dehydrogenase 3 was significantly up-regulated in LB segregants in the FST stage (Tukey test, p < 0.05), with lower expression in both groups of segregants at the B68 stage (Fig. 5b). Interestingly, the gene coding for stilbene synthase 6 showed a tendency to be up-regulated in SB segregants in the three evaluated stages. Significant differences were confirmed during FL and B68 (Tukey test, p < 0.05), being higher in the latter stage (Fig. 5c).
In addition, the gene coding for HSP 17.9 kDa class II (HSP17.9-D) showed similar expression in FL in LB and SB segregants. However, in the FST and B68 stages it was significantly up-regulated in LB segregants (Fig. 5d). Considering that HSP17.9-D was also highly correlated with component 1 of the PCA, it could be considered as a potential candidate gene for berry weight.
Participation of bHLH proteins in plant organ size determination has been described. In particular in V. vinifera, the cell elongation protein bHLH (VvCEB1) has been recently associated with berry weight in Cabernet Sauvignon, possibly involved in cell expansion during berry development [31]. Therefore, in order to evaluate the possible role of members of this transcription factor family in the differences between LB and SB segregants, three DE genes coding for bHLH60, bHLH93 and bHLH96 were selected to be experimentally confirmed by real-time qPCR experiments.
The results showed that in the case of genes coding for transcription factors (TFs), TF-bHLH60 was significantly up-regulated in SB segregants during the FL stage. However, in the FST stage it was up-regulated in LB segregants (p < 0.05) (Fig. 5e). There was an inflexion in the FST stage, with maximum expression for LB segregants and minimum for SB segregants. The same tendency was detected for TF-bHLH93, but with a significant differential expression during the FST and B68 stages, up-regulated in LB segregants (Fig. 5f). A similar expression profile was observed in the gene coding for TF-bHLH96, which was up-regulated in LB segregants at the FST stage (Fig. 5g).
Interestingly, our results differ from previous reports that proposed the gene VvCEB1 as a candidate marker for berry size, whose transcripts are predominantly accumulated in berries, especially with minimum auxin content [31]. Indeed, the three TFs evaluated showed higher expression in the FST stage comparing LB vs. SB segregants, suggesting a possible role in early stages of development. Experimental validation in advanced phenological stages would confirm their expression profile in berries in order to determine if, as with VvCEB1, these TFs plays a role in cell expansion in a wider genetic background.
Conclusions
We have carried out the first transcriptome analysis focused on seedless table grape segregants with contrasting phenotypes for berry weight. A group of 526 differentially expressed genes potentially associated with berry size was identified, 101 genes in the FST stage and 463 genes in the B68 stage.
The integration of differential expression, PCA, correlation and network analysis provided a wide characterization of overall regulation and dynamic remodeling of the gene expression in berry development in pre-véraison stages. A survey of candidate genes was also performed, and expression profiles of seven candidate genes were validated.
Methods
Plant material
The ‘Ruby’ x ‘Sultanina’ (RxS) population (n = 139) is planted at La Platina Experimental Station of the Instituto de Investigaciones Agropecuarias (INIA), located in Santiago, Chile (Latitude 33°34’23.3”S, longitude 70°37’35.73”W). This population is managed using a trendil system known as ‘spanish parron’, grafted over cv. Sultanina, and two to four replicates are available per segregant (clones). Segregants were managed under standard conditions for watering, fertilization, pest and diseases control and pruning. Both parents are publicly available resources and the segregants belong to the table grape breeding program of INIA.
The segregants and both parents were sampled in order to determine a number of quality-related traits; sugar content and titratable acidity, berry and seed weight and volume were the relevant traits for this study. Phenotype robustness was evaluated over three seasons (2009–2010 to 2011–2012), as well as health condition and vigor.
Experimental design and sample collection
A group of six segregants of the RxS cross (N = 139), named 19, 27, 112, 117, 151 and 359, plus both parents, Ruby Seedless and Sultanina, were selected for transcriptome analysis. These segregants exhibited contrasting phenotypes (Fisher test, p < 0.05) for berry size and weight, i.e., small (SB) and large (LB), all of them seedless (Fig. 1). Berry samples were collected in the 2009–2010 season 30 and 45 days after flowering, in phenological stages of E-L 27 and E-L 31 [70, 71], corresponding to fruit setting (FST) and berries of 6–8 mm diameter (B68), considered as early stages of berry development. Each genotype was sampled in two or three replicates (clones), which were later considered as technical replicates. Samples were collected in the field, frozen in liquid N2 and stored at −80 °C until RNA extraction.
RNA isolation from contrasting segregants for berry weight, library construction and mRNA sequencing
For RNA-Seq experiments, pericarp and mesocarp tissues were homogenized and analyzed together. Total RNA was isolated from 3 to 4 g of frozen tissue using the modified hot borate method [72]. The quantity and quality of RNA was assessed by measuring the A260/280 ratio using a Nanodrop ACT GeneASP-2680 equipment, and by agarose gel electrophoresis. RNA samples with 260/280 ratios between 1.8 and 2.2 were selected. Prior to sequencing, RNA integrity values were evaluated using a BioAnalyzer. Selected samples reported an RNA Integrity Number (RIN) ≥ 7.0. RNAs were sequenced after the corresponding cDNA synthesis, as described by [73]. Sequencing was performed using an Illumina sequencing platform (Genome Analyzer II) (IGA, Udine, Italy).
qPCR analysis followed the same RNA isolation protocol described above and cDNA were obtained by reverse transcription reactions with 2 ug of total RNA as template, using MMLV-RT reverse transcriptase (Promega, Madison, WI) and oligo dT primers according to standard procedures. The concentration of cDNA was assessed by measuring the absorbance at 260 nm, using a Nanodrop ACT Gene ASP-2680 equipment, finally diluting each cDNA to 50 ng/uL prior to use in qPCR experiments.
