
Abstract
Background
The Gram-positive facultative methylotrophic bacterium Bacillus methanolicus uses the sedoheptulose-1,7-bisphosphatase (SBPase) variant of the ribulose monophosphate (RuMP) cycle for growth on the C1 carbon source methanol. Previous genome sequencing of the physiologically different B. methanolicus wild-type strains MGA3 and PB1 has unraveled all putative RuMP cycle genes and later, several of the RuMP cycle enzymes of MGA3 have been biochemically characterized. In this study, the focus was on the characterization of the transaldolase (Ta) and its possible role in the RuMP cycle in B. methanolicus.
Results
The Ta genes of B. methanolicus MGA3 and PB1 were recombinantly expressed in Escherichia coli, and the gene products were purified and characterized. The PB1 Ta protein was found to be active as a homodimer with a molecular weight of 54 kDa and displayed KM of 0.74 mM and Vmax of 16.3 U/mg using Fructose-6 phosphate as the substrate. In contrast, the MGA3 Ta gene, which encodes a truncated Ta protein lacking 80 amino acids at the N-terminus, showed no Ta activity. Seven different mutant genes expressing various full-length MGA3 Ta proteins were constructed and all gene products displayed Ta activities. Moreover, MGA3 cells displayed Ta activities similar as PB1 cells in crude extracts.
Conclusions
While it is well established that B. methanolicus can use the SBPase variant of the RuMP cycle this study indicates that B. methanolicus possesses Ta activity and may also operate the Ta variant of the RuMP.
Similar content being viewed by others

Background
There are three groups of methylotrophic bacteria using the ribulose monophosphate (RuMP) cycle for the fixation of formaldehyde: Gram-negative obligate and facultative methylotrophs and Gram-positive facultative methylotrophs [3, 4]. Bacillus methanolicus, a facultative methylotroph, belongs to the latter group and utilizes methanol as its sole source of energy and carbon, assimilating formaldehyde via the RuMP cycle [33, 53]. B. methanolicus is a valuable host for industrial fermentations using methanol as sole carbon source [12, 14, 27, 44]. The oxidation of methanol to formaldehyde, the first step of methanol utilization in B. methanolicus, is catalyzed by NAD-dependent methanol dehydrogenase [6, 15, 24]. It has been shown that B. methanolicus possesses three different genes, all encoding active Mdhs [23, 36].
The RuMP cycle consists of three main parts: fixation, cleavage, and rearrangement (Fig. 1). In the fixation part, ribulose 5-phosphate (Ru5P) is condensed with formaldehyde by 3-hexulose-6-phosphate synthase (Hps) to yield hexulose 6-phosphate (H6P), which is subsequently isomerized to fructose 6-phosphate by 6-phospho-3-hexuloisomerase (Phi). Mdh, Hps, and Phi are specific enzymes of methylotrophy, whereas the other involved enzymes are also part of other pathways, such as glycolysis, Calvin cycle, and pentose phosphate pathway. In the cleavage part, F6P is converted by phosphofructokinase (Pfk) to fructose 1,6-bisphosphatase (FBP), which is further cleaved into the triose phosphates glyceraldehyde 3-phosphate (GAP) and dihydroxyacetone phosphate (DHAP) by fructose-1,6-bisphosphate aldolase (Fba).

Schematic representation of the RuMP as present in B. methanolicus. Reactions are represented by arrows and the genes encoding the respective enzymes are depicted next to them. For abbreviations of the genes see the text. Abbreviations for metabolites are H6-P, 3-hexulose 6-phosphate; F6-P, fructose-6-phosphate; FBP, fructose-1,6-bisphosphate; GAP, glyceraldehyde 3-phosphate; DHAP, dihydroxyacetone phosphate; E4-P, erythrose 4-phosphate; SBP, sedoheptulose 1,7-bisphosphate; S7-P, sedoheptulose-7-phosphate; Ri5-P, ribose 5-phosphate; X5P, xylulose 5-phosphate; Ru5P, ribulose 5-phosphate
Genome sequencing of the two physiologically different B. methanolicus wild-type strains MGA3 and PB1 that showed considerable differences with respect to growth, amino acid production, and respiration profiles in fed-batch methanol cultivations [23] provided genetic insight into all methylotrophic pathways of this species ([23]; Irla et al. 2014). Both strains carry the methylotrophy plasmids pBM19 and pBM20, respectively, and MGA3 in addition carry the natural and cryptic plasmid pBM69. B. methanolicus has two Fba enzymes, one encoded on the chromosome and one plasmid encoded variant (genes fbaC and fbaP, respectively). They catalyze opposite reactions, since FbaC is the major glycolytic Fba, whereas FbaP catalyzes the aldol condensation in gluconeogenesis [62]. Interestingly, B. methanolicus possesses enzymes for the sedoheptulose-1,7-bisphosphatase (SBPase) variant as well as the transaldolase (Ta) variant of the rearrangement part of the RuMP cycle. In this part, F6P and the triosephosphates are converted into Ru5P, which is thereby regenerated for the fixation of formaldehyde. In the SBPase variant, erythrose 4-phosphate (E4P) and DHAP are condensed to sedoheptulose 1,7-bisphosphate (SBP), catalyzed by either FbaP or FbaC. Therefore, these enzymes function also as sedoheptulose-1,7-bisphosphate aldolases [61]. In the following step, SBPase dephosphorylates SBP to yield S7P. Both the chromosomally encoded FBPase GlpXC and the plasmid encoded FBPase/SBPase GlpXP are active as bisphosphatases with FBP, whereas only the plasmid encoded GlpXP was found to function as SBPase [61]. In the Ta variant, E4P and F6P are directly converted into GAP and S7P. Both variants of the RuMP cycle involve the enzymes transketolase (Tkt) [39], ribose-5-phosphate isomerase (Rpi), and ribulose-phosphate 3-epimerase (Rpe). Previous studies on gene regulation and extensive enzyme characterizations has concluded that B. methanolicus MGA3 uses the SBPase variant, and the biological significance of the annotated Ta genes is unknown.
The genes of the rearrangement part are present in two versions in B. methanolicus; one encoded on the chromosome and the other one on the naturally occurring plasmid pBM19. The Ta and Rpi genes are exceptional as they are only present on the chromosome [11, 23].
Ta catalyzes the transfer of a dihydroxyacetone moiety from a ketose donor onto an aldehyde acceptor. This enzyme does not require any known cofactors and performs a base-catalyzed aldol cleavage reaction via a Schiff base intermediate. Several Tas have been purified and characterized from various sources, such as bacteria [60], archaea [59], yeasts [18], fungi [35], plants [41], and mammals, including humans [9]. The only thermostable Ta characterized to date is from Methanocaldococcus jannaschii [59]. Five subfamilies can be distinguished, based on the phylogeny of Ta [50]. Subfamily I is considered as the classical Ta that occurs in all domains of life, for example, TalB from E. coli [60], but not in plants. Subfamily II and subfamily III Tas are present in plants and in the case of subfamily III also in bacteria. Subfamily IV consists of small Tas found in bacteria and archaea. To the best of our knowledge, biochemical evidence for Ta enzyme activity has not been shown yet for enzymes belonging to either subfamily II or V [51]. Interestingly, enzymes of subfamily V are active as F6P aldolases as shown for respective enzymes from E. coli [57]. However, certain organisms, like Zymomobas mobilis [17], and Entamoeba histolytica [65], as well as certain mammalian tissues do not express Ta [21] and the non-oxidative branch can function without this enzyme [40, 45].
