
Abstract
Background
ClpP is important for bacterial growth and plays an indispensable role in cellular protein quality control systems by refolding or degrading damaged proteins, but the physiological significance of ClpP in Enterococcus faecalis remains obscure. A clpP deletion mutant (△clpP) was constructed using the E. faecalis OG1RF strain to clarify the effect of ClpP on E. faecalis. The global abundance of proteins was determined by a mass spectrometer with tandem mass tag labeling.
Results
The ΔclpP mutant strain showed impaired growth at 20 °C or 45 °C at 5% NaCl or 2 mM H2O2. The number of surviving ΔclpP mutants decreased after exposure to the high concentration (50× minimal inhibitory concentration) of linezolid or minocycline for 96 h. The ΔclpP mutant strain also demonstrated decreased biofilm formation but increased virulence in a Galleria mellonella model. The mass spectrometry proteomics data indicated that the abundances of 135 proteins changed (111 increased, 24 decreased) in the ΔclpP mutant strain. Among those, the abundances of stress response or virulence relating proteins: FsrA response regulator, gelatinase GelE, regulatory protein Spx (spxA), heat-inducible transcription repressor HrcA, transcriptional regulator CtsR, ATPase/chaperone ClpC, acetyl esterase/lipase, and chaperonin GroEL increased in the ΔclpP mutant strain; however, the abundances of ribosomal protein L4/L1 family protein (rplD), ribosomal protein L7/L12 (rplL2), 50S ribosomal protein L13 (rplM), L18 (rplR), L20 (rplT), 30S ribosomal protein S14 (rpsN2) and S18 (rpsR) all decreased. The abundances of biofilm formation-related adapter protein MecA increased, while the abundances of dihydroorotase (pyrC), orotate phosphoribosyltransferase (pyrE), and orotidine-5′-phosphate decarboxylase (pyrF) all decreased in the ΔclpP mutant strain.
Conclusion
The present study demonstrates that ClpP participates in stress tolerance, biofilm formation, antimicrobial tolerance, and virulence of E. faecalis.
Similar content being viewed by others
Background
Enterococcus faecalis has emerged as a significant cause of nosocomial infections in the last two decades, resulting in urinary tract infections, bacteremia, prosthetic joint infection, abdominal-pelvic infections, and endocarditis [1]. E. faecalis has resistance to many commonly used antimicrobial agents, and vancomycin-resistant enterococci (VRE) has emerged as a major cause of nosocomial infection outbreaks in recent years [2]. In addition to drug resistance, E. faecalis carries a high capacity for biofilm formation; more than 40% of clinical E. faecalis isolates can form biofilms [3,4,5,6,7]. Several virulence factors have been associated with E. faecalis biofilm formation. For example, the enterococcal surface protein (esp) was found to adhere to and colonize abiotic surfaces that participate in E. faecalis biofilm formation, and gelatinase (gelE) that can hydrolyze gelatin, collagen, and hemoglobin was also implicated in the adherence and biofilm formation of E. faecalis [6, 8,9,10]. However, esp and gelE were found to have no association with biofilm formation in other extensive collections of E. faecalis isolates [11,12,13]. Thus, the genes involved in the E. faecalis biofilm formation remain controversial and obscure. Other unknown factors may also participate in this important process.
The Hsp100/Clp family protein ClpP is important for bacterial growth and plays an indispensable role in cellular protein quality control systems by refolding or degrading damaged proteins in stressed cells [14]. ClpP was also associated with biofilm formation in some pathogenic species. For example, the biofilms of Streptococcus mutans, Staphylococcus epidermidis, Pseudomonas aeruginosa, and Actinobacillus pleuropneumoniae decreased when clpP was mutated [15,16,17,18]. However, the capacities to form biofilms were enhanced when clpP was mutated in Staphylococcus aureus, Haemophilus parasuis, and Porphyromonas gingivalis [19,20,21]. The roles of clpP in bacterial biofilm formation are not been fully understood. RNA levels of clpP of S. epidermidis were decreased by the agr quorum-sensing system, but in S. aureus Newman and USA300 strains, agrA and agrC RNA levels were significantly reduced in clpP mutants [16, 21]. clpP affected the expression of the transcriptional regulators csrA and rpoD and a possible biofilm repressor luxS to enhance H. parasuis biofilm formation, and it negatively adjusted the surface exposure of the minor fimbrial (Mfa) protein that promotes the biofilm formation of P. gingivalis [19, 20]. The role of clpP on E. faecalis biofilm formation remains unknown to date.
In addition to bacterial growth, stress response, and biofilm formation, ClpP also influences the virulence and antibacterial tolerance of several pathogenic organisms. clpP mutation significantly attenuated Streptococcus pneumoniae virulence in a murine intraperitoneal infection model. Expression of the virulence-related pneumolysin and pneumococcal antigen were dependent on the ClpP protease [22]. Michel found that the abundance of the agr system and agr-dependent extracellular virulence factors were diminished in the S. aureus 8325 △clpP strain [23]. In Legionella pneumophila, the clpP-deficient mutant strain was unable to escape the endosome-lysosomal pathway in host cells [24]. The clpP deletion mutation also attenuated Salmonella Typhimurium virulence through dysregulation of RpoS and indirect control of CsrA and the SPI genes [25]. In S. aureus, in addition to the stress response, biofilm formation, and virulence, the truncating mutation in clpP is responsible for the raised vancomycin resistance in VISA strain LR5P1-V3 [26]. Bæk found that inactivation of the components of the ClpXP protease substantially increased β-lactam resistance in the S. aureus USA300 strain, while the clpP mutant strain displayed significantly thicker cell walls, increased peptidoglycan cross-linking, and altered composition of monomeric muropeptide species compared to wild type [27]. As mentioned above, E. faecalis shows resistance to many antimicrobial agents; however, whether the clpP is involved in E. faecalis resistance to antimicrobials, especially vancomycin (VRE), is still unclear.
To obtain a more comprehensive understanding of the role of ClpP protease in the E. faecalis stress response, biofilm formation, virulence, and antimicrobial tolerance, a △clpP strain was constructed in E. faecalis strain OG1RF. The global abundance of proteins was detected with an Orbitrap Q Exactive HF-X mass spectrometer with tandem mass tag (TMT) labeling.