Sequencing data analysis
A total of approximately 10 million single-end reads were obtained per sequenced library, with an average length of 50 bp. Reads were trimmed by sequencing quality (Q20) and a minimum length of 30 bp. Trimmed, good-quality reads were aligned to the grapevine reference genome (PN40024 12X.v1) [74] using Tophat software [75], with a maximum of two mismatches per read. Multiple reads with more than 20 hits were discarded. Reads were then normalized as fpkm expression values, defined as reads per kilobase of exon per million reads mapped, to make them comparable across experiments. The reference grapevine genome and the gene annotation were downloaded from the GENOSCOPE database [76]. The RNA-Seq data used in this study are available at the NCBI’s Sequence Read Achieve [77] with SRA Study accession number SRX366617 [73].
Differential expression analysis
In order to identify differentially expressed genes, libraries derived from LB segregants (19, 112, and 117) were compared with SB segregant libraries (91, 151 and 359) in phenological stages FST and B68. Segregants exhibiting the same phenotype for berry weight were considered as biological replicates in the analysis [78]. Differential expression analysis was done using Cuffdiff2 (v. 2.0.2) software [79], using a geometric data normalization of library sizes (including replicates), multi-reads and fragment bias correction. Significant differences with p < 0.01 and a False Discovery Rate (FDR) of 0.05 were considered in this analysis.
Cluster analysis and gene ontology assignment
Hierarchical clustering (HCL) was performed using Pearson’s correlation distance and GENE-E software [80]. A gene ontology (GO) enrichment analysis was performed considering 526 differentially expressed genes (DE genes) grouped in five clusters, obtained from comparison of LB vs. SB segregants in the FST and B68 stages. The frequency of query genes was compared with the complete reference genome for V. vinifera (PN40024), searching for possible enrichment in biological processes. Analyses were performed using agriGO tool [81], with the singular enrichment analysis and complete GO options. Significant GO terms (p < 0.05) were calculated using the hypergeometric distribution and the Yekutieli multi-test adjustment method [42].
Principal components analysis
A principal components analysis with the group of 526 DE genes obtained from comparison of LB and SB segregants in the FST and B68 stages was performed using the FactoMineR library [82] and R statistical software [83]. Then, in order to identify candidate genes, i.e., transcripts whose absence, presence or expression level could be able to discriminate between segregants, a correlation analysis between DE genes and components 1 and 2 derived from PCA was performed. Thus significantly correlated DE genes (p < 0.05) were selected as candidate genes.
Gene co-expression network analysis
To perform the network analysis a matrix of Pearson correlations was developed, based on average values for each phenotype, which was later represented in a correlogram using the corrplot library [84] and R software [83]. Subsequently, a partial correlation analysis was performed using the PCIT library [85] and R software. Significant correlations were plotted in a correlogram. Later, correlations were considered for a network analysis using R qgraph [86]. Network analysis consisted of the representation of correlations between variables in a set of nodes connected by edges, which showed the correlation between variables [87].
Gene expression analysis by qPCR
Quantitative real-time PCR expression analysis (qPCR) of the seven selected genes was performed in the group of six segregants with contrasting phenotypes for berry weight, in the phenological stages of anthesis (FL), fruit-setting (FST) and berry of 6–8 mm (B68), corresponding respectively to E-L 23, E-L 27 and E-L 31 [70, 71]. qPCR were carried out using StepOne™Real-Time PCR System equipment (Applied Biosystems, Carlsbad, California). The qPCR amplification reactions were performed in a total volume of 10 μl containing 1 μL cDNA (50 ng/μL), 1 μL primer mix (from 400 nM to 600 nM depending on the gene), 5 μL FastStart Essential DNA Green Master (2X) (Roche, Mannheim, Germany), and 3 μL nuclease-free water. The thermal cycling conditions were denaturation at 95 °C for 10 min, followed by 40 cycles of template denaturation at 95 °C for 15 s, primer annealing at 60 °C for 1 min and extension at 72 °C for 25 s. The amplicon specificity was verified through melting curve analysis, 60 °C to 95 °C, with a gradient of 0.3 °C after 40 cycles. For each segregant three biological replicates (clones) were used, with three technical replicates per point. In addition, three segregants representative of SB or LB phenotypes were used per point, corresponding to a total of 27 observations. Values were normalized based on the housekeeping gene TCPb, which codes for a putative protein complex T subunit ß (GSVIVG01008708001) [73]. Statistical analysis of qPCR results involved ANOVA and Tukey tests (p < 0.05), and were performed using the statistical package Infostat (v2012) [88].
