
Abstract
Eukaryotic messenger RNAs (mRNAs) are often modified with methyl groups at the N6 position of adenosine (m6A), and these changes are interpreted by YTH domain-containing proteins to regulate the metabolism of m6A-modified mRNAs. Although alfalfa (Medicago sativa) is an established model organism for forage development, the understanding of YTH proteins in alfalfa is still limited. In the present investigation, 53 putative YTH genes, each encoding a YT521 domain-containing protein, were identified within the alfalfa genome. These genes were categorized into two subfamilies: YTHDF (49 members) and YTHDC (four members). Each subfamily demonstrates analogous motif distributions and domain architectures. Specifically, proteins encoded by MsYTHDF genes incorporate a single domain structure, while those corresponding to MsYTH5, 8, 12, 16 who are identified as members of the MsYTHDC subfamily, exhibit CCCH-type zinc finger repeats at their N-termini. It is also observed that the predicted aromatic cage pocket that binds the m6A residue of MsYTHDC consists of a sequence of two tryptophan residues and one tyrosine residue (WWY). Conversely, in MsYTHDF, the binding pocket comprises two highly conserved tryptophan residues and either one tryptophan residue (WWW) or tyrosine residue (WWY) in MsYTHDF.
Through comparative analysis of qRT-PCR data, we observed distinct expression patterns in specific genes under abiotic stress, indicating their potential regulatory roles. Notably, five genes (MsYTH2, 14, 26, 27, 48) consistently exhibit upregulation, and two genes (MsYTH33, 35) are downregulated in response to both cold and salt stress. This suggests a common mechanism among these YTH proteins in response to various abiotic stressors in alfalfa. Further, integrating qRT-PCR with RNA-seq data revealed that MsYTH2, MsYTH14, and MsYTH16 are highly expressed in leaves at various development stages, underscoring their potential roles in regulating the growth of these plant parts. The obtained findings shed further light on the biological functions of MsYTH genes and may aid in the selection of suitable candidate genes for future genetic enhancement endeavors aimed at improving salt and cold tolerance in alfalfa.
Similar content being viewed by others


Introduction
Posttranscriptional regulation is essential for plant growth, development, and stress response [13]. RNA-binding proteins are key players in these processes, either through direct interaction with RNA or by indirectly modulating other regulatory factors [27]. Among the various post-transcriptional modifications, N6-methyladenosine (m6A) is the most prevalent and influential modification in eukaryotic mRNAs and noncoding RNAs which is deposited by the writer complex (MTA-MTB-FIP37-VIR-HAKAI in plants) and can be deleted by erasers (ALKBH proteins) [19, 55]. The processes of m6A methylation and demethylation illustrate the reversibility of m6A modification, leading to dynamic changes of m6A levels in vivo [52]. Reader proteins can recognize the m6A mark and participate in a wide range of processes involving RNA metabolism, such as splicing, translation efficiency, stability, alternative polyadenylation, structure, nuclear export, and also exert epigenetic effects on noncoding RNAs [3, 6, 33, 41, 42, 49, 57]. According to Patil et al. [38] and Reichel et al. [41], reader proteins are responsible for determining the fate of m6A-modified mRNA. It is therefore essential to identify the m6A readers to unravel the mechanism by which m6A functions in cells.
The YT521-B homology (YTH) domain was first identified by matching known protein sequences with the splicing factor YT521-B, which is involved in RNA processing and interacts with other proteins [25, 53]. The YTH domain serves as the module for recognizing m6A in a methylation-dependent manner. There are five known YTH domain-containing proteins in humans, including YTHDC1, YTHDC2, YTHDF1, YTHDF2, and YTHDF3. The YTH domain-containing proteins extensively participate in post-transcriptional regulation. They also influence splicing, translation, localization, and RNA lifetime. Those processes involve targeting different complexes to specific sites through direct binding to m6A [50, 54]. The crystal structures of YTHDC2 and YTHDC1 domains have been compared, revealing a conserved hydrophobic pocket and positively charged surface for recognizing m6A RNA [30]. Similarly, YTHDF1, YTHDF2, and YTHDF3 also possess aromatic cages and specific residues for m6A recognition, while a basic patch contributes to RNA backbone binding [44]. In turn, the conservation of these amino acid residues across evolution and their analysis in Arabidopsis ECT protein mutants suggest that these residues likely have the same function in plant YTH proteins [4]. Scutenaire et al. [43] performed a systematic evolutionary analysis of YTH domains in Viridiplantae. They demonstrated that vascular plants display both YTHDF- and YTHDC-type motifs. These motifs carry the amino acids necessary for RNA binding and to accommodate m6A, and are predicted to adopt the same structural fold as YTH domains in animals and yeast.