Results
Construction of the clpP deletion mutant and complemented strain
To explore the role of ClpP in E. faecalis, we constructed a clpP deletion mutant in the E. faecalis OG1RF strain using the temperature-sensitive plasmid pJRS233. The deletion mutant strain was verified by polymerase chain reaction (PCR) and direct sequencing and was termed the OG1RF ΔclpP mutant strain. The complemented ΔclpP strain (ΔclpP/pIB166::clpP) was constructed using shuttle vector pIB166 and also verified by PCR and direct sequencing. The ΔclpP strain containing the empty vector pIB166 was designated as OG1RF ΔclpP/pIB166. clpP RNA levels of all the above four E. faecalis OG1RF strains were determined by quantitative reverse transcription PCR (RT-qPCR) as shown in Additional file 1: Figure S1.
ΔclpP mutant strain showed impaired growth at 20 °C, 45 °C, 5%NaCl, or 2 mM H2O2
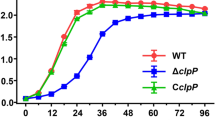
Previous research indicated that ClpP participated in the S. aureus stress response to low or high temperature and the oxidative stress response [23]; however, these issues have not been studied in E. faecalis. Thus, we first investigated the effects of clpP deletion on E. faecalis growth under the stresses of low or high temperature, hyperosmotic pressure, low pH, and oxidative stress. At 37 °C, there were no significant growth differences between the E. faecalis OG1RF parent strain and its ΔclpP mutant. However, under 20 °C or 45 °C, the ΔclpP mutant strain showed a lower optical density at 600 nm (OD600) than was observed for the wild-type strain after entering logarithmic phase growth (Fig. 1). As shown in Fig. 2, ΔclpP mutant strain growth was also impaired under 5% NaCl (logarithmic phase) or 2 mM H2O2 (later logarithmic phase or stationary phase).

Effect of clpP deletion on E. faecalis growth at 37 °C, 20 °C, and 45 °C. Three independent experiments were performed, and the data represent means ± SD

Sensitivity of the ΔclpP mutant to hyperosmotic pressure, low pH, oxidative stress, and SDS. a Overnight cultures of the E. faecalis strains were diluted in TSB containing 5% NaCl or with pH 5.5 and then incubated at 37 °C for 16 h, or in TSB containing 2 mM H2O2 incubated at 37 °C for 10 h. Three independent experiments were performed, and the data represent means ± SD. b The E. faecalis strains were spotted onto TSB agar plates containing 0.008% SDS and incubated for 24 h at 37 °C. Three independent experiments were performed, and the representative results are shown
clpP deletion leads to decreased biofilm formation
Polystyrene microtiter plate assays were performed to evaluate the role of clpP in the biofilm formation of E. faecalis under static conditions. The biofilm formation of E. faecalis OG1RF parent strain and its ΔclpP mutant was monitored at 12, 24, and 48 h on microtiter plates stained with crystal violet (CV), and OD570 values were determined. The biofilms of the ΔclpP mutant strain (OD570, 0.835 ± 0.091) were significantly decreased compared with that of the parent strain (OD570, 2.247 ± 0.138, P < 0.001, Student’s t test) after incubation for 48 h, and this outcome was also observed after incubation for 12 or 24 h (Fig. 3 a). We further investigated extracellular DNA (eDNA) release during E. faecalis biofilm formation but found no differences between the ΔclpP mutant and its parent strain (Fig. 3 b).

Effects of the ΔclpP mutant on E. faecalis biofilm formation and eDNA release. a The biofilms of E. faecalis strains were stained with crystal violet, and OD570 values were measured. *P < 0.05, **P < 0.01, ***P < 0.001 (Student’s t test). b PI-bound eDNA of E. faecalis strains was measured by a Varioskan™ LUX multimode microplate reader. Three independent experiments were performed, and the data represent means ± SD
Antimicrobial tolerance of the ΔclpP mutant strain
The minimal inhibitory concentrations (MICs) of eight antimicrobials for E. faecalis were detected by the broth microdilution method, and the MICs for the ΔclpP mutant strain were similar to those of the parent strain (Additional file 4: Table S1). To determine which antimicrobial concentrations ensured that only drug-tolerant bacterial cells survived, we performed time-killing assays for six antimicrobials. Based on previous research [28] and our preliminary results, the concentrations of six antimicrobials were set at 50× MIC. As shown in Fig. 4, the surviving bacteria of the ΔclpP mutant strain (log10 colony-forming units [CFU]/mL, under the detection limit) were significantly decreased compared with those of the parent strain (log10CFU/mL, 2.873 ± 0.243, P < 0.001, Student’s t test) after 96-h exposure to linezolid. After 96-h exposure to minocycline, the surviving bacteria of the ΔclpP mutant strain (log10CFU/mL, 1.477 ± 0.171) were also decreased compared with the parent strain (log10CFU/mL, 3.078 ± 0.303, P < 0.01, Student’s t test).

Survival of the ΔclpP mutant and the parent strain with antimicrobial exposure over time. Three independent experiments were performed, and the data represent means ± SD. The dashed line indicates the assay’s detection limit
ΔclpP mutant leads to increased E. faecalis virulence
The virulence of E. faecalis strains was detected by the infection of Galleria mellonella larvae. The survival of G. mellonella larvae infected with the ΔclpP mutant strain (15/40, 37.5%) significantly decreased compared with the parent strain (28/40, 70.0%, P < 0.01, log-rank test) at 72 h post infection (p.i.) (Fig. 5). The complemented △clpP/pIB166::clpP strain (23/40, 57.5%) showed a partially restored survival ability.

Deletion of clpP leads to increased virulence of E. faecalis. G. mellonella were infected with 20 μL inocula of E. faecalis strains containing 5 × 106 CFU/mL, and the survival of G. mellonella larvae was recorded at 12-h intervals for 72 h p.i. Data were collected from three independent experiments, and representative results are shown. **P < 0.01 (log-rank test)
Comparison of the global protein abundances of the ΔclpP mutant and parent strain
We compared the global protein abundances of the ΔclpP mutant and parent strain. The total proteins were extracted from logarithmic phase (4 h) and stationary phase (12 h) bacteria, and their abundances were determined on an Orbitrap Q Exactive HF-X mass spectrometer with TMT labeling. The protein quantitation results were statistically analyzed by Mann-Whitney tests, and the significant ratios, defined as P < 0.05 and ratio > 1.2 or < 0.83 (fold change, FC), were used to screen differential abundance proteins (DAPs). The protein quantitation results are given as the means from two independent experiments, and the repeatability of the two independent experiments was evaluated by the coefficient of variation (CV). As shown in Additional file 2: Figure S2, the CV for the two independent experiments was very low. All DAPs are summarized in Table 1. The abundances of 135 proteins changed in the ΔclpP mutant strain, of which 111 increased and 24 decreased.