Primer design
Specific primers for genes being analyzed were designed using PRIMER three software [89], according to parameters described by [90], and checked in silico using the Operon software [91]. Primers were synthesized by Integrated DNA Technologies, Inc. (Coralville, Iowa). The nucleotide sequences of the genes of interest were downloaded from a private database maintained at [92]. Primers used in real-time experiments (qRT-PCR) are summarized in Additional file 16: Table S10.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Availability of data and material
The RNA-Seq data used in this study is available at the NCBI’s Sequence Read Achieve (http://www.ncbi.nlm.nih.gov/sra) with the SRA Study accession number SRX366617.
Abbreviations
- B68:
-
berry 6–8 mm stage
- DE:
-
differentially expressed
- FST:
-
fruit set stage
- GO:
-
gene ontology
- HSP:
-
heat shock protein
- INIA:
-
Instituto de Investigaciones Agropecuarias
- LB:
-
large berry segregants
- PAL:
-
phenylalanine-ammonium lyase
- PCA:
-
principal components analysis
- qPCR:
-
Real-Time PCR
- QTL:
-
quantitative trait loci
- SB:
-
small berry segregants
- STS:
-
stilbene synthases
References
Food and Agriculture Organization of United Nations. Statistics division. http://faostat3.fao.org. Accessed 05 Oct 2015.
Aradhya MK, Dangl GS, Prins BH, Boursiquot JM, Walker A, Meredith CP, Simon CJ. Genetic structure and differentiation in cultivated grape Vitis vinifera L. Genet Res. 2003;81:179–92. doi:10.1017/S0016672303006177.
Salmaso M, Faes G, Segala C, Stefanini M, Salakhutdinov L, Zyprian E, Toepfer R, Grando MS, Velasco R. Genome diversity and gene haplotypes in the grapevine (Vitis vinifera), as revealed by single nucleotide polymorphisms. Mol Breed. 2004;14:385–95.
Myles S, Boyko AR, Owens CL, Brown PJ, Grassi F, Aradhya MK, Prins B, Reynolds A, Chia JM, Ware D, Bustamante CD, Buckler ES. Genetic structure and domestication history of the grape. Proc Natl Acad Sci U S A. 2011;108:3530–5. doi:10.1073/pnas.1009363108.
Costenaro da Silva D, Passaia G, Henriques JAP, Margis R, Pasquali G, Revers L. Identification and expression analysis of genes associated with the early berry development in the seedless grapevine (Vitis vinifera L.) cultivar Sultanine. Plant Sci. 2010;179:510–9.
Doligez A, Bertrand Y, Farnos M, Grolier M, Romieu C, Esnault F, Dias S, Berger G, Francois P, Pons T, Ortigosa P, Roux C, Houel C, Laucou V, Bacilieri R, Péros JP, This P. New stable QTLs for berry weight do not colocalize with QTLs for seed traits in cultivated grapevine (Vitis vinifera L.). BMC Plant Biol. 2013;13:217. doi:10.1186/1471-2229-13-217.
Houel C, Martin-Magniette ML, Nicolas SD, Lacombe T, Le Cunff L, Franck D, Torregrosa L, Conéjéro G, Lalet S, This P, Adam-Blondon AF. Genetic variability of berry size in the grapevine (Vitis vinifera L.). Aust J Grape Wine Res. 2013;19:208–20. doi:10.1111/ajgw.12021.
Correa J, Mamani M, Muñoz-Espinoza C, Laborie D, Muñoz C, Pinto M, Hinrichsen P. Heritability and identification of QTLs and underlying candidate genes associated with the architecture of the grapevine cluster (Vitis vinifera L.). Theor Appl Genet. 2014;127:1143–62. doi:10.1007/s00122-014-2286-y.
Correa J, Ravest G, Laborie D, Mamani M, Torres E, Muñoz C, Pinto M, Hinrichsen P. Quantitative trait loci for the response to gibberellic acid of berry size and seed mass in tablegrape (Vitis vinifera L.). Aust J Grape Wine Res. 2015;21:496–507. doi:10.1111/ajgw.12141.
Fanizza G, Lamaj F, Costantini L, Chaabane R, Grando MS. QTL analysis for fruit yield components in table grapes (Vitis vinifera). Theor Appl Genet. 2005;111:658–64. doi:10.1007/s00122-005-2016-6.
Barba P, Cadle-Davidson L, Harriman J, Glaubitz JC, Brooks S, Hyma K, Reisch B. Grapevine powdery mildew resistance and susceptibility loci identified on a high-resolution SNP map. Theor Appl Genet. 2014;127:73–84. doi:10.1007/s00122-013-2202-x.
Amrine KCH, Blanco-Ulate B, Riaz S, Pap D, Jones L, Figueroa-Balderas R, Walker MA, Cantu D. Comparative transcriptomics of Central Asian Vitis vinifera accessions reveals distinct defense strategies against powdery mildew. Hortic Res. 2015;2:15037. doi:10.1038/hortres.2015.37.
Bogrë L, Okresz L, Henriques R, Anthony R. Growth signaling pathways in Arabidopsis and the AGC protein kinases. Trends Plant Sci. 2003;8:424–31.
Coombe BG, McCarthy MG. Dynamics of grape berry growth and physiology of ripening. Aust J Grape Wine R. 2000;6:131–5.