In this study we investigated the biochemical properties and biological function of the putative Ta proteins in B. methanolicus MGA3 and PB1 [47]. The MGA3 Ta gene encodes a truncated and non-active protein (UNIPROT IDI3EBM5) while the PB1 Ta gene product (I3E1B6) displayed Ta activity, tested both in vitro and in vivo. Interestingly, genetically repaired full-length versions of the MGA3 Ta gene showed Ta activity. The PB1 enzyme was biochemically characterized and our results may indicate that B. methanolicus has an operative TA variant of the RuMP cycle for methylotrophic growth.
Results
The genomes from B. methanolicus PB1 and MGA3 encode transaldolase
Inspection of the genome sequences of B. methanolicus PB1 and MGA3 revealed that each strain possesses a single chromosomally encoded transaldolase, while no Ta gene was found on the plasmids pBM20 and pBM19. Bioinformatics comparison of the deduced primary amino acid sequences of these Ta proteins, MGA3 (TaMGA3) and PB1 (TaPB1) [23, 25], with Ta proteins from other Bacillus species (Fig. 2a) revealed that they can be associated with group IV family of Ta enzymes. This comparison surprisingly shows that TaMGA3 lacks about 80 N-terminal amino acids that are present in TaPB1 and the other Tas compared here. Aspartate residue D20 is part of the catalytic triad D20-E84-K107 of Ta enzymes [50, 55] and this triad is present in TaPB1, but D20 is absent from TaMGA3. Thus, TaMGA3 likely lacks Ta activity.

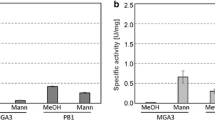
a Schematic comparison of transaldolases from B. methanolicus strains MGA3 and PB1 with transaldolases from Bacillus subtilis, Bacillus cereus, and Bacillus licheniformis. Shaded boxes represent the protein sequence, the number in front of and behind the shaded boxes represent the first and last amino acid. Residues of the catalytic active center are given in one letter code with their position in the protein in superscript. D, E, and K represent the amino acids aspartate, glutamate, and lysine. b Ta activities measured in crude extracts of B. methanolicus strains MGA3 and PB1
Closer inspection of the gene sequences revealed several single nucleotide polymorphisms (SNPs) and suggested that the putative translation initiation codon ATG present in PB1 is mutated to ATA in MGA3 (Fig. 3). An alternative GTG start codon only leads to a peptide with two amino acids (TaMGA3put). These SNPs could be confirmed when the corresponding region was PCR amplified from MGA3 total DNA and sequenced in several parallels (data not shown). Thus, these SNPs were not simply due to previous sequencing errors.

Comparison of the nucleotide sequences of the Ta genes from B. methanolicus PB1 and MGA3 as well as those of recreated MGA3 Ta genes. MGA3put represents an alternative Ta coding sequence of MGA3. For MGA3rec the ATA was changed to ATG and adjusted to that of PB1. For the variants M1-M6 the third codon of the sequence upstream of taMGA3 was changed from a stop TAA codon to different codons. The dashes indicate the same nucleotide as for PB1 and the dots show that the sequence continues. The red letters indicate a stop codon and the pink letter shows an additional nucleotide at that position. A rough screen showed that crude extracts of E. coli BL21 (DE3) expressing all of these proteins except for TaMGA3contained at least 20 mU/mg, i.e. two fold of the activity present in the parent strain (about 10 mU/mg)
Enzyme assays in crude extracts demonstrated Ta activities for both strains
In order to test if Ta activity can be detected in strains B. methanolicus PB1 and MGA3, crude extracts were prepared from cells growing in defined methanol media. Surprisingly, both strains displayed Ta activities although MGA3 crude extracts contained significantly lower Ta activities (0.11 ± 0.02 and 0.24 ± 0.03 U/mg for MGA3 and PB1, respectively; Fig. 2b). We also tested just the assay mixture without added crude extracts or with crude cell extracts of a Ta negative C. glutamicum mutant ∆tal in order to exclude any background activity of the assay mixture. Ta activity was not detected when assaying just the assay mixture without added crude extracts or with crude cell extracts of a Ta negative C. glutamicum mutant ∆tal. A possible explanation for the observed Ta activity in B. methanolicus PB1 and MGA3 could be the presence of potential bifunctional enzymes with transaldolase activity, such as glucose-6-phosphate isomerase/transaldolase described for Gluconobacter oxydans [64]. However, a BLAST search with the nucleotide and amino acid sequences of Ta from B. methanolicus PB1 and sequences of other Ta variants against the MGA genome sequence did not reveal any putative Ta genes or proteins. In addition, also enzymes like fructose-6-phosphate aldolase, that might use the substrates of our Ta enzyme assay, were not found to be encoded in the MGA3 genome sequence.
Construction of mutant taMGA3 genes expressing full-length Ta proteins
In order to test if the TaMGA3 sequence can be corrected to recreate a full-length TA, the ATA was changed to ATG to generate a translational start codon and the downstream T (indicated in pink in Fig. 3) was deleted to correct for a reading-frame shift. The corresponding protein was named TaMGA3rec. As alternative, full-length TaMGA3 proteins based on using the GTG as start codon (TaMGA3rec) were constructed (named M1 to M6; Fig. 3). These variants differ with regard to the change of the TAA stop codon to different sense codons (Fig. 3).
Heterologous expression of different MGA3 ta variants in E. coli
To test for Ta activity in crude cell extracts of E. coli DH5α, genes for the Ta variants M1-M6 and taMGA3rec (Fig. 3) were expressed. As suggested based on the bioinformatics analysis, TaMGA3rec showed Ta activity, but TaMGA3 did not. This is likely due to the fact that TaMGA3 lacks 80 amino acids that are present in the N-terminus of TaPB1 and TaMGA3rec. Recreation of functional Ta proteins was successful since TaMGA3rec and all six M1-M6 variants were found to be catalytically active (data not shown).
Overexpression and purification of the TaMGA3rec and TaPB1 proteins
The coding sequences for TaPB1 and TaMGA3rec were cloned into the vector pET28b for production of the corresponding enzymes with a C-terminal His-tag in E. coli BL21 (DE3). Recombinant protein production was induced by the addition of IPTG to exponentially growing cell cultures. Stationary growth phase cells were harvested, crude extracts were prepared, and the proteins were purified by Ni-NTA chromatography. A buffer exchange to 50 mM Tris–HCl (pH 7.8) was performed and average concentrations of about 2.5 mg/mL were obtained. A total protein amount of about 7.5 mg was obtained per purification from 500 mL of culture broth. To determine the oligomeric state of the purified enzymes gel filtration and Ta activity assays of the eluted fractions were performed. The enzymes are active as homodimers with a molecular weight of 54 kDa since they eluted in a single fraction corresponding to that molecular weight. In the next steps we chose to focus on the biochemical characterization of wild-type TaPB1, whereas TaMGA3rec was not studied any further.