Gene ontology (GO) and Kyoto encyclopedia of genes and genomes (KEGG) analysis of DAPs
DAPs between the △clpP mutant and parent strain were analyzed by GO and KEGG analyses. As shown in Fig. 6, GO analysis revealed that increased DAPs in the △clpP mutant strain (logarithmic phase) were mainly concentrated in the following molecular functions: N-acetyltransferase activity, coenzyme binding, cofactor binding, ATPase activity, nucleoside-triphosphatase activity, hydrolase activity, ATP binding, kinase activity, nucleotide binding, organic cyclic compound binding, heterocyclic compound binding, DNA binding, and nucleic acid binding. Decreased DAPs were mainly included in the following molecular functions: structural constituent of ribosome, rRNA binding, orotidine-5′-phosphate decarboxylase activity, hydrolase activity, organic cyclic compound binding, heterocyclic compound binding, and nucleic acid binding. KEGG analysis demonstrated that the functions of most DAPs in the △clpP mutant (logarithmic phase) belonged to the ribosome, fructose and mannose metabolism, pyrimidine metabolism, purine metabolism, pentose phosphate pathway, glycolysis/gluconeogenesis, and ABC transporters (Fig. 7). The functions of DAPs in the stationary phase of △clpP mutant strain were similar to those in the logarithmic phase (Additional file 3: Figure S3).

Gene Ontology (GO) analysis of differential abundance proteins (DAPs). The molecular functions of DAPs were classified by GO analysis

KEGG (Kyoto Encyclopedia of Genes and Genomes) analysis of differential abundance proteins (DAPs) (logarithmic phase). The protein families and pathways were analyzed using the KEGG database
DAPs associated with the stress response, virulence, or biofilm formation of E. faecalis
Based on the literature, we selected DAPs that may be associated with stress response, virulence, or biofilm formation of E. faecalis for a thorough analysis. The abundance of DAPs associated with the stress response or virulence of E. faecalis increased in the △clpP mutant strain, including the FsrA response regulator and gelatinase GelE; ATPase/chaperone ClpC; chaperonin GroEL, acetyl esterase/lipase; and transcriptional regulator proteins, HrcA, CtsR, and Spx (Table 2). However, the abundances of ribosomal proteins L4/L1, L7/L12, L13, L18, L20, S14, and S18 decreased in the △clpP mutant strain. The abundance of the biofilm formation of E. faecalis-associated DAPs and adapter protein MecA increased in the △clpP mutant strain, while the abundances were lower for orotate phosphoribosyltransferase, orotidine-5′-phosphate decarboxylase, and dihydroorotase (Table 2). The RNA levels of all the above DAPs were verified by RT-qPCR and were consistent with protein abundance changes in the △clpP mutant strain.
Discussion
ClpP is a protease of the Hsp100/Clp family that is very important for bacterial growth and plays an irreplaceable role in cellular protein quality control systems by refolding or degrading damaged proteins in stressed cells [14]. To date, ClpP has been implicated in many essential bacterial activities such as stress responses to abnormal temperature, hyperosmotic pressure, low pH, oxidative stress, virulence, and biofilm formation. However, the global abundances of proteins affected by ClpP in bacteria are still little known. Feng et al. found that the abundance of transcriptional regulators CtsR and Spx, the ClpC adaptor proteins McsB and MecA, and the cell division protein FtsZ were clearly affected by ClpP in S. aureus strains NCTC8325–4, COL, SA564, and Newman using a two-dimensional difference gel electrophoresis (2-D DIGE) technique [29, 30]. However, the abundances of only 80 proteins changed in their studies, a result that may be due to the low sensitivity of 2-D DIGE. In the present study, we found 135 DAPs in the △clpP mutant strain. These included the transcriptional regulators CtsR and Spx, the ClpC adaptor proteins MecA and FtsZ-interacting cell division protein YlmF, as previously described in S. aureus strains. Interestingly we also found other new proteins, such as acetyl esterase/lipase, ribosomal protein, orotidine-5′-phosphate decarboxylase, and others.
ClpP has been shown to participate in stress tolerance by refolding or degrading damaged proteins during bacteria growth, and several studies have indicated that the ΔclpP mutant strain showed a growth defect over a broad range of temperatures including high (40, 42, 45 °C) or low (20, 30 °C) temperatures, and even under 37 °C [19, 23, 31, 32]. However, this study showed altered growth of E. faecalis OG1RF ΔclpP mutant strain at 45 °C and 20 °C but not 37 °C. Previous studies also demonstrated the ΔclpP strain is more vulnerable to oxidative stress, osmotic stress, acid, or sodium dodecyl sulfate (SDS) [19, 33,34,35]. We found the growth of OG1RF ΔclpP was impaired under osmotic or oxidative stress conditions. The ribosomal protein L9 plays a significant role in the Escherichia coli response to starvation stress [36]. The present study found that in E. faecalis OG1RF, the abundance of many ribosomal proteins decreased, including both 50S and 30S ribosomal proteins. Thus, ClpP may participate in the stress response of E. faecalis by affecting the abundance of ribosomal proteins.
Previous studies have found that ClpP can significantly affect bacteria biofilm formation, but its effects in different genera vary [15, 16, 18, 19, 21]. This study provides the first evidence that biofilm formation decreased when the clpP of OG1RF strain was deleted. The adapter protein MecA can decrease the RNA level of eps, which encodes synthesis of the biofilm matrix exopolysaccharide, thus inhibiting biofilm formation by Bacillus subtilis [37]. The present study showed MecA abundance increased in the ΔclpP mutant strain, and this contribute to the decreased biofilm formation of the clpP deleted strain. Another reason for decreased biofilm formation of the ΔclpP mutant strain may be the reduced abundances of orotate phosphoribosyltransferase (pyrE) and orotidine-5′-phosphate decarboxylase (pyrF), proteins that promote the biofilm formation of Streptococcus sanguinis and E. faecalis, respectively [38, 39].