It is of particular interest that plants possess a higher number of YTH domain proteins compared to other organisms, as suggested by Shi et al. [44]. The Arabidopsis genome harbors 13 YTH domain proteins which can be categorized into two groups. The larger group, with 11 YTHDF proteins, is classified as EVOLUTIONARY CONSERVED C-TERMINAL REGION proteins, owing to their conserved C-terminal region. The smaller group consists of 2 YTHDC proteins, At4g11970 (also known as AtYTH11) and CPSF30 (also known as AtYTH03) as reported by Scutenaire et al. [43] and Arribas-Hernández et al. [4].
A recent study by Govindan et al. [16] demonstrated that the increased RNA methylation, specifically the m6A modification, significantly contributes to the regulation of cold tolerance. Under salt stress, m6A is dynamically deposited on transcripts, stabilizing the genes that encode proteins involved in salt and osmotic stress responses [2]. In rice, cadmium treatment results in differential m6A modifications in thousands of transcripts in the root, implying that m6A might be linked to abnormal root development caused by cadmium stress [9]. In wheat, genes encoding the m6A reader protein TaYTHs display noticeable expression changes in response to abiotic stresses like water and drought. Additionally, salt stress in sweet sorghum leads to significant changes in the m6A methylome, increasing m6A modification and mRNA stability of salt-resistance genes, which in turn positively regulates tolerance to salt stress [56]. Significant alterations in the m6A methylome profile, along with its correlation with mRNA abundance, have been identified in pak choi, tomato, and apple leaf [28, 32, 51]. These findings suggest that m6A modification also plays a role in modulating crops’ responses to temperature and humidity-induced stresses.
While significant research has been conducted on the YTH domain proteins of several plant species, our understanding of the YTH family members in forage species like alfalfa remains limited. Alfalfa is a highly valuable feed crop due to its high protein content and capacity to fix atmospheric nitrogen. A comprehensive genome-wide understanding of the YTH gene family in alfalfa is crucial for deciphering the molecular processes underpinning RNA modification and gene regulation in this important crop. Therefore, this study aims to identify and analyze the YTH gene family in alfalfa using a comprehensive genome-wide approach. By employing bioinformatics methods, we analyzed the whole genome sequence of alfalfa to systematically identify YTH genes. The discovered YTH genes were further investigated for their structural characteristics, conserved domains, evolutionary linkages, and expression patterns under various organs and abiotic stresses. The findings from this study could pave the way for future genetic engineering and breeding initiatives focused on enhancing alfalfa quality and yield by targeting the identified YTH genes.
Material and methods
Plant growth and sample collection
The alfalfa cultivar used in this study for qRT-PCR analysis was Xinjiang Daye, aligning with the genome data for this investigation. The Xinjiang Daye seeds were provided by Dr. Rui Dong (Guizhou University). For each treatment, three seeds were sown in each plastic pot, and this process was replicated three times. Subsequently, from each pot, a seedling was selected based on identical growth characteristics observed within that specific pot, while the other two seedlings from the same pot were discarded. The plants were grown with a mixture of soil (vermiculite/humus = 1:1) under a 16/8 h day/night cycle, 25°/20 °C day/night temperatures, and a light intensity of 300 mol m−2 s−1. To validate the gene expression patterns across different plant organs, which are stem, root, leaf and flower, samples were collected from 8-week-old plants during the flowering stage. To assess gene expression under cold and salt treatments, seedlings were subjected to cold treatment at 4 °C and salinity treatment with 200 mM NaCl for 6 h. Subsequently, leaf samples were collected for gene expression analysis. Control groups were implemented for each treatment, involving seedlings maintained under normal growth conditions at 25 °C (day) / 20 °C (night), without exposure to salinity stress. Upon harvest, all samples were promptly frozen in liquid nitrogen and stored at -80 °C until use.