Carmona MJ, Chaïb J, Martínez-Zapater JM, Thomas MR. A molecular genetic perspective of reproductive development in grapevine. J Exp Bot. 2008;59:2579–96. doi:10.1093/jxb/ern160.
Dal Santo S, Tornielli GB, Zenoni S, Fasoli M, Farina L, Anesi A, Guzzo F, Delledonne M, Pezzotti M. The plasticity of the grapevine berry transcriptome. Genome Biol. 2013;14:R54. doi:10.1186/gb-2013-14-6-r54.
Nitsch JP, Pratt C, Nitsch C, Shaulis NJ. Natural growth substances in concord and concord seedless grapes in relation to Berry development. Am J Bot. 1960;47:566–76. doi:10.2307/2439435.
Coombe BG. Research on development and ripening of the grape berry. Am J Enol Viticult. 1992;43:101–10.
Friend AP, Trought MCT, Creasy GL. The influence of seed weight on the development and growth of berries and live green ovaries in Vitis vinifera L. cvs. Pinot Noir and Cabernet Sauvignon. Aust J Grape Wine Res. 2009;15:166–74. doi:10.1111/j.1755-0238.2009.00050.x.
Barticevic M, Zavala K, De Felice S, Valenzuela C, Muñoz C, Hinrichsen P. Caracterización fenotípica de segregantes identificados con marcadores de microsatélites, con énfasis en apirenia y respuesta a ácido giberélico en crecimiento de bayas de uva. Agric Téc (Chile). 2004;64:3–16.
Cabezas JA, Cervera MT, Ruiz-Garcia L, Carreño J, Martinez-Zapater JM. A genetic analysis of seed and berry weight in grapevine. Genome. 2006;49:1572–85. doi:10.1139/g06-122.
Mejía N, Gebauer M, Muñoz L, Hewstone N, Muñoz C, Hinrichsen P. Identification of QTLs for seedlessness, berry size, and ripening date in a seedless x seedless table grape progeny. Am J Enol Vitic. 2007;58:499–507.
Costantini L, Battilana J, Lamaj F, Fanizza G, Grando MS. Berry and phenology-related traits in grapevine (Vitis vinifera L.): from quantitative trait loci to underlying genes. BMC Plant Biol. 2008;8:38. doi:10.1186/1471-2229-8-38.
Mejía N, Soto B, Guerrero M, Casanueva X, Houel C, Miccono MA, Ramos R. Le Cunff L, Boursiquot JM, Hinrichsen P, Adam-Blondon AF. Molecular, genetic and transcriptional evidence for a role of VvAGL11 in stenospermocarpic seedlessness in grapevine. BMC Plant Biol. 2011;11:57. doi:10.1186/1471-2229-11-57.
Moreno D, Berli F, Piccoli P, Bottini R. Gibberellins and abscisic acid promote carbon allocation in roots and berries of grapevines. J Plant Growth Regul. 2011;30:220–8. doi:10.1007/s00344-010-9186-4.
Pratt C. Reproductive anatomy in cultivated grapes - a review. Am J Enol Viticult. 1971;22:92–109.
Ledbetter CA, Ramming DW. Seedlessness in grapes. Hort Rev. 1989;11:159–84.
Cramer GR, Ghan R, Schlauch KA, Tillent R, Heymann H, Ferrarini A, Delledonne M, Zenoni S, Fasoli M, Pezzotti M. Transcriptomic analysis of the late stages of grapevine (Vitis vinifera cv. Cabernet Sauvignon) berry ripening reveals significant induction of ethylene signaling and flavor pathways in the skin. BMC Plant Biol. 2014;14:370. doi:10.1186/s12870-014-0370-8.
Conde C, Silva P, Fontes N, Dias A, Tavares R, Sousa A, Delrot S, Geros H. Biochemical changes throughout grape berry development and fruit and wine quality. Food. 2007;1:1–22.
Fischer BM, Salakhutdinov I, Akkurt M, Eibach R, Edwards KJ, Töpfer R, Zyprian EM. Quantitative trait locus analysis of fungal disease resistance factors on a molecular map of grapevine. Theor Appl Genet. 2004;108:501–15. doi:10.1007/s00122-003-1445-3.
Nicolas P, Lecourieux D, Gomès E, Delrot S, Lecourieux F. The grape berry-specific basic helix–loop–helix transcription factor VvCEB1 affects cell size. J Exp Bot. 2013;64:991–1003. doi:10.1093/jxb/ers374.
Tello J, Torres-Pérez R, Grimplet J, Carbonell-Bejerano P, Martínez-Zapater JM, Ibáñez J. Polymorphisms and minihaplotypes in the VvNAC26 gene associate with berry size variation in grapevine. BMC Plant Biol. 2015;15:253. doi:10.1186/s12870-015-0622-2.
Ali MB, Howard S, Chen S, Wang Y, Yu O, Kovacs LG, Qiu W. Berry skin development in Norton grape: distinct patterns of transcriptional regulation and flavonoids biosynthesis. BMC Plant Biol. 2011;11:7. doi:10.1186/1471-2229-11-7.