ClpP participates in bacterial virulence, and the virulence of S. pneumoniae, S. aureus and L. pneumophila was attenuated in clpP mutation strains [22,23,24]. Liu et al. recently reported that the clpP mutant strain showed increased biofilm formation and reduced virulence in S. aureus [21]. However, we found that the ΔclpP mutant strain decreased biofilm formation and increased virulence in a G. mellonella model. A previous study proposed that the CtsR regulator controlled the expression of clpC, clpE, and clpP and was required for the virulence of E. faecalis V583, but the role of clpP in the virulence of E. faecalis was still unclear [40]. The FsrABDC signal transduction system and GelE are major virulence factors in E. faecalis [41, 42]. Thus, it may be that the increased abundances of FsrA and GelE leads enhance virulence of the ΔclpP mutant strain. The abundance of acetyl esterase/lipase, another E. faecalis virulence factor, was also increased in the ΔclpP mutant strain and may contribute to the enhanced virulence of the ΔclpP mutant strain [43].
This study also found that the tolerance to linezolid or minocycline of the ΔclpP mutant strain decreased. Linezolid is an inhibitor of bacterial protein synthesis that acts on the 50S ribosome subunit of gram-positive bacteria, and minocycline is a synthetic tetracycline derivative that acts on the 30S ribosome subunit of gram-positive or -negative bacteria [44, 45]. The abundances of 50S ribosomal proteins L13, L18, and L20 and 30S ribosomal proteins S14 and S18 were decreased in the △clpP mutant strain, thus might lead to the decrease of the tolerance of the △clpP mutant strain to linezolid or minocycline.
In B. subtilis, Spx plays a significant role in protecting against oxidative stresses [46]. Recently Rojas-Tapias and Helmann found that Spx is a regulator of the ctsR operon, and the ctsR operon regulates the expression of clpC and clpP [47]. The present study showed that when clpP was deleted in E. faecalis OG1RF, the abundance of ClpC, CtsR, and Spx all increased, which was similar to observations in S. aureus [30]. In S. aureus, the RNA levels of the clpC operon (ctsR-mcsA-mcsB-clpC), groE, and dnaK were induced in response to accumulation of misfolded proteins, which supported the hypothesis that ClpP proteases degrade misfolded proteins [30]. Our study found that the abundances of ClpC, GroEL, and DnaB (but not DnaK) increased in the △clpP mutant strain, possibly due to the accumulation of misfolded proteins.
It is easy to understand how ClpP, as a protease, can significantly affect the abundance of proteins, but not RNA levels. In the present study, the abundances of many transcription regulation-related proteins changed in the △clpP mutant strain, such as regulatory protein Spx (spxA), heat-inducible transcription repressor HrcA, transcriptional regulator CtsR, as reported previously [29, 30]. Transcriptional regulators usually control the transcription and RNA levels of their functional genes. So, ClpP may affect the abundance of transcriptional regulators alter the RNA levels of the genes. The RNA levels of many genes changed in the ΔclpP mutant strain in this study, and similar results were reported in other studies [23, 30]. Since ClpP is a protease involved in protein degradation, its absence should lead to protein accumulation, and this is consistent with our result that the abundance of most DAPs increased in the △clpP mutant strain. However, the abundances of some proteins and their corresponding RNA levels decreased in the △clpP mutant strain, and similar results were also found in another study [30]. As mentioned above, the reason may be that ClpP reduced the transcription and expression of those genes by regulating the abundance of transcriptional regulators.
Conclusion
The present study indicates that ClpP may affect the abundance of ribosomal proteins L4/L1, L7/L12, L13, L18, L20, S14, and S18 that participate in the stress response and linezolid or minocycline tolerance of E. faecalis. ClpP participates in E. faecalis biofilm formation by affecting the abundances of adapter protein MecA, orotate phosphoribosyltransferase (pyrE), and orotidine-5′-phosphate decarboxylase (pyrF). Our results also suggest that ClpP may modulate the abundances of FsrA, GelE, and acetyl esterase/lipase to participate in E. faecalis virulence.
Methods
Bacterial strains, plasmids, growth conditions, and chemicals
All of the bacterial strains and plasmids used in this study are shown in Table 3. E. faecalis ATCC 47077 (OG1RF; GenBank accession number CP002621.1) and ATCC 29212 were purchased from the American Type Culture Collection (ATCC; Manassas, VA, USA). E. faecalis strains were cultured in tryptic soya broth (TSB; Oxoid, Basingstoke, UK) as previously described [28]. TSBG (TSB medium added 0.25% glucose) for biofilm formation detection. Electroporation was used for plasmid transformation, and B2 medium was used for bacteria recovery [28]. The antibiotics used in this study were purchased from Sigma Chemical Co. (St Louis, MO, USA) and used at concentrations of 20 mg/L for chloramphenicol and 750 or 25 mg/L for erythromycin.
Construction of △clpP mutants and complemented strains
The clpP deletion mutant of the OG1RF strain was constructed by in-frame deletion using the temperature-sensitive plasmid pJRS233 as previously described [48]. Briefly, the upstream and downstream fragments of OG1RF_10505 (gene: clpP; product: ATP-dependent Clp protease proteolytic subunit), which is highly homologous (86.8%) to SA0723 (product as the ClpP protease) of S. aureus N315 strain [23], were amplified from OG1RF by PCR and separately cloned into the pJRS233 vector to generate pJRS233-ΔclpP. The recombinant plasmid pJRS233-ΔclpP was successively transferred and electroporated into wildtype OG1RF strain, then the pJRS233-ΔclpP clones were selected by variable temperature screening as previously described [28]. The complemented ΔclpP mutant strain was constructed using the E. coli -Streptococcus shuttle vector pIB166. The clpP gene was amplified by PCR and cloned into the pIB166 vector to produce pIB166:: clpP. The recombinant plasmid pIB166:: clpP was transformed by electroporation into the ΔclpP mutant strain, forming the complemented ΔclpP/pIB166:: clpP strain. The ΔclpP strain containing the empty vector pIB166 was designated the ΔclpP/pIB166 mutant. The ΔclpP mutant and complemented ΔclpP mutant strain were identified by PCR, RT-qPCR, and direct sequencing. The primers used in this assay are listed in Table 4.