In-silico identification of the YTH gene family
The genome and annotation data for the alfalfa cultivar “Xinjiang daye” were retrieved from the Figshare data repository (https://figshare.com/projects/whole_genome_sequencing_and_assembly_of_Medicago_sativa/66380) [8]. To identify YTH domain homologs in alfalfa, the Hidden Markov model of the HMMER 3.0 software was used (PF04146). The Arabidopsis thaliana YTH members identified by Li et al. [24] were retrieved from The Arabidopsis Information Resource (http://www.arabidopsis.org/index.jsp) and were used as queries in a BLASTp search against all the protein sequences of the alfalfa genome with an E-value threshold of 1e−5. The genes identified using both approaches were compiled, and duplicate genes were manually removed. The presence of the YTH domain was verified using the CDD (https://www.ncbi.nlm.nih.gov/cdd) and Interpro (https://www.ebi.ac.uk/interpro) databases [39, 47]. Sequences that lacked the whole YTH domain were eliminated. The discovered genes were named according to their chromosomal position. The ProtParam tool (http://web.expasy.org/protparam/) was used to determine the number of amino acids (AA) characteristics, molecular weights (MW), and isoelectric points (pIs), grand average of hydropathy (GRAVY) and Cell-PLoc (http://www.csbio.sjtu.edu.cn/bioinf/Cell-PLoc/) was used to estimate the subcellular localizations [10, 15].
Phylogeny and chromosomal location
Sequences of YTH protein in Arabidopsis and alfalfa were aligned using Clustal X with default parameters [21]. The phylogenetic tree of YTH family members in alfalfa and Arabidopsis was generated using MEGA X software. The unrooted phylogenetic tree was built using the Neighbor-joining (NJ) technique with a bootstrap value of 1000 iterations, and the phylogenetic trees were drawn using the iTOL (Interactive Tree of Life) web program (http://itol.embl.de/) [23]. The chromosomal location of all discovered genes was determined from the M. sativa genomic annotation file GFF3 and named based on their locations on chromosome. We plotted the chromosomal distribution of MsYTH genes using the TBtools program [8].
Gene duplication events and gene selection pattern
Gene duplication events in MsYTH genes were detected and identified utilizing collinear scanning toolkits (MCScanX) with an e-value of 10–5 [48]. Non-synonymous (Ka) / synonymous (Ks) ratios for each gene pair were calculated using the DnaSP5 to analyze the evolutionary constraints of the MsYTH genes [26].
Gene structure and functional prediction
MEME (http://meme-suite.org/tools/meme) was used to identify conserved motifs in MsYTH family members and the WebLogo tool was utilized to construct the theme Logos [5, 11]. Using the TBtools program, the exon–intron structure of MsYTH genes was visualized by constructing a gene structure based on the CDS and the associated full-length sequence. The MsYTH genes’ upstream regions (-1500 bp) were explored for regulatory elements. Plantcare (http://bioinformatics.psb.ugent.be/webtools/plantcare/html/) was used to identify and visualize the cis-acting regulatory elements in the promoter [22]. ESPrint3.0 (https://espript.ibcp.fr/ESPript/cgi-bin/ESPript.cgi) was used to analyze the protein domain. The 3D structure of MsYTH proteins was constructed using AlphaFold2 v1.5.1 with the default parameters [34].
Expression pattern of MsYTH in different organs by RNA-seq
The expression of MsYTH members was examined using RNA-seq data from various organs, obtained from alfalfa database LegumeIP V3 (https://www.zhaolab.org/LegumeIP) [12]. Six different organs of CADL alfalfa (Cultivated Alfalfa at the Diploid Level), namely root, nodule, leaf, flower, pre-elongated stem, elongated stem, were included in the analysis. Three replicates were performed for each organ. The FPKM values obtained from the RNA-seq were normalized using the log10(FPKM) treatment, and a heatmap was generated using HemI software [35].
Validation of MsYTH genes expression in various organs and under abiotic stress using qRT-PCR
For further validation, the expression of the MsYTHs genes was analyzed using quantitative real-time polymerase chain reaction (qRT-PCR). Total RNA was extracted from the harvested samples using the Trizol reagent according to the manufacturer’s instructions. The quality and quantity of RNA were assessed using a NanoDropTM UV spectrophotometer (Thermo Fisher Scientific, Lenexa, KS, USA). To ensure the accuracy of the analysis, genomic DNA contamination was eliminated, and first-strand cDNA synthesis was performed using the PrimeScriptTM RT reagent Kit with gDNA Eraser on 1 µg of total RNA (Takara, Japan). The qRT-PCR study was conducted using a CFX96 TouchTM Real-Time PCR Detection System (Bio-Rad, USA). The housekeeping gene EF-1α was used as a reference. Primer details are provided in Table S2. The relative expression values for YTH family genes were calculated using the 2−∆∆Ct method, as described by Livak and Schmittgen [29].