Guillaumie S, Fouquet R, Kappel C, Camps C, Terrier N, Moncomble D, Dunlevy JD, Davies C, Boss PK, Delrot S. Transcriptional analysis of late ripening stages of grapevine berry. BMC Plant Biol. 2011;11:165. doi:10.1186/1471-2229-11-165.
Lijavetzki D, Carbonell-Bejerano P, Grimplet J, Bravo G, Flores P, Fenoll J, Hellín P, Okiveros JC, Martínez-Zapater JM. Berry flesh and skin ripening features in Vitis vinifera assessed by transcriptional profiling. PLoS ONE. 2012;7, e39547. doi:10.1371/journal.pone.0039547.
Zenoni S, Ferrarini A, Giacomelli E, Xumerle L, Fasoli M, Malerba G, Bellin D, Pezzotti M, Delledonne M. Characterization of transcriptional complexity during berry development in Vitis vinifera using RNA-Seq. Plant Physiol. 2010;152:1787–95. doi:10.1104/pp.109.149716.
Sweetman C, Wong DCJ, Ford CM, Drew DP. Transcriptome analysis at four developmental stages of grape berry (Vitis vinifera cv. Shiraz) provides insights into regulated and coordinated gene expression. BMC Genomics. 2012;13:691. doi:10.1186/1471-2164-13-691.
da Silva FG, Iandolino A, Al-Kayal F, Bohlmann M, Cushman MA, Lim H, Ergul A, Figueroa R, Kabuloglu EK, Osborne C, Rowe J, Tattersall E, Leslie A, Xu J, Baek J, Cramer GR, Cushman JC, Cook D. Characterizing the grape trascriptome. Analysis of expressed sequence tags from multiple vitis species and development of a compendium of gene expression during berry development. Plant Physiol. 2005;139:574–97. doi:10.1104/pp.105.065748.
Deluc L, Grimplet J, Wheatley M, Tillett RL, Quilici D, Osborne C, Schooley DA, Schlauch KA, Cushman JC, Cramer GR. Transcriptomic and metabolite analysis of Cabernet Sauvignon grape berry development. BMC Genomics. 2007;8:429. doi:10.1186/1471-2164-8-429.
Fasoli M, Del Canto S, Zenoni S, Tornielli JB, Farina L, Zamboni A, Porceddu A, Venturini L, Bicego M, Murino V, Ferrarini A, Delledonne M, Pezzotti M. The grapevine expression atlas reveals a deep transcriptome shift driving the entire plant into a maturation program. Plant Cell. 2012;24:3489–505. doi:10.1105/tpc.112.100230.
Pérez FJ, Viani C, Retamales J. Bioactive gibberellins in seeded and seedless grapes: identification and changes in content during berry development. Am J Enol Viticult. 2000;51:315–8.
Du Z, Zhou X, Ling Y, Zhang Z, Su Z. agriGO: a GO analysis toolkit for the agricultural community. Nucleic Acids Res. 2010;38:W64–70. doi:10.1093/nar/gkq310.
Yang K, Rong W, Qi L, Li J, Wei X, Zhang Z. Isolation and characterization of a novel wheat cysteine-rich receptor-like kinase gene induced by Rhizoctonia cerealis. Scientific Reports. 2013;3:3021. doi:10.1038/srep03021.
Chen K, Fan B, Du L, Chen Z. Activation of hypersensitive cell death by pathogeninduced receptorlike protein kinases from Arabidopsis. Plant Mol Biol. 2004;56:271–83.
Negri AS, Prinsi B, Rossoni M, Failla O, Scienza A, Cocucci M, Espen L. Proteome changes in the skin of the grape cultivar Barbera among different stages of ripening. BMC Genomics. 2008;9:378. doi:10.1186/1471-2164-9-378.
Martínez-Esteso MJ, Sellés-Marchart S, Lijavetzky D, Pedreño MA, Bru-Martinez R. A DIGE-based quantitative proteomic analysis of grape berry flesh development and ripening reveals key events in sugar and organic acid metabolism. J Exp Bot. 2011;62:2521–69. doi:10.1093/jxb/erq434.
Guo J, Su G, Zhang J, Wang G. Genetic analysis and QTL mapping of maize yield and associate agronomic traits under semi-arid land condition. Afr J Biotechnol. 2008;7:1829–38.
Yokotani N, Ichikawa T, Kondou Y, Matsui M, Hirochika H, Iwabuchi M, Oda K. Expression of rice heat stress transcription factor OsHs- fA2e enhances tolerance to environmental stresses in transgenic Arabidopsis. Planta. 2008;227:957–67.
Dyda F, Klein DC, Hickman AB. GCN5-related N-acetyltransferases: a structural overview. Annu Rev Biophys Biomol Struct. 2000;29:81–103. doi:10.1146/annurev.biophys.29.1.81.
Wang N, Zheng Y, Xin H, Fang L, Li S. Comprehensive analysis of NAC domain transcription factor gene family in Vitis vinifera. Plant Cell Rep. 2013;32:61–75. doi:10.1007/s00299-012-1340-y.
Fernandez L, Ageorges A, Torregrosa L. A putative NAP homolog specifically expressed during grapevine flower and berry development. Vitis. 2006;45:51–2.
Carrari F, Fernie AR. Metabolic regulation underlying tomato fruit development. J Exp Bot. 2006;57:1883–97.