Growth analysis of the △clpP mutant strain
The OG1RF, ΔclpP, ΔclpP/pIB166:: clpP, and ΔclpP/pIB166 strains were cultured in TSB at 37 °C with shaking for 12 h and diluted in the same medium to an OD600 value of 1.5, then 50 μL aliquot of the diluted suspension was inoculated into 10 mL fresh TSB and incubated at either 37 °C, 45 °C or 20 °C with circular agitation (220 rpm). The diluted suspension was also inoculated into fresh TSB with 5% NaCl pH 5.5 or 2 mM H2O2 and incubated at 37 °C with circular agitation (220 rpm). OD600 values for the cultures were determined using an Eppendorf Biospectrometer (Eppendorf, Hamburg, Germany) at 1-h intervals. Three independent experiments were performed.
The sensitivity of the △clpP mutant strain to SDS
Overnight cultures of E. faecalis strains were diluted 1:200 in fresh TSB medium and incubated at 37 °C for 4 h until an OD600 of 1.0 was reached. After 10-fold serial dilution, 5 μL of the aliquot was spotted onto a TSB agar plate containing 0.008% SDS and incubated at 37 °C for 24 h. Bacterial colonies on the plates were photographed and counted [28]. Three independent experiments were performed, and representative results are shown.
Microtiter plate assay of biofilm formation
The biofilm-forming ability of E. faecalis isolates was detected as previously described with modifications [49]. Overnight cultures were diluted 1:200 in 200 μL of TSBG (TSB with 0.25% glucose) and inoculated into 96-well polystyrene microtiter plates. After 12, 24, or 48 h of static incubation at 37 °C, the supernatant was discarded, and plates were washed thrice with deionized water to remove unattached cells, stained with 1% CV for 20 min at room temperature, and rinsed with distilled water. Finally, the CV was solubilized in ethanol-acetone (80:20, vol/vol), and absorbance at OD570 was determined. Three independent experiments were performed.
Quantification of eDNA
eDNA was quantified as described previously [50]. Overnight cultures of E. faecalis strains were diluted to OD600 = 0.001 in AB medium supplemented with 0.5% glucose, 0.05 mM propidium iodide (PI) and 10% TSB. The diluted cultures were transferred to polystyrene microtiter plates (200 μL/well) and incubated for 24 h at 37 °C. The cell density was measured at OD600 using a microtiter plate reader (Bio-Rad Laboratories, Hercules, CA, USA). The fluorescence of PI-bound eDNA was measured by a Varioskan™ LUX multimode microplate reader (Thermo Fisher, Waltham, MA, USA) with the excitation/emission wavelength at 535/610 nm. Relative amounts of eDNA per OD600 unit were determined. Three independent experiments were performed.
Determination of MIC and antimicrobial tolerance of strains
The MICs of the antimicrobials against E. faecalis isolates were determined by the broth microdilution method according to Clinical and Laboratory Standards Institute (CLSI) guideline CLSI-M100-S26 with CLSI-recommended MIC breakpoints. E. faecalis ATCC29212 served as the quality control standard strain. The antimicrobial-tolerance of strains was detected as described previously with modifications [28]. Antimicrobials (at 50× MIC) were added to the stationary-phase cultures (16 h) of the E. faecalis strains, then the cultures were incubated at 37 °C for 120 h without shaking. Every 24 h, 1-mL aliquots were sampled and washed twice with ice-cold saline. Ten-fold dilutions were then plated on Muller-Hinton agar, and the numbers of CFUs were determined. Three independent experiments were performed.
Virulence of E. faecalis in G. mellonella
Infection of G. mellonella larvae with E. faecalis strains was performed as described previously for other pathogens [51]. G. mellonella larvae in groups of 40 were infected in the left posterior proleg with 20 μL inocula of E. faecalis strains containing 5 × 106 CFU/mL. Survival of G. mellonella larvae was recorded at 12 h intervals for 72 h p.i. Every trial included a group of 20 G. mellonella larvae injected with saline as a control. Experiments were performed in at least three independent tests, and representative results are shown.
Protein extraction and detection by a mass spectrometer with TMT labeling
E. faecalis strain OG1RF and the ΔclpP mutant were inoculated into TSB and cultured at 37 °C for 4 h to logarithmic phase or for 12 h to stationary phase. The cells were harvested at 4 °C centrifugation, minced individually with liquid nitrogen, lysed in lysis buffer, and ultrasonicated for 5 min on ice. Protein concentration was determined again with Bradford protein assays. The supernatant from each sample, containing precisely 0.1 mg of protein, was digested with Trypsin Gold (Promega, Madison, WI, USA) at 1:50 enzyme-to-substrate ratio. After 16 h of digestion at 37 °C, peptides were desalted with a C18 cartridge to remove urea, and desalted peptides were dried by vacuum centrifugation. Desalted peptides were labeled with TMT6/10-plex reagents (TMT6/10plex™ Isobaric Label Reagent Set, Thermo Fisher) as previously described [52]. TMT-labeled peptide mix was fractionated using a C18 column (Waters BEH C18 4.6 × 250 mm, 5 μm; Waters Corporation, Milford, MA, USA) on a Rigol L3000 high-performance liquid chromatographer operating at 1 mL/min, and the column oven was set at 50 °C. Shotgun proteomics analyses were performed using an EASY-nLCTM 1200 ultra high-performance liquid chromatography system (Thermo Fisher) coupled with an Orbitrap Q Exactive HF-X mass spectrometer (Thermo Fisher) operated in the data-dependent acquisition mode. The Q Exactive HF-X mass spectrometer was operated in positive polarity mode with a spray voltage of 2.3 kV and capillary temperature of 320 °C. Two independent experiments were performed.
Global protein abundance analysis
The resulting spectra from each fraction were searched separately against the NCBI E. faecalis strains OG1RF (CP002621.1) database (https://www.ncbi.nlm.nih.gov/nuccore/CP002621.1) using the search engine Proteome Discoverer 2.2 (PD 2.2, Thermo). The searched parameters were as follows: mass tolerance of 10 ppm for precursor ion scans and mass tolerance of 0.02 Da for production scans. Carbamidomethyl was specified in PD 2.2 as a fixed modification. Oxidation of methionine, acetylation of the N-terminus, and TMT of lysine were specified in PD 2.2 as variable modifications. A maximum of 2 miscleavage sites was allowed. For protein identification, a protein with at least one unique peptide was identified at a false discovery rate FDR < 1.0% on peptide and protein levels. Proteins containing similar peptides that could not be distinguished based on MS/MS analysis were grouped as separate protein groups. The protein quantitation results were statistically analyzed by Mann-Whitney tests, and the significance ratios defined as P < 0.05 and ratio > 1.2 or < 0.83 (FC) were used to screen DAPs. GO and InterPro (IPR) analyses were conducted using the interproscan-5 program against the non-redundant protein database (including Pfam, PRINTS, ProDom, SMART, ProSiteProfiles, and PANTHER). The databases of COG (Clusters of Orthologous Groups) and KEGG were used to analyze protein families and pathways. The enrichment pipeline was used to perform the enrichment analyses of GO, IPR, and KEGG.