Relative gene expression levels were assessed after normalization against the expression levels of the housekeeping gene. Each conditions’s value represents the mean expression level, and error bars indicate the standard deviation (SD) calculated from three independent biological replicates. Statistical significance between different groups was determined by one-way ANOVA with posterior Duncan test (p < 0.05), with different lowercase letters marking significantly different groups.
Results
Identification of the YTH gene family in alfalfa
A total of 53 putative YTH genes were discovered and designated by their chromosomal position. Table 1 lists gene IDs, chromosomal locations, chromosome start, and end positions, amino acid counts, molecular weights, pIs, GRAVY, and potential subcellular localizations of proteins. The sequences of all the MsYTH proteins are provided in Supplemental Table S1. Members of the YTH family are distributed unevenly on alfalfa chromosomes. The number of composing amino acid residues was variable as well. This variability ranged from the shortest/smallest being 169 amino acids to the longest/biggest being 853 amino acids. The MW ranges from 18.83 kDa (MsYTH30) to 96.61 kDa (MsYTH13). The pIs vary from 4.97 (MsYTH17) to 9.57 (MsYTH43). The majority of YTH proteins are likely to be found in the nucleus, with nine members predicted to be located in both the cell wall and nucleus, one likely in the cell wall, and one predicted in the chloroplast.
Phylogenetic analysis and conserved domain of YTH members
To gain insights into the evolutionary dynamics of YTH domain-containing proteins in alfalfa, an unrooted phylogenetic tree was constructed using a dataset comprising the 53 sequences from alfalfa and 13 YTH protein sequences from Arabidopsis retrieved from the TAIR database.
Results showed that MsYTH proteins were divided into two distinct clades: YTHDF and YTHDC subfamilies (Fig. 1). According to the evolutionary tree, no close relatives were found in Arabidopsis and Alfalfa in any clade. There are 22, 3 and 24 protein sequences in the further subclades YTHDFa, YTHDFb and YTHDFc, respectively. The YTHDC subfamily was divided into two subclades, YTHDCa, which included four alfalfa sequences, and YTHDCb. Notably, the YTHDCb subclade did not contain any MsYTH protein, indicating a possible evolutionary divergence between alfalfa and other species in the YTHDC subfamily. Together, these results indicate that the YTH family proteins were highly conserved in alfalfa, and that there was a slight evolutionary divergence in the subfamily or subclade.

Phylogenetic relationship between YTH proteins identified in alfalfa and Arabidopsis. The phylogenetic tree was constructed using the Neighbor-jointing method with 1000 bootstrap replications. The five subclades of YTHDCa, YTHDCb, YTHDFa, YTHDFb and YTHDFc are labeled with different colors
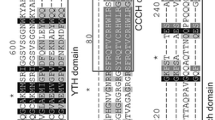
Additional multiple sequence alignment of YTH proteins showed that many functional sites were conserved and displayed a conserved aromatic cage (Fig. 2), suggesting that MsYTHs might have a similar m6A read mechanism to human YTH proteins especially in subfamily YTHDFs. Our research uncovered that the aromatic cage that binds the m6A residue within MsYTHDC proteins are highly conserved and consist of two tryptophan residues and one tyrosine residue (WWY). In contrast, the aromatic cage in MsYTHDFa and MsYTHDFb proteins are composed of three conserved tryptophan residues (WWW), whereas in MsYTHDFc, it consists of two highly conserved tryptophan residues and either one tryptophan residue (WWW) or tyrosine (WWY). Specifically, proteins encoded by MsYTHDF genes incorporate a single domain structure, while those corresponding to MsYTH5, 8, 12, 16 identified as members of the MsYTHDC subfamily, exhibit CCCH-type zinc finger repeats at their N-termini.

Multiple sequence alignment showing the conserved domains of the alfalfa YTH protein sequences. The secondary structural elements predicted with AlphaFold DB model of A0A3Q7Y049_CICAR are shown above. The red triangles represent the key functional residues WWW/WWY, and the red dot represent the key functional residues CCC-H within the YTHDC subfamily
Gene chemical structure and conserved motif analysis
The MEME Suite was employed to detect conserved motifs in MsYTH proteins. We found ten conserved motifs in YTH proteins, and the size of the motif varied widely with motif 8 being the largest and motif 2 being the smallest. Amino acid conservation per motif also varied widely. For example, AF, ERK, SGA, YY, F and GR were the most conserved amino acids in motifs 1 to 6, respectively, while motifs 7 and 8 had the most conserved amino acids (Fig. 3).