Biochemical characterization of TaPB1
Optimal conditions for TaPB1 activity
A coupled spectrometric assay for measuring the formation of GAP from F6P and E4P (as described in Materials & Methods) was used to determine optimal assay conditions for the TaPB1. The auxiliary enzymes TIM and G3PDH were added in excess and found not to be limiting under the different conditions. TaPB1 showed a pH optimum of pH 7.8. The highest activity was measured in the range of 60 °C to 65 °C (data not shown). The physiological temperature of 50 °C was chosen for further experiments and for determination of kinetic parameters.
Kinetic parameters of TaPB1
We chose the conversion of F6P and E4P to S7P and GAP to determine the kinetic parameters of TaPB1 (Table 1). All assays were performed in 50 mM Tris-HCl, pH 7.8, at 50 °C. A KM of 740 μM and Vmax of 16.3 U/mg was determined for the substrate F6P. The catalytic efficiency for F6P of 9.98 s− 1 mM− 1 could be derived. The corresponding kinetic constants for substrate E4P were KM 2,5 mM and Vmax 8.9 U/mg.
Heterologous expression of taPB1 and TaMGA3rec complemented Ta-deficiency in C. glutamicum ∆tal
A genetic complementation experiment was performed to detect activity of TaPB1 and TaMGA3rec in vivo. Since methods for gene deletion in B. methanolicus are lacking, a defined deletion mutant lacking Ta encoding gene tal was constructed in C. glutamicum. This bacterium possesses functional transaldolase (encoded by cg1776), which operates in the pentose phosphate pathway. Comparative growth analysis of C. glutamicum ∆tal and wild type revealed that C. glutamicum requires Ta for maximum growth rate in minimal medium with ribose as sole carbon source. C. glutamicum ∆tal could be complemented by expression of the endogenous tal from plasmid pVWEx1. Strain C. glutamicum ∆tal (pVWEx1-talCG) grew with an even slightly higher specific growth rate than the wild type in CGXII minimal medium with 2% ribose. By contrast, the strain carrying the empty vector showed a reduced specific growth rate (Fig. 4). Thus, the deletion of tal, which is part of the tkt-tal-zwf-opcA-pgl operon in this bacterium, did not result in any secondary effects, such as polar effects on cotranscribed genes downstream of tal. In order to test if Ta from B. methanolicus PB1 is active in C. glutamicum, taPB1 was heterologously expressed in the ∆tal strain. As this strain showed a similar specific growth rate as C. glutamicum ∆tal (pVWEx1-talCG) (Fig. 4), expression of taPB1 complemented C. glutamicum Δtal. Since the same effect was observed for heterologous expression of taMGA3rec, both Ta from B. methanolicus PB1 and the reconstructed version of the B. methanolicus MGA3 Ta protein are functionally active in vivo in C. glutamicum.

Genetic complementation of C. glutamicum ∆tal by expression of taPB1, taMGA3rec and talCG. The mean values and standard deviations for the specific growth rate of three replicates are shown for growth in CGXII minimal medium supplemented with 2% ribose
Overexpression of taPB1 in B. methanolicus MGA3
To test if overexpression of taPB1 in B. methanolicus MGA3 would affect growth with methanol as sole carbon source, strains B. methanolicus MGA3(pHP13) and MGA3(pTH1mp-taPB1) were constructed. However, growth in MeOH medium supplemented with different methanol concentrations was comparable. Thus, overexpression of taPB1 in B. methanolicus MGA3 did not result in a growth advantage on methanol minimal media.
Size exclusion chromatography, SDS-PAGE and MALDI-TOF analysis of MGA3 crude extracts
At this point, collected data and results from MGA3 were contradictory; i) the MGA3 genome has one Ta gene encoding a truncated and inactive protein, ii) MGA3 crude extracts display Ta activity, and iii) single point mutations can be introduced in the Ta gene restoring a full-length and catalytically active Ta protein. We therefore proceeded to investigate what could be the genetic and enzymatic cause of Ta activity in MGA3. First, Ta activity measured in a crude cell extract of B. methanolicus MGA3 (0.284 U) was used for size exclusion chromatography (gel filtration), in order to determine if Ta activity can be assigned to a particular fraction. Ta activity was confirmed for fractions 2 (0.174 U) and 8 (0.110 U) of a total of 18 fractions received, leading to 100% of the initial activity found in the crude cell extract. The two fractions showing Ta activity were separated using a SDS-PAGE and then every band was extracted from the gel and used for analysis by MALDI-TOF. However, peptide mass fingerprinting analysis did not allow to identify these proteins. This may be due to technical limitations in our laboratory, since, by contrast, the label-free quantitative proteomics analysis performed by [42] comparing growth of MGA3 with mannitol or methanol revealed that transaldolase I3EBM5 was not affected, whereas strong upregulation of fructose-1,6-bisphosphatase/sedoheptulose-1,7-bisphosphatase (GlpX) was observed [42]. Thus, there is evidence that MGA3 synthesizes transaldolase I3EBM5 or a variant sharing the MS detected peptides.
Identification of a possible transcription start site upstream of the MGA3 ta gene
Since purified protein corresponding to the annotated Ta of B. methanolicus MGA3 (TaMGA3) showed no activity when expressed in E. coli, however, crude extracts of MGA3 did show Ta activity, the available transcriptome data from RNA-sequencing [26] were used to search for a possible transcription start site (TSS) upstream of the start codon of our proposed Ta coding sequence taMGA3put (Fig. 3). Indeed, a putative TSS preceded by a − 10 region (TTTCAA(T)) and a − 35 region (TTGAAA) were found. The − 35 region represents the consensus sequence [26], while the − 10 region shares only a low similarity with the B. methanolicus consensus sequence (TATAAT) [26].
Proteome data indicating biosynthesis of functional Ta encoded by taMGA3put
A search with the amino acid sequence of TaMGA3put against a database including proteome data from B. methanolicus MGA3 [42] identified peptides upstream of the annotated Ta (Müller, personal communication), which indicates transcription and translation of taMGA3put in MGA3. Taken together, our analysis of the available transcriptome and proteome data may indicate transcription of the proposed taMGA3put-coding sequence, leading to the translation of a functional Ta protein responsible for the measured Ta activity in B. methanolicus MGA3. We did not try to determine kinetic constants of this putative Ta since the low activity present in the crude extracts was expected not to yield reliable data.