Schenck D, Christian M, Jones A, Lüthen H. Rapid auxin- induced cell expansion and gene expression: a four-decade-old question revisited. Plant Physiol. 2010;152:1183–5. doi:10.1104/pp.109.149591.
Athauda SBP, Matsumoto K, Rajapakshe S, Kuribayashi M, Kojima M, Kubomura-Yoshida N, Iwamatsu A, Shibata C, Inoue I, Takahashi K. Enzymic and structural characterization of nepenthesin, a unique member of a novel subfamily of aspartic proteinases. Biochem J. 2004;381:295–306. doi:10.1042/BJ20031575.
Palumbo MC, Zenoni S, Fasoli M, Massonnet M, Farina L, Castiglione F, Pezzotti M, Paci P. Integrated network analysis identifies fight-club nodes as a class of hubs encompassing Key putative switch genes that induce major transcriptome reprogramming during grapevine development. Plant Cell. 2014;26:4617–35. doi:10.1105/tpc.114.133710.
Rienth M, Torregrosa L, Luchaire N, Chatbanyong R, Lecourieux D, Kelly MT, Romieu C. Day and night heat stress trigger different transcriptomic responses in green and ripening grapevine (Vitis vinifera) fruit. BMC Plant Biol. 2014;14:108. doi:10.1186/1471-2229-14-108.
Pastore C, Zenoni S, Fasoli M, Pezzotti M, Tornielli GB, Filippetti I. Selective defoliation affects plant growth, fruit transcriptional ripening program and flavonoid metabolism in grapevine. BMC Plant Biol. 2013;13:30. doi:10.1186/1471-2229-13-30.
Neudecker P, Schweimer K, Nerkamp J, Scheurer S, Vieths S, Sticht H, Rösch P. Allergic cross-reactivity made visible - Solution structure of the major cherry allergen Pru av 1. J Biol Chem. 2001;276:22756–63. doi:10.1074/jbc.M101657200.
Lu M, Han YP, Gao JG, Wang XJ, Li WB. Identification and analysis of the germin-like gene family in soybean. BMC Genomics. 2010;11:620. doi:10.1186/1471-2164-12-16.
Langcake P, Pryce RJ. The production of resveratrol by Vitis vinifera and other members of the Vitaceae as a response to infection or injury. Physiol Mol Plant Pathol. 1976;9:77–86.
Bais AJ, Murphy PJ, Dry IB. The molecular regulation of stilbene phytoalexin biosynthesis in Vitis vinifera during grape berry development. Funct Plant Biol. 2000;27:723–723.
Zamboni A, Di Carli M, Guzzo F, Stocchero M, Zenoni S, Ferrarini A, Tononi P, Toffali K, Desiderio A, Lilley K, Pé ME, Benvenuto E, Delledonne M, Pezzotti M. Identification of putative stage-specific grapevine berry biomarkers and omics data integration into networks. Plant Physiol. 2010;154:1439–59. doi:10.1104/pp.110.160275.
Schumann N, Navarro-Quezada A, Ullrich K, Kuhl C, Quint M. Molecular evolution and selection patterns of plant F-Box proteins with C-Terminal Kelch repeats. Plant Physiol. 2011;155:835–50. doi:10.1104/pp.110.166579.
Chakrabarti M, Zhang N, Sauvage C, Muños S, Blanca J, Cañizares J, Diez MJ, Schneider R, Mazourek M, McClead J, Causse M, van der Knaap E. A cytochrome P450 regulates a domestication trait in cultivated tomato. Proc Natl Acad Sci U S A. 2013;110:17125–30. doi:10.1073/pnas.1307313110.
Xu J, Yuan Y, Xu Y, Zhang G, Guo X, Wu F, Wang Q, Rong T, Pan G, Cao M, Tang Q, Gao S, Liu Y, Wang J, Lan H, Lu Y. Identification of candidate genes for drought tolerance by whole-genome resequencing in maize. BMC Plant Biol. 2014;14:83. doi:10.1186/1471-2229-14-83.
Chen X, Goodwin SM, Boroff VL, Liu X, Jenks MA. Cloning and characterization of the WAX2 gene of Arabidopsis involved in cuticle membrane and wax production. Plant Cell. 2003;15:1170–85. doi:10.1105/tpc.010926.
Sekito T, Fujiki Y, Ohsumi Y, Kakinuma Y. Novel families of vacuolar amino acid transporters. IUBMB Life. 2008;60:519–25. doi:10.1002/iub.92.
Chen C, Song Y, Zhuang K, Li L, Xia Y, Shen Z. Proteomic Analysis of Copper-Binding Proteins in Excess Copper-Stressed Roots of Two Rice (Oryza sativa L.) Varieties with Different Cu Tolerances. PLoS ONE. 2015;10, e0125367. doi:10.1371/journal.pone.0125367.
Debeaujon I, Peeters AJM, Léon-Kloosterziel KM, Koornneef M. The transparent testa12 gene of Arabidopsis encodes a multidrug secondary transporter-like protein required for flavonoid sequestration in vacuoles of the seed coat endothelium. Plant Cell. 2001;13:853–71. doi:10.1105/tpc.13.4.853.