Conserved motif compositions and gene structure of MsYTH domain-containing proteins. A Graphical representation of the distributions of conserved motifs in MsYTH proteins. Each distinct motif is represented by a different color and associated number. B Schematic diagram of the exon/intron organization of MsYTH genes. Green boxes denote exons, and black lines indicate introns. C Detailed sequence logo analysis of the conserved motifs within the MsYTH proteins. Each motif is represented by a sequence logo with a corresponding number
The proteins MsYTH5, MsYTH8, MsYTH12, and MsYTH16, which collectively constitute the YTHDCa subclass, each possess only four specific motifs—namely motifs 2, 3, 5, and 6. This reduced motif representation is considerably lower compared to the remaining members of the gene family. Notably, motifs 2 and 6 are the most common motifs, being present in 52 and 51 proteins, respectively. We also observed a noteworthy divergence between the MsYTHDC and MsYTHDF subfamilies. Specifically, in the MsYTHDC subfamily, we identified the presence of CCCH-type zinc fingers, a prominent structural motif crucial for protein-DNA and protein–protein interactions, characterized by the pattern C-X7-C-X5-C-X3-H. This motif is notably absent in the members of the MsYTHDF subfamily. The absence or presence of this zinc finger motif could signify distinct functional roles and regulatory mechanisms within these subfamilies.
Next, we assessed the variance in the quantity of exons and UTRs as shown in Fig. 3b. The number of exons and UTRs exhibited significant divergence across the examined gene samples. For instance, MsYTH19 presented only one exon while MsYTH6 and MsYTH13 exhibited as many as 19. It is hypothesized that the increased intron count within alfalfa YTHs could potentially bolster the repertoire of YTH proteins. Such an increase would, in theory, enable plants to embody more complex and varied biological functions.
Protein 3D structure and conserved domain of MsYTH
To understand the protein structure, we used the default parameter for the structural creation of MsYTH proteins on a web platform (alphafold2: https://colab.research.google.com/github/sokrypton/ColabFold/blob/main/AlphaFold2.ipynb). Given the complexity, we strategically selected nine representative proteins from the four subclade within the gene family. These selected proteins cover the functional and structural diversity of the family, ensuring a comprehensive insight into the family’s characteristics. The general structure displays a globular fold with a four-stranded sheet center encircled by four helices, flanking areas on both sides, and a central core. This fundamental structure of a protein dictates its characteristics, whereas the higher structure controls its physiological activity [40]. The MsYTH models revealed that the binding sites of YTH domain-containing proteins are in the center of these folds, implying that YTH proteins in alfalfa include m6A binding sites and carry out their biological tasks by binding m6A (Fig. 4). The YTH domain is highly conserved, whereas the other regions don’t exhibit a fixed or ordered 3D structure. These areas, also known as intrinsically disordered proteins and intrinsically disordered regions (IDPs/IDRs), perform significant biological activities despite their lack of structural order. Their dynamic and disordered nature has been associated with key processes like enzyme catalysis and allosteric regulation. Moreover, they play crucial roles in essential physiological functions such as cell signaling and transcription [14, 46].

A three-dimensional schematic of the selected YTH protein structure predicted using AlphaFold2. The α-helix and β-sheet in the protein’s structure are represented by ochre and red colors, respectively, visualized using PyMOL2.5
Chromosome location, synteny, and gene duplication analysis
In alfalfa, the 53 YTH genes were clustered across 21 chromosomes. The number of genes per chromosome varied widely. For instance, while chr2.1 had 12 genes, chr8.1 had only one gene. MsYTH1/MsYTH2, MsYTH3, and MsYTH4 are members of the MsHDF subfamily and are found on chromosome 2.1. No relationship was observed between chromosomal length and the number of MsYTH genes. We further developed a comparative syntenic map between alfalfa and Arabidopsis to learn more about the evolutionary processes of the YTH family. On chr2.4, four genes MsYTH14, MsYTH15, MsYTH17 and MsYTH18 belonging to the YTHDFc subfamily are aggregated into one tandem duplication event area. Except for MsYTH49 and MsYTH19, all MsYTH genes have a syntenic connection with Arabidopsis, indicating that these orthologous pairings may have existed before the divergence. The collinear gene pair associated with MsYTH28 was not found in Arabidopsis, indicating that this gene might have been generated through gene duplication or segmental duplication events (Fig. 5).