Discussion
The focus of this study was a genetic and biochemical characterization of the transaldolase (Ta) from B. methanolicus [47] and to unravel its possible role in the RuMP cycle and thus methylotrophy. B. methanolicus wild-type strains MGA3 and PB1 display considerable physiological differences (growth rate, methanol consumption rate, respiration) in fed-batch methanol cultivations and these phenotypes are accompanied by certain genetic differences [23]. These two strains together therefore represent a valuable model system to investigate and understand methylotrophy, although MGA3 has been and still is the host for all metabolic engineering research of this organism. Both MGA3 and PB1 have one chromosomal putative Ta gene, and bioinformatics analysis presented here, indicated that the MGA3 gene has acquired point mutations in the 5′-region leading to a truncated gene product with a defective active site. Recombinant expression of the truncated protein in E. coli confirmed that it displays no catalytic activity. PB1 on the other hand encodes a full-length Ta gene and this gene was shown to express Ta activity when tested both in vitro and in vivo. Surprisingly, Ta activity was still measured in cell free extracts of both MGA3 and PB1. Totally seven genetically engineered versions of the MGA3 Ta gene were then constructed with different point mutations repairing the full-length version of the gene, and all seven gene products displayed Ta activity in vitro. Attempts to understand and explain this discrepancy in MGA3 as well as the biological role of Ta in B. methanolicus is further discussed below. Primary sequence alignments indicated that the B. methanolicus TaPB1 protein belongs to the subfamily IV of Tas, similar as the Ta from Bacillus subtilis. Other well-characterized Ta proteins, such as TalB from E. coli and Ta from C. glutamicum belong to subfamily I and III, respectively.
Since the TaMGA3 protein displayed no catalytic activity, the full-length and catalytically active TaPB1 protein was used as B. methanolicus model protein for further purification and biochemical characterization in vitro. The kinetic constants for TaPB1were calculated based on enzyme assays at the B. methanolicus relevant physiological conditions of 50 °C and pH 7.8. The reported kinetic constants for Ta of E. coli, B. subtilis, and C. glutamicum have been determined at 30 °C and pH 8.5 [51]. The measured KM and Vmax for substrates F6P (0.74 mM and 16 U/mg) and E4P (2.5 mM and 8.9 U/mg) of TaPB1 are in the same range as those reported determined for the B. subtilis Ta (1.4 mM and 28 U/mg; 1.2 mM and 19 U/mg, respectively) [56]. While the KM for F6P of TalB from E. coli (1.2 mM) and of Ta from C. glutamicum (1.3 mM) are in the same range as for TaPB1, the Vmax values of the latter enzymes are slightly higher (60 U/mg and 110 U/mg, respectively) [51, 60]. For the substrate E4P the KM of TalB from E. coli (0.09 mM) and of Ta from C. glutamicum (0.7 mM) are lower, whereas their Vmax values areslightly higher (80 U/mg and 84 U/mg, respectively) [51, 60], compared to TaPB1.
Furthermore, the purified TaPB1was found to have a temperature optimum of 60 °C in vitro, which is high compared to both TalB from E. coli (40 °C) [60] and Ta from C. glutamicum (40 °C) [51]. This was expected since B. methanolicus is a thermophilic bacterium with an optimal growth temperature of 50 °C to 55 °C [5, 53]. Further, TalB of E. coli and Ta of C. glutamicum are both reported to lose all catalytic activity at temperatures above 50 °C, whereas Ta of B. subtilis displayed increased activity when temperature was raised from 20 °C to 55 °C [51, 56, 60]. Thermostability is a common feature of Tas belonging to subfamily IV and V and not only restricted to thermophilic organisms [51]. It is proposed that a compact protein structure and a tight packing of the subunits causes this thermostability [66]. Summarized, TaPB1 displays similar KM and Vmax values as Ta from B. subtilis and both proteins have temperature optima above 50 °C. TaPB1 is however slightly different from TalB of E. coli and Ta of C. glutamicum with respect to KM for substrate E4P, Vmax values, and temperature optimum. Moreover, TaPB1 was here demonstrated to form an active homodimer, whereas Ta of B. subtilis is reported to form a decamer (dimer of pentamers) [56], TalB of E. coli is a homodimer, and Ta of C. glutamicum is monomeric [51].
The active site is conserved among bacterial Ta proteins and it is also proposed that the catalytic mechanism is similar [51]. The important and highly conserved amino acid residue aspartate (Fig. 2a) acts as follows: The aspartate residue assists in the deprotonation of the C4 hydroxyl group of the enzyme-bound imine during the reaction catalyzed by Ta. This leads to the cleavage of the imine and releasing the first product, glyceraldehyde 3-phosphate (GAP). The resulting Schiff base intermediate is stabilized by resonance until the acceptor molecule is bound at the active site [30, 31, 50, 54, 55]. Since the truncated TaMGA3 protein lacks this active site aspartate residue, this likely explains why no Ta activity was detected when expressed recombinant in E. coli.
Combined genomic and experimental analyses in MGA3 for identification of any other potential proteins with Ta activity, or that utilizes metabolites of the Ta assay, gave no results, and re-inspection of previous RNA-sequencing and proteome data indicated that a full-length Ta protein presumably using an upstream and in-frame GTG start codon is expressed in this strain. This is peculiar as the third codon in this putative taMGA3put gene would be the translational stop codon TAA, and the biological explanation to this remains unknown. Plausible explanation could be that this putative gene is subject to rare phenomena such as translational recoding which refers to alternative “translational decoding” [19, 58]. There are three different classes of such translational recoding described in nature: i) programmed-frame-shifting, ii) translational bypassing, and iii) translational redefinition of codons [7, 20]. Recoding is caused by special signals on the mRNA and is characterized by an unchanged genetic code as well as the synthesis of more than one protein from one mRNA [8]. In case of translational bypassing the ribosome stops translation at a certain point, skips some nucleotides, and then continues translation downstream [10]. For example, in Bacillus firmus two open reading frames have been found that are separated by a UGA stop codon and might be subject to read through [28]. In theory, the general mechanisms of translational bypassing or stop codon read through might serve as an explanation for the translation of a full-length and active Ta protein in MGA3. However, this remains to be experimentally tested and this was not within the scope of this study.
It has previously been shown that B. methanolicus uses the SBPase variant of the RuMP cycle [61] and three separate metabolomics studies have identified S7P in methanol grown MGA3 cells [13, 34, 43]. Our results presented here may indicate that B. methanolicus also possesses an active Ta variant of the RuMP cycle for regeneration of the formaldehyde acceptor Ru5P. The metabolomics analysis by [34] revealed similar labeling kinetics of SBP and S7P, which may indicate that the SBPase variant of the RuMP cycle is dominant in MGA3 grown in 13C-MeOH. Data from metabolic flux analysis in MGA3 have indicated that both RuMP variants are used in parallel (Carnicer, personal communication), and at the same time Ta expression is not upregulated during growth on methanol, neither on the transcriptome nor the proteome level [23, 42]. It has been shown that plants can use Ta besides SBPases [48]; however, to date no bacterium has been reported that uses the Ta and SBPase variant of the RuMP pathway in parallel.
Conclusions
Evidence for an active transaldolase operating in B. methanolicus is presented based on biochemical characterization of the enzyme purified from recombinant E. coli. While it is well established that B. methanolicus can use the SBPase variant of the RuMP cycle this study indicates that B. methanolicus possesses Ta activity and may also operate the Ta variant of this cycle.