RNA isolation and RT-qPCR
RNA isolation of E. faecalis strains was performed as described previously with some modifications [28]. The E. faecalis strain OG1RF and the ΔclpP mutant were inoculated into TSB and cultured at 37 °C for 4 h to logarithmic phase or for 12 h to stationary phase, and the following operations were performed at 4 °C for centrifugation or on ice. Bacterial cultures were centrifuged at 12,000 rpm for 5 min, and then the pellets were washed twice with 0.9% saline; the culture was homogenized 5 times using 0.1-mm zirconia-silica beads in a mini-BeadBeater (Biospec, Bartlesville, OK, USA) at 5000 rpm for 60 s at 1-min intervals; the samples were centrifuged at 15,000 rpm, and the bacterial RNA in the supernatant was purified using an RNeasy minikit (Qiagen, Hilden, Germany) and quantified using an ND-1000 spectrophotometer (NanoDrop Technologies, Wilmington, DE, USA). RNA samples that had a 260/280 ratio between 2.0 and 2.2 were used for RT-qPCR.
Total RNA extracted from strains OG1RF and the ΔclpP mutant were reverse transcribed with the PrimeScript RT Reagent Kit (TaKaRa Biotechnology, Dalian, China), and RT-qPCR was performed with the SYBR Premix Ex Taq II Kit (TaKaRa Biotechnology) on the Mastercycler ep realplex system (Eppendorf), with an initial incubation at 95 °C for 2 min, followed by 40 cycles of 15 s at 95 °C, and 60 s at 60 °C. Each sample was analyzed in triplicate. For all samples, the internal control gene recA was used to normalize the abundance of E. faecalis strains OG1RF genes [53]. The threshold cycle (Ct) numbers were confirmed by the detection system software, and the data were analyzed based on the 2−△△Ct method. The RT-qPCR primers are listed in Additional file 4: Table S2.
Statistical analysis
Experimental data were analyzed with SPSS software (version 16.0; SPSS, Chicago, IL, USA) and compared using Student’s t tests, one-way analysis of variance, Mann-Whitney tests, or the log-rank tests. Differences with a P value < 0.05 were considered statistically significant.
Availability of data and materials
All data generated or analyzed during this study are included in this published article [and its supplementary information files].
The mass spectrometry proteomics data were deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD014211.
Abbreviations
- CV:
-
Crystal violet
- DAP:
-
Differential abundance protein
- eDNA:
-
Extracellular DNA
- MIC:
-
Minimal inhibitory concentration
- TMT:
-
Tandem mass tags
- VRE:
-
Vancomycin-resistant enterococci
References
Ali L, Goraya MU, Arafat Y, Ajmal M, Chen JL, Yu D. Molecular Mechanism of Quorum-Sensing in Enterococcus faecalis: Its Role in Virulence and Therapeutic Approaches. Int J Mol Sci. 2017;18(5):.
Flokas ME, Karageorgos SA, Detsis M, Alevizakos M, Mylonakis E. Vancomycin-resistant enterococci colonisation, risk factors and risk for infection among hospitalised paediatric patients: a systematic review and meta-analysis. Int J Antimicrob Agents. 2017;49(5):565–72.
Dupre I, Zanetti S, Schito AM, Fadda G, Sechi LA. Incidence of virulence determinants in clinical Enterococcus faecium and Enterococcus faecalis isolates collected in Sardinia (Italy). J Med Microbiol. 2003;52(Pt 6):491–8.
Sandoe JA, Witherden IR, Cove JH, Heritage J, Wilcox MH. Correlation between enterococcal biofilm formation in vitro and medical-device-related infection potential in vivo. J Med Microbiol. 2003;52(Pt 7):547–50.
Seno Y, Kariyama R, Mitsuhata R, Monden K, Kumon H. Clinical implications of biofilm formation by Enterococcus faecalis in the urinary tract. Acta Med Okayama. 2005;59(3):79–87.
Toledo-Arana A, Valle J, Solano C, Arrizubieta MJ, Cucarella C, Lamata M, et al. The enterococcal surface protein, Esp, is involved in Enterococcus faecalis biofilm formation. Appl Environ Microbiol. 2001;67(10):4538–45.
Zheng JX, Wu Y, Lin ZW, Pu ZY, Yao WM, Chen Z, et al. Characteristics of and virulence factors associated with biofilm formation in clinical Enterococcus faecalis isolates in China. Front Microbiol. 2017;8:2338.
Paganelli FL, Willems RJ, Leavis HL. Optimizing future treatment of enterococcal infections: attacking the biofilm? Trends Microbiol. 2012;20(1):40–9.
Kayaoglu G, Orstavik D. Virulence factors of Enterococcus faecalis: relationship to endodontic disease. Crit Rev Oral Biol Med. 2004;15(5):308–20.
Park SY, Kim KM, Lee JH, Seo SJ, Lee IH. Extracellular gelatinase of Enterococcus faecalis destroys a defense system in insect hemolymph and human serum. Infect Immun. 2007;75(4):1861–9.
Kristich CJ, Li YH, Cvitkovitch DG, Dunny GM. Esp-independent biofilm formation by Enterococcus faecalis. J Bacteriol. 2004;186(1):154–63.
Mohamed JA, Murray BE. Lack of correlation of gelatinase production and biofilm formation in a large collection of Enterococcus faecalis isolates. J Clin Microbiol. 2005;43(10):5405–7.
Anderson AC, Jonas D, Huber I, Karygianni L, Wolber J, Hellwig E, et al. Enterococcus faecalis from food, clinical specimens, and Oral sites: prevalence of virulence factors in association with biofilm formation. Front Microbiol. 2015;6:1534.
Frees D, Savijoki K, Varmanen P, Ingmer H. Clp ATPases and ClpP proteolytic complexes regulate vital biological processes in low GC, Gram-positive bacteria. Mol Microbiol. 2007;63(5):1285–95.