A Chromosomal distribution of MsYTH genes in alfalfa and B Syntenic analysis in alfalfa genome. The colored lines connecting the genes denote potential gene duplication pairs. The grey lines indicate syntenic blocks across the genome
In order to elucidate the evolutionary selection patterns within the YTH gene family in alfalfa, we quantitatively assessed the synonymous and non-synonymous substitution rates (Ka/Ks) of corresponding gene pairs (Table S3). Remarkably, only a minority of pairs (4 out of 72) displayed Ka/Ks values exceeding 1. This suggests that the majority of the YTH gene family has undergone negative selection, indicative of a relatively slow rate of evolutionary change.
Cis-regulatory element analysis of MsYTH genes
Multiple hormone-related elements were discovered in MsYTH promoters, including methyl jasmonate (MeJA) responsiveness (MsYTH2,4,6,7,9), abscisic acid responsiveness (MsYTH2-4,6–9), auxin responsiveness (MsYTH1,3–6,8), salicylic acid responsiveness (MsYTH4-6), and gibberellin responsiveness (MsYTH1,5,6). Furthermore, each promoter contained at least two categories of phytohormone-related elements. Multiple cis-acting regulatory elements implicated in abiotic stressors were also discovered, including drought-inducibility (MsYTH1,3–7,9), wound responsiveness (MsYTH1,7,9), defense and stress response (MsYTH1,3,4,6), and low-temperature responsiveness (MsYTH3) (Fig. 6). A circadian regulatory element was discovered in the promoters of MsYTH1, MsYTH7, and MsYTH9. MsYTH7 and MsYTH9 have the same cis-elements, suggesting that these two members may have comparable transcriptional regulation. These cis-acting regulatory elements found in MsYTH promoters suggested that the MsYTH family may be involved in controlling alfalfa plant response to hormone and abiotic stressors.

Identification of Predicted Cis-Acting Elements in Alfalfa Genes. Cis-acting elements were predicted using the PlantCARE database. The scale at the bottom allows inference of the distance upstream to the translation start site, providing insight into potential gene regulation locations
RNA-seq expression profile
While attempting to examine the expression patterns of MsYTH genes in alfalfa, we encountered challenges in acquiring direct transcriptome data. As a workaround, we turned to the transcriptome data from CADL (Cultivated Alfalfa at the Diploid Level). Using this data, we investigated the homologs of 12 MsYTHs in CADL that exhibited the highest homology with those in alfalfa.
Our analysis revealed distinct expression patterns across nine different organs. In senescent leaves, the homologs of MsYTH7, 34 and 40 demonstrated low abundance, whereas the homologs of MsYTH9, 16, and 17 showed high abundance. This expression pattern was consistent in young and mature leaves, with these genes clustering together. Notably, these homologs presented almost inverse expression patterns in the root, elongating stem, and post-elongating stem.
The homologs of six genes, namely MsYTH6, 7, 22, 27, 34 and 40 showed lower mRNA levels in senescent, young, and mature leaves as compared to their transcript accumulation in the post-elongation and elongation stem phases. The MsYTH2 homolog exhibited the highest mRNA abundance in the leaf, whereas the homologs of MsYTH9, 16, and 17 showed the highest expression levels during the early growth stages, as indicated in Fig. 7.

Expression analysis of 12 MsYTH gene homologs across various organs in CADL based on transcriptome data. FPKM values of genes were transformed by log10 and the heatmap was constructed by HemI software. Red represents high expression and green represents low expression level
Moreover, the homologs of the MsYTH family displayed a variety of dynamic expression patterns throughout the flowering process. For instance, the MsYTH16 homolog expression was consistently and significantly downregulated from post-flowering to maturity, while the MsYTH7 homolog was upregulated exclusively during the leaf flower and nodule stages.
Response of MsYTH genes to abiotic stresses and organ-specific expression patterns by qRT-PCR
When the expression of MsYTH in the root is compared to that in the flower, some MsYTHs are expressed significantly higher in the root, some MsYTHs are expressed lower int the root, and some MsYTHs show comparable expression in the. MsYTH2, MsYTH14, MsYTH26, MsYTH37 and MsYTH42 are all downregulated in the roots while maintaining high expression in the leaves. 12 of the genes (MsYTH2, MsYTH7, MsYTH14, MsYTH16, MsYTH21, MsYTH27, MsYTH34, MsYTH37, MsYTH40, MsYTH42, MsYTH49, and MsYTH50) are expressed significantly higher in flower when compared to that in roots.