Methods
Microorganisms and cultivation conditions
Bacillus methanolicus strains were grown at 50 °C in the following media. SOBsuc medium is SOB medium (Difco) supplemented with 0.25 M sucrose. Methanol growth of B. methanolicus was performed in MeOH200 medium, containing salt buffer, 1 mM MgSO4, vitamins, trace metals, 0.025% yeast extract, and 200 mM methanol [29, 53]. The medium pH was adjusted to 7.2 unless stated otherwise. Bacterial growth was performed in shake flasks (500 mL) in 100 mL medium at 200 rpm and monitored by measuring the absorbance at 600 nm (A600). Chloramphenicol (5 μg/mL) was added to the medium when appropriate. The inoculation of the precultures for all growth experiments with B. methanolicus strains was performed with frozen ampoules of B. methanolicus as a starter culture. Ampoules of B. methanolicus cells were prepared from exponentially growing cultures (A600 of 1.0 to 1.5) and stored at − 80 °C in 15% (vol/vol) glycerol. Transformation of B. methanolicus MGA3 was performed by electroporation as described previously [29]. The E. coli strain DH5α was used as a standard cloning host. Recombinant cells were grown in lysogeny broth (LB) medium at 37 °C supplemented with kanamycin (25 μg/mL), spectinomycin (100 μg/mL), chloramphenicol (15 μg/mL), and 1 mM isopropyl-β-d-thiogalactopyranoside (IPTG) when appropriate and standard recombinant DNA procedures were performed as described elsewhere [49]. C. glutamicum cells were grown in lysogeny broth (LB) medium or CgXII minimal medium [32] and incubated at 30 °C. Recombinant protein production was carried out using E. coli BL21 (DE3) as host. Bacterial strains and plasmids used in this work are listed in Table 2 and oligonucleotides for polymerase chain reaction (PCR) and cloning are listed in Table 3.
The C. glutamicum wild type (WT) strain (ATCC 13032) and the derived Δtal mutant, lacking transaldolase activity, were used for the heterologous expression of the Ta genes taPB1 and taMGA3rec from B. methanolicus PB1 and MGA3, respectively. The vector pVWEx1 was used for IPTG-inducible expression of taPB1. For growth experiments, C. glutamicum cells were harvested from cultures grown in LB medium overnight by centrifugation (4000 g for 10 min), washed in CgXII minimal medium, and used to inoculate fresh CgXII minimal medium. All growth experiments with C. glutamicum were carried out with 50 ml medium in 500 mL baffled shake flasks at 30 °C and 120 rpm. Growth was monitored by determination of the absorbance at 600 nm (A600) until the stationary phase was reached.
Construction of tal (cg1776) deletion mutant in C. glutamicum
PCR products from chromosomal C. glutamicum DNA were obtained using primer pairs tal_cg_del_A; tal_cg_del_B and tal_cg_del_C; tal_cg_del_D. The resulting PCR products were used as template-DNA in a crossover PCR using primer pair tal_cg_del_A; tal_cg_del_D. The resulting PCR product with a shortened tal gene was then phosphorylated and ligated into the SmaI restricted vector pK19mobsacB [52]. Chromosomal deletion was performed as described elsewehere [16], and correct deletion was confirmed by sequencing using primers tal_cg_del_E; tal_cg_del_E. The constructed C. glutamicum Δtal strain was used for the heterologous and homologous expression of different Ta variants.
Vector constructions for heterologous expression of taPB1 and taMGA3rec of B. methanolicus and homologous expression of talCG in C. glutamicum
The PCR products from taPB1 and taMGA3rec were generated from genomic DNA of B. methanolicus PB1 and MGA3 by PCR, using the oligonucleotide primer pairs ta_PB1-RBS-fw; ta_PB1-rv and ta_MGA3rec-RBS-fw; ta_MGA3rec-rv (Table 3). The PCR product from talCG was generated from genomic DNA of C. glutamicum ATCC 13032 by PCR using the oligonucleotide primer pair tal_CG-RBS-fw; tal_CG-rv (Table 3). The amplified PCR products were digested with BamHI and KpnI and then ligated with the BamHI and KpnI restricted vector pVWEx1 [46]. The resulting vectors were named pVWEx1-taPB1, pVWEx1-taMGA3rec, and pVWEx1-talCG. The vector pVWEx1 allows IPTG-inducible gene expression in C. glutamicum and E. coli. All resulting vector inserts were sequenced using the primer pair pVWEx1_fw; pVWEx1_rv to confirm their sequence integrity.
Vector construction for heterologous expression of different taMGA3 variants in E. coli
For the heterologous expression of the different taMGA3put variants M1-M6 in E. coli DH5α, the primers ta_MGA3,M1_BamHI-fw to ta_MGA3,M6_BamHI-fw; ta_MGA3,M_BamHI-rv (Table 3) were used for amplification by PCR from chromosomal DNA of B. methanolicus MGA3. The PCR product of the annotated taMGA3 gene was generated using primers ta_MGA3_fw; ta_MGA3_rv. The resulting PCR products were digested with BamHI and ligated into the BamHI restricted vector pET28b (Table 3). Ta activity in crude cell extracts of recombinant E. coli DH5α strains was determined as described below.
Vector construction for homologous overexpression of taPB1 in B. methanolicus MGA3
The expression vector pTH1mp was used to allow methanol-inducible expression of the B. methanolicus PB1 Ta gene. This vector is analogous to the plasmid pHP13, in which the strong mdh promoter was cloned in-frame with the mdh RBS region to allow methanol-inducible expression in B. methanolicus. The DNA fragment of the taPB1-coding region was amplified from DNA of B. methanolicus by the primer pair ta_PB1-TH-fw; ta_PB1-TH-rv (Table 3). The resulting PCR product was digested with PciI/KpnI and ligated with the PciI/KpnI digested vector pTH1mp. The resulting vector was named pTH1mp-taPB1 and the correct insert was verified by sequencing using the primer pair pTH1_fw; pTH1_rv. Crude cell extracts were prepared based on the protocol described elsewhere [11]. The cells were inoculated from a glycerol stock and grown in MeOH200 medium overnight before they were transferred to fresh MeOH200 medium and grown to an A600 of 1.5 to 2.0. 40 mL of the cell culture was harvested by centrifugation (4.000 g, 30 min, 4 °C), washed in 50 mM potassium phosphate buffer (pH 7.8), and stored at − 20 °C. The cells were disrupted by sonication. Cell debris was removed by centrifugation (14.000 g, 60 min, 4 °C) and the supernatant was collected as crude extract. Ta activity was measured according to the conditions of the assay described below.