Coombe BG. Adoption of a system for identifying grapevine growth stages. Aust J Grape Wine R. 1995;1:100–10.
Lorenz DH, Eichhorn KW, Bleiholder H, Klose R, Meier U, Weber E. Phenological growth stages of the grapevine (Vitis vinifera L.ssp. vinifera)- Codes and descriptions according to the extended BBCH scale. Aust J Grape Wine Res. 1995;1:100–10. doi:10.1111/j.1755-0238.1995.tb00085.x.
Gudenschwager O, González-Agüero M, Defilippi BG. A general method for high-quality RNA isolation from metabolite-rich fruits. S Afr J Bot. 2012;83:186–92. doi:10.1016/j.sajb.2012.08.004.
González-Agüero M, García-Rojas M, Di Genova A, Correa J, Maass A, Orellana A, Hinrichsen P. Identification of two putative reference genes from grapevine suitable for gene expression analysis in berry and related tissues derived from RNA-Seq data. BMC Genomics. 2013;14:878. doi:10.1186/1471-2164-14-878.
Jaillon O, Aury JM, Noel B, Policriti A, Clepet C, Casagrande A, Choisne N, Aubourg S, Vitulo N, Jubin C, Vezzi A, Legeai F, Hugueney P, Dasilva C, Horner D, Mica E, Jublot D, Poulain J, Bruyere C, Billault A, Segurens B, Gouyvenoux M, Ugarte E, Cattonaro F, Anthouard V, Vico V, Del Fabbro C, Alaux M, Di Gaspero G, Dumas V, et al. The grapevine genome sequence suggests ancestral hexaploidization in major angiosperm phyla. Nature. 2007;449:463–7. doi:10.1038/nature06148.
Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25:1105–11. doi:10.1093/bioinformatics/btp120.
GENOSCOPE database. http://www.genoscope.cns.fr/externe/GenomeBrowser/Vitis. Accesed 12 May 2013.
NCBI’s Sequence Read Achieve. http://www.ncbi.nlm.nih.gov/sra. Accesed 09 June 2013.
Almeida AM, Urra C, Moraga C, Jego M, Flores A, Meisel L, González M, Infante R, Defilippi BG, Campos-Vargas R, Orellana A. Proteomic analysis of a segregant population reveals candidate proteins linked to mealiness in peach. J Proteomics. 2016;131:71–81. doi:10.1016/j.jprot.2015.10.011.
Roberts A, Trapnell C, Donaghey J, Rinn JL, Pachter L. Improving RNA-Seq expression estimates by correcting for fragment bias. Genome Biol. 2011;12:R22. doi:10.1186/gb-2011-12-3-r22.
GENE-E. http://www.broadinstitute.org. Accesed 01 Dec 2014.
agriGO tool. http://bioinfo.cau.edu.cn/agriGO . Accesed 09 Jan 2015.
Lê S, Josse J, Husson F. FactoMineR: An R Package for Multivariate Analysis. J Stat Softw. 2008;25:1–18.
R Core Team. R: A Language and Environment for Statistical Computing. Vienna: R Foundation for Statistical Computing; 2012. http://www.R-project.org/.
Wei T. corrplot: Visualization of a correlation matrix, R package version 0.72. 2013. http://CRAN.R-project.org/package=corrplot.
Watson-Haigh NS, Kadarmideen HN, Reverter A. PCIT: an R package for weighted gene co-expression networks based on partial correlation and information theory approaches. Bioinformatics. 2010;26:411–3. doi:10.1093/bioinformatics/btp674.
Epskamp S, Cramer AOJ, Waldorp LJ, Schmittmann VD, Borsboom D. qgraph: Network Visualizations of Relationships in Psychometric Data. J Stat Softw. 2012;48:1–18. http://www.jstatsoft.org/v48/i04/.
Ursem R, Tikunov Y, Bovy A, Berloo R, Eeuwijk F. A correlation network approach to metabolic data analysis for tomato fruits. Euphytica. 2008;161:181–93. doi:10.1007/s10681-008-9672-y.
Infostat (v2012). http://www.infostat.com.ar/. Accesed 20 Aug. 2014.
PRIMER 3. http://frodo.wi.mit.edu/. Accesed 15 Mar 2014.
Taylor S, Wakem M, Dijkman G, Alsarraj M, Nguyen M. A practical approach to RT-qPCR-Publishing data that conform to the MIQE guidelines. Methods. 2010;50:S1–5. doi:10.1016/j.ymeth.2010.01.005.
Operon software. http://www.operon.com/tools/oligo-analysis-tool.aspx . Accesed 01 April 2014.
http://vitisdb.cmm.uchile.cl/ . Accesed 08 Jan 2014.
Acknowledgements
This work was mainly supported by FONDEF-Chile Genoma Program, grant G07I-1002, and Fondecyt grants 1120888 and 3150519. CME was a recipient of a doctoral fellowship from Mecesup Program. We are also grateful to the Fondap project 15090007, Basal project PB-16 and the National Laboratory for High Performance Computing at the Center for Mathematical Modeling, Santiago, Chile.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interest
The authors declare that they have no competing interests.