Under abiotic stresses, a dramatic change in the expression of MsYTH genes in alfalfa is also observed (Fig. 8). For example, the highest upregulation of MsYTH under cold stress is observed in MsYTH2 and MsYTH14, while under salinity conditions, MsYTH2 has an absolute advantage expression. This suggests that these genes may play important roles in both salt and cold tolerance in alfalfa. Most of the genes are upregulated more under cold conditions than under salt conditions. Overall, the findings shed light on the possible involvement of MsYTH proteins in alfalfa cold tolerance. The RNA-Seq data acquired from the CADL showed that the expression profiles of YTH genes were mainly consistent with our qRT-PCR results.

Bar plots showing the expression of MsYTH genes under cold and salt treatments. The x-axis represents the different treatments, while the y-axis represents the relative gene expression calculated using the 2−∆∆Ct method. Each bar represents the mean ± standard deviation (SD) of three biological replicates. Lowercase letters above the bars indicate significant differences (p < 0.05) according to one-way ANOVA
Discussion
Based on the previous studies on the erasers and writers of m6A, it was uncovered that m6A modification has notable effects on the regulation of gene expression at the post-transcriptional level, and plays key roles in various physiological processes. To understand the regulatory mechanism of m6A, recognizing m6A sites by reader proteins is pivotal. These recognitions perform multiple functions of m6A modifications, determining the fate of their target RNAs and influencing physiological aspects [38].
The YTH (YT521-B homology) domain, a 100–150 amino acid long gene family, was the first identified m6A reader in mammals, including humans, and has been shown to be substantially conserved across the five kingdoms [1, 36]. Of the five kingdoms, plants have the most abundant YTH genes [31]. Among plants, Arabidopsis is the most studied species. In this plant, the protein reader m6A modification has been found to affect developmental timing and morphogenesis under abiotic stress [3]. The Arabidopsis genome contains 13 YTH domain proteins that may be split into two clades based on the C-terminal area, two members in the YTHDC clade (Evolutionarily Conserved C-Terminal region) and 11 members in the YTHDF clade [3].
The chromosomal-level genome of the tetraploid plant alfalfa, an important forage species, was not published until 2020, providing a significant advantage for understanding its evolution, genetics, and breeding. In this study, a total of 53 putative YTH domain-containing protein family genes were identified in alfalfa. This number exceeds that of all the 26 species reported by Ouyang [37], which include six Chlorophyta, one moss, one Lycopodiatae, four monocot species, and 15 eudicot species. The author in that study asserted that the number of YTH genes is not directly proportional to genome size. In our study, all 53 identified YTH domain-containing protein family genes were found to be distributed across 21 chromosomes in alfalfa. Notably, the distribution is heterogeneous, with some chromosomes harboring only one gene (chr8.4), while some others harboring six genes (chr2.1). This observation indicates the significance of both genome size and the occurrence of tandem and segmental duplications in shaping the YTH domain-containing protein family genes landscape of alfalfa.
The role of tandem and segmental duplication, recognized as crucial events in gene family expansion [7], emerges prominently in our study. For example, three MsYTH1/MsYTH2/MsYTH3 genes cluster within 15 kb on chromosome 2.1, showing that both tandem and segmental duplication events were likely involved in the development of the MsYTH gene (Fig. 5). The pattern of functional redundancy among paralogs is similar to Arabidopsis, while segmental duplication dominated the evolution of YTH genes in rice [24] and cucumber [58]. These differences indicate potentially more complex regulatory mechanisms or functional redundancy within the YTH family in alfalfa, which require additional experimental validation in future studies.
The distribution of MsYTH within various groups displays a unique pattern when compared with Arabidopsis. For instance, the 13 YTH genes in Arabidopsis are broadly distributed across five groups, whereas in alfalfa, the majority of the YTH genes are primarily clustered within two groups, YTHDFa (22 genes) and YTHDFc (24 genes). Our study did not identify any alfalfa genes that belong to YTHDCb group. This gene distribution pattern deviates significantly from the typical configurations reported in the 26 species analyzed by Ouyang et al. [37], where no species had more than 20 YTH genes. Conversely, this distribution aligns more closely with the pattern reported in wheat, which possesses 39 YTH genes [45]. The distinct concentration of YTH genes in the YTHDFa and YTHDFc groups in alfalfa suggests possible functional redundancy within these specific gene subsets. This conjecture supports the theory of gene duplication and diversification being a critical evolutionary strategy in plants.