Purification and molecular mass determination of Ta proteins
For protein production in E. coli BL21 (DE3) [63], taPB1 and taMGA3rec were amplified by PCR using the primer pairs ta_PB1_NcoI-fw; ta_PB1_Xho-rv and ta_MGA3rec_NcoI-fw; ta_MGA3rec_Xho-rv (Table 3). After restriction with NcoI and XhoI the resulting PCR products were ligated into NcoI and XhoI restricted pET28b (Novagen, Madison, Wisconsin, USA), resulting in pET28b-taPB1 and pET28b-taMGA3rec. The pET28b vector allows the production of a C-terminal hexahistidine (His)-tagged Ta in E. coli BL21 (DE3). Protein production and purification were performed as described previously [38]. The enzyme was purified to homogeneity as verified by a 12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) [37]. The protein concentration was measured according to the method of Bradford using the Sigma Bradford reagent with bovine serum albumin (BSA) as a standard. The dimeric structures of the Ta proteins were determined by gel filtration as described previously [38] using 1 mg Ta dissolved in 2 ml of 20 mM Tris-HCl, pH 7.8.
Enzyme assays for the purified Ta proteins
To study the thermal stability of the Ta proteins, the assay mixture described below was prepared in 1.5 mL reaction tubes and incubated for up to 2 h at 30 °C to 80 °C. Samples were taken periodically and the residual enzyme activity was measured under standard conditions in a separate reaction mixture.
The Ta activity in the direction of S7P and GAP from E4P and F6P was determined. The standard reaction mixture (final volume 1 mL) contained 50 mM Tris-HCl buffer (pH 7.8), 0.25 mM nicotinamide adenine dinucleotide (NADH), triosephosphate isomerase (TIM), glycerol 3-phosphate dehydrogenase (G3PDH) and purified Ta protein which was preheated for 3 min at 50 °C. NADH (ε340nm = 6.22 mM− 1 cm− 1) oxidation was followed at 340 nm on a Shimadzu UV1700 spectrophotometer. The reaction was initiated by the addition of E4P or F6P respectively (final concentration varied 0.05–10 mM).
Tryptic digestion of proteins and mass spectrometry analysis
Protein samples displaying Ta activity were separated using a SDS-PAGE and the bands were excised from the gel and put in clean reaction tubes. The tubes were treated with an aqueous solution containing 60% (v/v) acetonitrile and 0,1% (v/v) trifluoroacetic acid (TFA) for removal of softener and dried overnight. The protein bands were treated two times with an aqueous solution containing 0.1 M ammonia carbonate and 30% (v/v) acetonitrile for 10 min each with soft shaking. The supernatant was removed and the fragments were dried in an Eppendorf SpeedVac. The dried fragments were treated with a trypsin solution (1 μL trypsin + 14 μL 10 mM ammonia carbonate, Promega) for 30 min at 21 °C. After addition of 20 μL 10 mM ammonia carbonate solution the samples were incubated for 12 h at 37 °C. The trypsin-digested samples were dried in the SpeedVac for 30 min. After the addition of 10 μL 50% (v/v) acetonitrile and 0.1% (v/v) TFA the samples were spotted onto a 800 μm Burker Anchor Chip, following the Burker Daltronics protocol. An Ultraflex matrix-assisted laser desorption/ionization time-of-flight mass spectrometer (MALDI-TOF-MS) (Bruker, Bremen, Germany) was used to obtain the corresponding peptide mass fingerprints, using the standard manufacturer’s parameters. The proteins were identified from a primary sequence database of B. methanolicus MGA3 using the Mascot (Matrix Science, London) search engine.
Computational analysis
Sequence comparisons were carried out using protein sequences obtained from the NCBI database (http://www.ncbi.nlm.nih.gov) and using BLAST (Basic Local Alignment Search Tool) (http://blast.ncbi.nlm.nih.gov/Blast.cgi) [2].
Availability of data and materials
All data generated or analysed during this study are included in this published article. The datasets used and/or analysed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- DHAP:
-
Dihydroxyacetone phosphate
- E4P:
-
Erythrose 4-phosphate
- Fba:
-
Fructose-1,6-bisphosphate aldolase
- FbaC :
-
Chromosomally encoded fructose-1,6-bisphosphate aldolase
- FbaP :
-
Plasmid encoded fructose-1,6-bisphosphate aldolase
- FBP:
-
Fructose 1,6-bisphosphatase
- GAP:
-
Glyceraldehyde 3-phosphate
- H6P:
-
Hexulose 6-phosphate
- Hps:
-
3-hexulose-6-phosphate synthase
- Mdh:
-
Methanol dehydrogenase
- Pfk:
-
Phosphofructokinase
- Phi:
-
6-phospho-3-hexuloisomerase
- Rpe:
-
Ribulose-phosphate 3-epimerase
- Rpi:
-
Ribose-5-phosphate isomerase
- Ru5P:
-
Ribulose 5-phosphate
- RuMP cycle:
-
Ribulose monophosphate cycle
- S7P:
-
Sedoheptulose 7-phosphate
- SBP:
-
Sedoheptulose 1,7-bisphosphate
- SBPase:
-
Sedoheptulose-1,7-bisphosphatase
- Ta:
-
Transaldolase
- Tkt:
-
Transketolase
References
Abe S, Takayama KI, Kinoshia S. Taxonomical studies on glutamic acid-producing bacteria. J Gen Appl Microbiol. 1967;13:279–301.
Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–402.
Anthony C. Biochemistry of methylotrophs. New York: Academic Press, London; 1982.
Arfman N, de Vries KJ, Moezelaar HR, Attwood MM, Robinson GK, van Geel M, Dijkhuizen L. Environmental regulation of alcohol metabolism in thermotolerant methylotrophic Bacillus strains. Arch Microbiol. 1992;157:272–8.
Arfman N, Dijkhuizen L, Kirchhof G, Ludwig W, Schleifer KH, Bulygina ES, Chumakov KM, Govorukhina NI, Trotsenko YA, White D, Sharp RJ. Bacillus methanolicus sp. nov., a new species of thermotolerant, methanol-utilizing, endospore-forming bacteria. Int J Syst Bacteriol. 1992;42:439–45.
Arfman N, Hektor HJ, Bystrykh LV, Govorukhina NI, Dijkhuizen L, Frank J. Properties of an NAD(H)-containing methanol dehydrogenase and its activator protein from Bacillus methanolicus. Eur J Biochem. 1997;244:426–33.
Atkins JF, Baranov PV, Fayet O, Herr AJ, Howard MT, Ivanov IP, Matsufuji S, Miller WA, Moore B, Prere MF, Wills NM, Zhou J, Gesteland RF. Overriding standard decoding: implications of recoding for ribosome function and enrichment of gene expression. Cold Spring Harb Symp Quant Biol. 2001;66:217–32.
Atkins JF, Baranov PV. The distinction between recoding and codon reassignment. Genetics. 2010;185:1535–6.
Banki K, Halladay D, Perl A. Cloning and expression of the human gene for transaldolase. A novel highly repetitive element constitutes an integral part of the coding sequence. J Biol Chem. 1994;269:2847–51.
Baranov PV, Gesteland RF, Atkins JF. Recoding: translational bifurcations in gene expression. Gene. 2002;286:187–201.