Authors’ contributions
CME, PH and AO conceived the experimental design; AM, AO and PH supervised the project; CME analyzed and interpreted the data, performed bioinformatic and statistical analyses, designed the qPCR experiments and wrote the first version of the manuscript; AD carried out bioinformatic analysis of NGS data; JC carried out experimental field work and statistical analyses; RS performed the qPCR experiments and statistical analyses; AD, AM, MGA, AO and PH critically reviewed the manuscript. All authors approved the final manuscript.
Additional files
Additional file 1: Table S1.
Phenotypical characterization of berry weight at harvest of six RxS segregants exhibiting contrasting phenotypes for berry weight, including parents cv Ruby Seedless and Sultanina. Analyses were performed during the 2009–2010 to 2011–2012 seasons. SEM is the standard error of the mean and CV is the coefficient of variation. (PDF 59 kb)
Additional file 2: Table S2.
Read quality summary considering the total 14 libraries. (PDF 46 kb)
Additional file 3: Table S3.
Read mapping summary. (PDF 54 kb)
Additional file 4: Table S4.
Read mapping distribution summary. (PDF 49 kb)
Additional file 5: Table S5.
Unmapped reads summary describing the total of contigs mapped to the reference genome using MegaBlast program. (PDF 57 kb)
Additional file 6: Table S6.
Differentially expressed (DE) genes identified in the comparison between LB and SB segregants, in the FST stage (Cuffdiff2, p < 0.01). (PDF 117 kb)
Additional file 7: Table S7.
Differentially expressed (DE) genes identified in the comparison between LB and SB segregants, in the B68 stage (Cuffdiff2, p < 0.01). (PDF 327 kb)
Additional file 8: Table S8.
List of 37 DE genes (p < 0.01) identified in the FST and B68 stages in the comparisons between LB and SB segregants. (PDF 69 kb)
Additional file 9: Figure S1.
A, B, C, D, E. GO enrichment of five clusters identified in hierarchical clustering of 526 DE genes, from comparison between LB and SB segregants. A. GO categories over-represented in Cluster 1 (Biological Process); B. GO categories over-represented in Cluster 3 (Biological Process); C. GO categories over-represented in Cluster 4 (Molecular Function); D. GO categories over-represented in Cluster 5 (Biological Process); E. GO categories over-represented in Cluster 5 (Molecular Function). Biological Process and Molecular Function categories are shown, and only significantly over-represented categories were considered (p < 0.05 and FDR < 0.05). The analysis was performed using the online agriGO tool and the GO complete category. The boxes contain the GO number, the p-value (in parentheses), the category description, the number of genes in each category associated with the GO term versus the total of query genes and the number of genes in each category out of 14,511 genes of the reference genome of Vitis vinifera (PN40024, 12X.v1), with associated GO terms. The arrows indicate the relationships among the GO categories, as follows: black solid arrows mean that a GO category is also included in the other one; red solid arrows mean that one GO category positively regulates the other; green solid arrows mean that the GO category negatively regulates the other; black dashed arrows indicate that there are two significant nodes related to the GO category; and black dotted arrows indicate that only one significant node is related to the GO category. (PDF 567 kb)
Additional file 10: Figure S2.
Functional characterization of 68 candidate genes significantly correlated with PCA component 1, associated with differences between LB and SB segregants. (PDF 96 kb)
Additional file 11: Figure S3.
Functional characterization of 16 candidate genes significantly correlated with PCA component 2, associated with differences between FST and B68 stages. (PDF 87 kb)
Additional file 12: Figure S4.
Correlogram representing a total of 4,950 partial correlations, including significant and non-significant correlations, found among the group of 100 DE genes with the highest significance, associated with differences between LB and SB segregants, in the FST and B68 stages. The color indicates the type of correlation i.e., negative significant correlations are in red while positive significant correlations are shown in blue. Intensity of colors indicates strength of correlations; darker shades represent higher or more negative values. (PDF 174 kb)
Additional file 13: Figure S5.
Correlogram representing 431 significant correlations (p < 0.05), found among the group of 100 DE genes with the highest significance, associated with differences between LB and SB segregants in the FST and B68 stages. Correlograms were plotted with the total of observed correlations. The color indicates the type of correlation i.e., negative significant correlations are in red while positive significant correlations are shown in blue. Intensity of colors indicates strength of correlations; darker shades represent higher or more negative values. (PDF 132 kb)
Additional file 14: Table S9.
Summary of significant partial correlations observed in the group of 100 DE genes with high significance, associated with differences between LB and SB segregants, in the FST and B68 stages. (PDF 295 kb)
Additional file 15: Figure S6.
Network analysis of co-expressed genes among LB and SB segregants. In blue and red are represented DE genes that are part of the network, which respectively present or not significant correlation with component 1 of the PCA analysis, selected as candidate genes for berry weight. Lines in red and blue represent negative and positive correlations, respectively. (PDF 64 kb)
Additional file 16: Table S10.
Primers designed for the analysis of the expression level of the seven candidate genes, based on real-time qPCR. (PDF 63 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Muñoz-Espinoza, C., Di Genova, A., Correa, J. et al. Transcriptome profiling of grapevine seedless segregants during berry development reveals candidate genes associated with berry weight. BMC Plant Biol 16, 104 (2016). https://doi.org/10.1186/s12870-016-0789-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12870-016-0789-1