Lemos JA, Burne RA. Regulation and physiological significance of ClpC and ClpP in Streptococcus mutans. J Bacteriol. 2002;184(22):6357–66.
Wang C, Li M, Dong D, Wang J, Ren J, Otto M, et al. Role of ClpP in biofilm formation and virulence of Staphylococcus epidermidis. Microbes Infect. 2007;9(11):1376–83.
Fernandez L, Breidenstein EB, Song D, Hancock RE. Role of intracellular proteases in the antibiotic resistance, motility, and biofilm formation of Pseudomonas aeruginosa. Antimicrob Agents Chemother. 2012;56(2):1128–32.
Xie F, Zhang Y, Li G, Zhou L, Liu S, Wang C. The ClpP protease is required for the stress tolerance and biofilm formation in Actinobacillus pleuropneumoniae. PLoS One. 2013;8(1):e53600.
Huang J, Wang X, Cao Q, Feng F, Xu X, Cai X. ClpP participates in stress tolerance and negatively regulates biofilm formation in Haemophilus parasuis. Vet Microbiol. 2016;182:141–9.
Capestany CA, Tribble GD, Maeda K, Demuth DR, Lamont RJ. Role of the Clp system in stress tolerance, biofilm formation, and intracellular invasion in Porphyromonas gingivalis. J Bacteriol. 2008;190(4):1436–46.
Liu Q, Wang X, Qin J, Cheng S, Yeo WS, He L, et al. The ATP-dependent protease ClpP inhibits biofilm formation by regulating Agr and Cell Wall hydrolase Sle1 in Staphylococcus aureus. Front Cell Infect Microbiol. 2017;7:181.
Kwon HY, Kim SW, Choi MH, Ogunniyi AD, Paton JC, Park SH, et al. Effect of heat shock and mutations in ClpL and ClpP on virulence gene expression in Streptococcus pneumoniae. Infect Immun. 2003;71(7):3757–65.
Michel A, Agerer F, Hauck CR, Herrmann M, Ullrich J, Hacker J, et al. Global regulatory impact of ClpP protease of Staphylococcus aureus on regulons involved in virulence, oxidative stress response, autolysis, and DNA repair. J Bacteriol. 2006;188(16):5783–96.
Zhao BB, Li XH, Zeng YL, Lu YJ. ClpP-deletion impairs the virulence of Legionella pneumophila and the optimal translocation of effector proteins. BMC Microbiol. 2016;16(1):174.
Knudsen GM, Olsen JE, Aabo S, Barrow P, Rychlik I, Thomsen LE. ClpP deletion causes attenuation of Salmonella Typhimurium virulence through mis-regulation of RpoS and indirect control of CsrA and the SPI genes. Microbiology. 2013;159(Pt 7):1497–509.
Shoji M, Cui L, Iizuka R, Komoto A, Neoh HM, Watanabe Y, et al. walK and clpP mutations confer reduced vancomycin susceptibility in Staphylococcus aureus. Antimicrob Agents Chemother. 2011;55(8):3870–81.
Bæk KT, Grundling A, Mogensen RG, Thogersen L, Petersen A, Paulander W, et al. Beta-lactam resistance in methicillin-resistant Staphylococcus aureus USA300 is increased by inactivation of the ClpXP protease. Antimicrob Agents Chemother. 2014;58(8):4593–603.
Wang X, Han H, Lv Z, Lin Z, Shang Y, Xu T, et al. PhoU2 but Not PhoU1 as an Important Regulator of Biofilm Formation and Tolerance to Multiple Stresses by Participating in Various Fundamental Metabolic Processes in Staphylococcus epidermidis. J Bacteriol. 2017;199(24):e00219–7.
Feng J, Michalik S, Varming AN, Andersen JH, Albrecht D, Jelsbak L, et al. Trapping and proteomic identification of cellular substrates of the ClpP protease in Staphylococcus aureus. J Proteome Res. 2013;12(2):547–58.
Frees D, Andersen JH, Hemmingsen L, Koskenniemi K, Baek KT, Muhammed MK, et al. New insights into Staphylococcus aureus stress tolerance and virulence regulation from an analysis of the role of the ClpP protease in the strains Newman, COL, and SA564. J Proteome Res. 2012;11(1):95–108.
Gaillot O, Pellegrini E, Bregenholt S, Nair S, Berche P. The ClpP serine protease is essential for the intracellular parasitism and virulence of Listeria monocytogenes. Mol Microbiol. 2000;35(6):1286–94.
Frees D, Qazi SN, Hill PJ, Ingmer H. Alternative roles of ClpX and ClpP in Staphylococcus aureus stress tolerance and virulence. Mol Microbiol. 2003;48(6):1565–78.
Hou XH, Zhang JQ, Song XY, Ma XB, Zhang SY. Contribution of ClpP to stress tolerance and virulence properties of Streptococcus mutans. J Basic Microbiol. 2014;54(11):1222–32.
Rajagopal S, Sudarsan N, Nickerson KW. Sodium dodecyl sulfate hypersensitivity of clpP and clpB mutants of Escherichia coli. Appl Environ Microbiol. 2002;68(8):4117–21.
Park CY, Kim EH, Choi SY, Tran TD, Kim IH, Kim SN, et al. Virulence attenuation of Streptococcus pneumoniae clpP mutant by sensitivity to oxidative stress in macrophages via an NO-mediated pathway. J Microbiol. 2010;48(2):229–35.
Pei H, Han S, Yang S, Lei Z, Zheng J, Jia Z. Phosphorylation of bacterial L9 and its functional implication in response to starvation stress. FEBS Lett. 2017;591(20):3421–30.
Prepiak P, Defrancesco M, Spadavecchia S, Mirouze N, Albano M, Persuh M, et al. MecA dampens transitions to spore, biofilm exopolysaccharide and competence expression by two different mechanisms. Mol Microbiol. 2011;80(4):1014–30.
Ge X, Kitten T, Chen Z, Lee SP, Munro CL, Xu P. Identification of Streptococcus sanguinis genes required for biofilm formation and examination of their role in endocarditis virulence. Infect Immun. 2008;76(6):2551–9.
Suriyanarayanan T, Qingsong L, Kwang LT, Mun LY, Truong T, Seneviratne CJ. Quantitative proteomics of strong and weak biofilm formers of Enterococcus faecalis reveals novel regulators of biofilm formation. Mol Cell Proteomics. 2018;17(4):643–54.