Brautaset T, Jakobsen OM, Flickinger MC, Valla S, Ellingsen TE. Plasmid-dependent methylotrophy in thermotolerant Bacillus methanolicus. J Bacteriol. 2004;186:1229–38.
Brautaset T, Jakobsen ØM, Josefsen KD, Flickinger MC, Ellingsen TE. Appl Microbiol Biotechnol. 2007;74:22–34.
Carnicer M, Vieira G, Brautaset T, Portais JC, Heux S. Quantitative metabolomics of the thermophilic methylotroph Bacillus methanolicus. Microb Cell Factories. 2016;15:92.
Drejer EB, Chan D, Haupka C, Wendisch VF, Brautaset T, Irla M. Green Chem. 2020;22:788–802.
Dijkhuizen L, Levering PR, de Vries GE. The physiology and biochemistry of aerobic methanol-utilizing gram-negative and gram-positive bacteria. In: Murrell CJ, Dalton H, editors. Methane and methanol utilizers. New York: Plenum Press; 1992. p. 149–81.
Eggeling L, Bott M. Handbook of Corynebacterium glutamicum. Boca Raton: CRC press; 2005.
Feldmann SD, Sahm H, Sprenger GA. Pentose metabolism in Zymomonas mobilis wild-type and recombinant strains. Appl Microbiol Biotechnol. 1992;38:354–61.
Fiki AE, Metabteb GE, Bellebna C, Wartmann T, Bode R, Gellissen G, Kunze G. The Arxula adeninivorans ATAL gene encoding transaldolase-gene characterization and biotechnological exploitation. Appl Microbiol Biotechnol. 2007;74:292–1299.
Gesteland RF, Weiss RB, Atkins JF. Recoding: reprogrammed genetic decoding. Science. 1992;257:1640–1.
Gesteland RF, Atkins JF. Recoding: dynamic reprogramming of translation. Annu Rev Biochem. 1996;65:741–68.
Grossman CE, Qian Y, Banki K, Perl A. ZNF143 mediates basal and tissue-specific expression of human transaldolase. J Biol Chem. 2004;279:12190–205.
Haima P, Bron S, Venema G. The effect of restriction on shotgun cloning and plasmid stability in Bacillus subtilis Marburg. Mol Gen Genet. 1987;209:335–42.
Heggeset TM, Krog A, Balzer S, Wentzel A, Ellingsen TE, Brautaset T. Genome sequence of thermotolerant Bacillus methanolicus: features and regulation related to methylotrophy and production of L-lysine and L-glutamate from methanol. Appl Environ Microbiol. 2012;78:5170–81.
Hektor HJ, Kloosterman H, Dijkhuizen L. Nicotinoprotein methanol dehydrogenase enzymes in gram-positive methylotrophic bacteria. J Mol Catal B-Enzym. 2000;8:103–9.
Irla M, Neshat A, Winkler A, Albersmeier A, Heggeset TMB, Brautaset T, Kalinowski J, Wendisch VF, Rückert C. Complete genome sequence of Bacillus methanolicus MGA3, a thermotolerant amino acid producing methylotroph. J Biotechnol. 2014;188:110–11. https://doi.org/10.1016/j.jbiotec.2014.08.013.
Irla M, Neshat A, Brautaset T, Rückert C, Kalinowski J, Wendisch VF. Transcriptome analysis of thermophilic methylotrophic Bacillus methanolicus MGA3 using RNA-sequencing provides detailed insights into its previously uncharted transcriptional landscape. BMC Genomics. 2015;16:73.
Irla M, Nærdal I, Brautaset T, Wendisch VF. Ind Crops Prod. 2017;106:12–20.
Ivey DM, Cheng J, Krulwich TA. A 1.6 kb region of Bacillus firmus OF4 DNA encodes a homolog OF Escherichia coli and yeast DNA topoisomerases and may contain a translational readthrough of UGA. Nucleic Acids Res. 1992;20:4928.
Jakobsen OM, Benichou A, Flickinger MC, Valla S, Ellingsen TE, Brautaset T. Upregulated transcription of plasmid and chromosomal ribulose monophosphate pathway genes is critical for methanol assimilation rate and methanol tolerance in the methylotrophic bacterium Bacillus methanolicus. J Bacteriol. 2006;188:3063–72.
Jia J, Huang W, Schörken U, Sahm H, Sprenger GA, Lindqvist Y, Schneider G. Crystal structure of transaldolase B from Escherichia coli suggests a circular permutation of the alpha/beta barrel within the class I aldolase family. Structure. 1996;4:715–24.
Jia J, Schörken U, Lindqvist Y, Sprenger GA, Schneider G. Crystal structure of the reduced Schiff-base intermediate complex of transaldolase B from Escherichia coli: mechanistic implications for class I aldolases. Protein Sci. 1997;6:119–24.
Keilhauer C, Eggeling L, Sahm H. Isoleucine synthesis in Corynebacterium glutamicum: molecular analysis of the ilvB-ilvN-ilvC operon. J Bacteriol. 1993;175:5595–603.
Kemp MB, Quayle JR. Microbial growth on C1 compounds. Uptake of 14C-formaldehyde and 14C-formate by methane-grown Pseudomonas methanica and determination of the hexose labelling pattern after brief incubation with 14C-methanol. Biochem J. 1967;102:94–102.
Kiefer P, Schmitt U, Müller JEN, Hartl J, Meyer F, Ryffel F, Vorholt JA. DynaMet: a fully automated pipeline for dynamic LC-MS data. Anal Chem. 2015;87:9679–86.
Kourtoglou E, Mamma D, Topakas E, Christakopoulos P. Purification, characterization and mass spectrometric sequencing of transaldolase from Fusarium oxysporum. Process Biochem. 2008;43:1094–101.
Krog A, Heggeset TM, Müller JE, Kupper CE, Schneider O, Vorholt JA, Ellingsen TE, Brautaset T. Methylotrophic Bacillus methanolicus encodes two chromosomal and one plasmid born NAD+ dependent methanol dehydrogenase paralogs with different catalytic and biochemical properties. PLoS One. 2013;8:e59188.
Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–5.
Lindner SN, Vidaurre D, Willbold S, Schoberth SM, Wendisch VF. NCgl2620 encodes a class II polyphosphate kinase in Corynebacterium glutamicum. Appl Environ Microbiol. 2007;73:5026–33.
Markert B, Stolzenberger J, Brautaset T, Wendisch VF. Characterization of two transketolases encoded on the chromosome and the plasmid pBM19 of the facultative ribulose monophosphate cycle methylotroph Bacillus methanolicus. BMC Microbiol. 2014;14:7.
McIntyre LM, Thorburn DR, Bubb WA, Kuchel PW. Comparison of computer simulations of the F-type and L-type non-oxidative hexose monophosphate shunts with 31P-NMR experimental data from human erythrocytes. Eur J Biochem. 1989;180:399–420.
Moehs CP, Allen PV, Friedman M, Belknap WR. Cloning and expression of transaldolase from potato. Plant Mol Biol. 1996;32:447–52.
Müller JEN, Litsanov B, Bortfeld-Miller M, Trachsel C, Grossmann J, Brautaset T, Vorholt JA. Proteomic analysis of the thermophilic methylotroph Bacillus methanolicus MGA3. Proteomics. 2014;14:725–37.
Müller JEN, Meyer F, Litsanov B, Kiefer P, Vorholt JA. Core pathways operating during methylotrophy of Bacillus methanolicus MGA3 and induction of bacillithiol-dependent detoxification pathway upon formaldehyde stress. Mol Microbiol. 2015;98:1089–100.
Nærdal I, Pfeifenschneider J, Brautaset T, Wendisch VF. Microb Biotechnol. 2015;8:342–350.
Perl A. The pathogenesis of transaldolase deficiency. IUBMB Life. 2007;59:365–73.
Peters-Wendisch P, Schiel B, Wendisch VF, Katsoulidis E, Möckel B, Sahm H, Eikmanns BJ. Pyruvate carboxylase is a major bottleneck for glutamate and lysine production by Corynebacterium glutamicum. J Mol Microbiol Biotechnol. 2001;3:295–300.
Pfeifenschneider J. Metabolic engineering of Bacillus methanolicus and Corynebacterium glutamicum for the production of cadaverine from methanol. Germany: PhD thesis, Bielefeld University; 2015.
Raines CA. The Calvin cycle revisited. Photosynth Res. 2003;75:1–10.
Sambrook J, Russell DW. Molecular cloning: a laboratory manual, Vol 3 set. New York: Cold Spring Harbor Laboratory Press, Cold Spring Harbor; 2001.
Samland AK, Sprenger GA. Transaldolase: from biochemistry to human disease. Int J Biochem Cell Biol. 2009;41:1482–94.
Samland AK, Baier S, Schürmann M, Inoue T, Huf S, Schneider G, Sprenger GA, Sandalova T. Conservation of structure and mechanism within the transaldolase enzyme family. FEBS J. 2012;279:766–78.
Schäfer A, Tauch A, Jäger W, Kalinowski J, Thierbach G, Pühler A. Small mobilizable multi-purpose cloning vectors derived from the Escherichia coli plasmids pK18 and pK19: selection of defined deletions in the chromosome of Corynebacterium glutamicum. Gene. 1994;145:69–73.
Schendel FJ, Bremmon CE, Flickinger MC, Guettler M, Hanson RS. L-lysine production at 50 degrees C by mutants of a newly isolated and characterized methylotrophic Bacillus sp. Appl Environ Microbiol. 1990;56:963–70.
Schneider S, Sandalova T, Schneider G, Sprenger GA, Samland AK. Replacement of a phenylalanine by a tyrosine in the active site confers fructose-6-phosphate aldolase activity to the transaldolase of Escherichia coli and human origin. J Biol Chem. 2008;283:30064–72.
Schörken U, Thorell S, Schürmann M, Jia J, Sprenger GA, Schneider G. Identification of catalytically important residues in the active site of Escherichia coli transaldolase. Eur J Biochem. 2001;268:2408–15.
Schürmann M. Biochemische Charakterisierung und Struktur-Funktionsbeziehungen bakterieller Transaldolasen und Fruktose-6-Phosphat Aldolasen. Dissertation, Forschungszentrum Jülich GmbH Zentralbibliothek, Verlag, Berichte des Forschungszentrums Jülich 3903. Jülich; 2001. p. 119.
Schürmann M, Sprenger GA. Fructose-6-phosphate aldolase is a novel class I aldolase from Escherichia coli and is related to a novel group of bacterial transaldolases. J Biol Chem. 2001;276:11055–61.
Sharma V, Firth AE, Antonov I, Fayet O, Atkins JF, Borodovsky M, Baranov PV. A pilot study of bacterial genes with disrupted ORFs reveals a surprising profusion of protein sequence recoding mediated by ribosomal frameshifting and transcriptional realignment. Mol Biol Evol. 2011;28:3195–211.
Soderberg T, Alver RC. Transaldolase of Methanocaldococcus jannaschii. Archaea. 2004;1:255–62.
Sprenger GA, Schörken U, Sprenger G, Sahm H. Transaldolase B of Escherichia coli K-12: cloning of its gene, talB, and characterization of the enzyme from recombinant strains. J Bacteriol. 1995;177:5930–6.
Stolzenberger J, Lindner SN, Persicke M, Brautaset T, Wendisch VF. Characterization of fructose 1,6-bisphosphatase and sedoheptulose 1,7-bisphosphatase from the facultative ribulose monophosphate cycle methylotroph Bacillus methanolicus. J Bacteriol. 2013;195:5112–22.
Stolzenberger J, Lindner SN, Wendisch VF. The methylotrophic Bacillus methanolicus MGA3 possesses two distinct fructose 1,6-bisphosphate aldolases. Microbiology. 2013;159:1770–81.
Studier FW, Rosenberg AH, Dunn JJ, Dubendorff JW. Use of T7 RNA polymerase to direct expression of cloned genes. Methods Enzymol. 1990;185:60–89.
Sugiyama M, Suzuki S, Tonouchi N, Yokozeki K. Transaldolase/glucose-6-phosphate isomerase bifunctional enzyme and ribulokinase as factors to increase xylitol production from D-arabitol in Gluconobacter oxydans. Biosci Biotechnol Biochem. 2003;67:2524–32.
Susskind BM, Warren LG, Reeves RE. A pathway for the interconversion of hexose and pentose in the parasitic amoeba Entamoeba histolytica. Biochem J. 1982;204:191–6.
Thorell S, Schürmann M, Sprenger GA, Schneider G. Crystal structure of decameric fructose-6-phosphate aldolase from Escherichia coli reveals inter-subunit helix swapping as a structural basis for assembly differences in the transaldolase family. J Mol Biol. 2002;319:161–71.
Acknowledgements
We thank Dr. Thomas Patschkowski for help with the MADLI-TOF analysis, Marina Gil Lopez for help with crude extract enzyme assays, Dr. Jonas Müller for the proteome data, and Dr. Marc Carnicer for providing us with data of the metabolic flux analysis.
Funding
This work was funded by the research grants ‘PROMYSE’ (EU FW7) and ‘MetApp’ (ERASysAPP-33). Support for the Article Processing Charge by the Deutsche Forschungsgemeinschaft and the Open Access Publication Fund of Bielefeld University is acknowledged. The funding bodies had no role in the design of the study or collection, analysis, or interpretation of data or in writing the manuscript.
Author information
Authors and Affiliations
Contributions
JP, BM, JS, TB and VFW designed the study. JP, BM, JS performed the experiments and analyzed the data. JP drafted the manuscript. JP, TB and VFW reviewed and finalized the manuscript. All authors approved the final version of the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Pfeifenschneider, J., Markert, B., Stolzenberger, J. et al. Transaldolase in Bacillus methanolicus: biochemical characterization and biological role in ribulose monophosphate cycle. BMC Microbiol 20, 63 (2020). https://doi.org/10.1186/s12866-020-01750-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12866-020-01750-6