Cassenego AP, de Oliveira NE, Laport MS, Abranches J, Lemos JA, Giambiagi-deMarval M. The CtsR regulator controls the expression of clpC, clpE and clpP and is required for the virulence of Enterococcus faecalis in an invertebrate model. Antonie Van Leeuwenhoek. 2016;109(9):1253–9.
Del Papa MF, Perego M. Enterococcus faecalis virulence regulator FsrA binding to target promoters. J Bacteriol. 2011;193(7):1527–32.
Armin S, Fallah F, Karimi A, Rashidan M, Shirdust M, Azimi L. Genotyping, antimicrobial resistance and virulence factor gene profiles of vancomycin resistance Enterococcus faecalis isolated from blood culture. Microb Pathog. 2017;109:300–4.
Elsner HA, Sobottka I, Mack D, Claussen M, Laufs R, Wirth R. Virulence factors of Enterococcus faecalis and Enterococcus faecium blood culture isolates. Eur J Clin Microbiol Infect Dis. 2000;19(1):39–42.
Zahedi Bialvaei A, Rahbar M, Yousefi M, Asgharzadeh M, Samadi KH. Linezolid: a promising option in the treatment of gram-positives. J Antimicrob Chemother. 2017;72(2):354–64.
Rosenblat JD, McIntyre RS. Efficacy and tolerability of minocycline for depression: a systematic review and meta-analysis of clinical trials. J Affect Disord. 2018;227:219–25.
Shiwa Y, Yoshikawa H, Tanaka T, Ogura M. Bacillus subtilis degSU operon is regulated by the ClpXP-Spx regulated proteolysis system. J Biochem. 2015;157(5):321–30.
Rojas-Tapias DF, Helmann JD. Identification of novel Spx regulatory pathways in Bacillus subtilis uncovers a close relationship between the CtsR and Spx regulons. J Bacteriol. 2019;201(13):e00151–19.
Kline KA, Kau AL, Chen SL, Lim A, Pinkner JS, Rosch J, et al. Mechanism for sortase localization and the role of sortase localization in efficient pilus assembly in Enterococcus faecalis. J Bacteriol. 2009;191(10):3237–47.
Mohamed JA, Huang W, Nallapareddy SR, Teng F, Murray BE. Influence of origin of isolates, especially endocarditis isolates, and various genes on biofilm formation by Enterococcus faecalis. Infect Immun. 2004;72(6):3658–63.
Dai L, Yang L, Parsons C, Findlay VJ, Molin S, Qin Z. Staphylococcus epidermidis recovered from indwelling catheters exhibit enhanced biofilm dispersal and "self-renewal" through downregulation of agr. BMC Microbiol. 2012;12:102.
Velikova N, Kavanagh K, Wells JM. Evaluation of Galleria mellonella larvae for studying the virulence of Streptococcus suis. BMC Microbiol. 2016;16(1):291.
Wu P, Shang Q, Huang H, Zhang S, Zhong J, Hou Q, et al. Quantitative proteomics analysis provides insight into the biological role of Hsp90 in BmNPV infection in Bombyx mori. J Proteome. 2019;203:103379.
Ruiz-Cruz S, Espinosa M, Goldmann O, Bravo A. Global regulation of gene expression by the MafR protein of Enterococcus faecalis. Front Microbiol. 2015;6:1521.
Acknowledgments
The authors thank Prof. Michael G. Caparon (Department of Molecular Microbiology, Washington University School of Medicine, Saint Louis, Missouri, USA) and Prof. Jingren Zhang (Center for Infectious Disease Research, School of Medicine, Tsinghua University, Beijing, China) for generously providing generously plasmids pJRS233 and pIB166, respectively. We also thank Ms. Cynthia Brast (University of Florida, Gainesville, Florida, USA) for reviewing the manuscript.
Funding
This work was supported by the following grants: the Sanming Project of Medicine in Shenzhen (grant number SMGC201705029); Science, Technology and Innovation Commission of Shenzhen Municipality of key funds (JCYJ20170412143551332 and JCYJ20180508162403996) and basic research funds (JCYJ20180302144721183 and JCYJ20180302144431923). Shenzhen Nanshan District Scientific Research Program of the People’s Republic of China (grant number 2019027).
Author information
Authors and Affiliations
Contributions
JZ participated in the design of the study, carried out the gene manipulation, biofilm and eDNA assay, analyzed and interpreted the proteomic data, and drafted the manuscript. YW participated in the gene manipulation, RT-qPCR, and proteomic data. ZL conducted the RNA extraction and RT-qPCR. GW performed antibiotic-susceptibility testing, antimicrobial tolerance experiments, and protein extraction. SJ, XS, and HT performed the gene manipulation, stress tolerance experiments, biofilm and eDNA tests, G. mellonella trials and RNA extraction, and RT-qPCR. ZY and DQ designed the study, participated in the data analysis, and provided critical revisions of the manuscript for valuable intellectual content. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1: Figure S1.
Relative RNA levels of clpP in E. faecalis strains. The RNA levels of clpP were determined by RT-qPCR, with the OG1RF parent strain as the reference strain (RNA level = 1). Three independent experiments were performed, and the data represent means ± SD.
Additional file 2: Figure S2.
Coefficient of variation (CV) distributions for the two independent repetitions of the four group samples. The proteins were extracted from the E. faecalis OG1RF and its ΔclpP mutant strains and divided into four groups: strains cultured at 37 °C for 4 h to logarithmic phase were marked as OG1RF_4 or OG1RF_clpP_4; strains cultured at 37 °C for 12 h to stationary phase were marked as OG1RF_12 or OG1RF_clpP_12. The proteins extracted from each group included two independent biological repetitions, and the peptides were labeled with TMT6/10-plex reagents, then sequenced with the Orbitrap Q Exactive HF-X mass spectrometer.
Additional file 3: Figure S3.
Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis of differential abundance proteins (DAPs) (stationary phase). The protein family and pathway were analyzed using the KEGG database.
Additional file 4: Table S1.
Antimicrobial susceptibility of E.faecalis determined by conventional broth macrodilution method. Table S2. Primers used for the RT-qPCR for the detection of RNA levels of differential abundance proteins.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Zheng, J., Wu, Y., Lin, Z. et al. ClpP participates in stress tolerance, biofilm formation, antimicrobial tolerance, and virulence of Enterococcus faecalis. BMC Microbiol 20, 30 (2020). https://doi.org/10.1186/s12866-020-1719-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12866-020-1719-9