Members of each YTH subfamily have completely distinct gene architectures in terms of the size and organization of exons and introns (Fig. 3), showing that these genes developed separately. Members of the YTHDC subfamily have comparable gene structures, suggesting that these genes are created through gene duplication (Fig. 3a).
A search utilizing the SMART algorithm revealed the existence of a typical functioning YTH domain in each MsYTH protein. YTH domain are generally found near the C-terminus in the alfalfa YTHDF group but found near the N-terminus in alfalfa YTHDFc subgroup. In the alfalfa YTHDCa subgroup, YTH domains are located at the midway point. The YTH domain and the motifs are distributed similarly among members of each subfamily or subclade, indicating that they are highly conserved. Detailed motif analysis further revealed the evolutionary connections between members of the MsYTH family. Certain amino acid-rich motifs in the aligned protein sequence, such as the leucine-rich repeat domain, might be connected to salt resistance (Fig. 3c). Motifs 1–4 of the YTHDFa subclades were found to be identical to those reported in previous research on YTH domains [45]. In addition to the YTH domain, we have also discovered the presence of CCCH-type zinc finger motifs exclusively within the YTHDC clade of alfalfa YTH genes (Fig. 2). This finding aligns with the pattern observed in wheat YTH proteins, further highlighting the evolutionary conservation of this motif within the YTHDC subfamily. The CCCH-type zinc finger motifs have been associated with RNA-binding activities and have been implicated in post-transcriptional regulation processes [18]. The identification of these motifs within the YTHDC clade suggests potential functional implications related to RNA-binding and post-transcriptional regulation in this specific subclades of YTH genes in both alfalfa and wheat.
Looking further into the amino acids of the motifs, the aromatic cage pocket in the YTH domain of YTHDF and YTHDC functions to bind the m6A residue [17]. This positively charged pocket is created by the side chains of three amino acids: tryptophan (W411), tryptophan (W465), and tryptophan (W470) in human YTHDF1 and tryptophan (W377), tryptophan (W428), and leucine (L439) in human YTHDC1 [25, 38]. Specifically, proteins encoded by MsYTHDF genes incorporate a single domain structure, while those corresponding to MsYTH8, MsYTH12, MsYTH16 are identified as members of the MsYTHDC subfamily, exhibit CCCH-type zinc finger repeats at their N-termini. It has also been observed that the predicted aromatic cage pocket that binds the m6A residue of MsYTHC primarily consists of a sequence of three tryptophan residues (WWW). Conversely, the binding pocket was composed of two highly conserved tryptophan residues and either one tryptophan residue (WWW) or tyrosine (WWY) in MsYTHDF. This pattern aligns with those observed in wheat, suggesting a potential divergence in m6A binding between plant and animal YTHDCs.
In animals, the YTH domain is centrally located in YTHDCa, while it is found at the C-terminus in YTHDCb. We identified similar variations in plant species. However, in plant’s YTHDCs, all YTH domains are observed to be centrally located, akin to the positioning in animal YTHDCa. This may imply that the molecular mechanisms in plant YTHDCs could parallel those in animal YTHDCa.
A study of the binding affinity of YTHDF1 and YTHDC1 for m6A indicated that the asparagine (N367) in YTHDC1 forms a stronger hydrogen bond with N1 of m6A than the comparable aspartic acid (D401) residue in YTHDFa [20]. We observed that YTHDFa, YTHDFb, and YTHDFc proteins use aspartic acid to form a hydrogen bond with N1 of m6A; however, aspartic acid has been replaced by asparagine in six YTHDFb, three YTHDFc, and three YTHDC proteins. Moreover, in three YTHDFb proteins, histidine replaceds the usual residue at the corresponding location, a pattern not identified in YTH proteins of other plant species. The N-termini of YTHDF proteins have low-complexity areas, including Y/P/Q-rich regions; however, the Y/P/Q-rich regions of YTHDCs were found between the zinc finger repeat (YTH1 superfamily domain) and the YTH domain.
Information about the promoter sequence may provide insight into the gene’s activities. In this study, it was observed that light responsiveness had the most cis-regulatory elements among all the genes, suggesting a photoperiodism-related function. The second most prevalent cis-regulatory element is linked to stress-signaling hormones such as MeJA, salicylic acid, abscisic acid, and gibberellins. Other notable features included zein metabolism and regulation, circadian control, MYB binding, and anaerobic control.
YTH genes were expressed in all the investigated organs and showed diverse expression patterns. Several members of the YTH family have organ-specific expression patterns indicating that they may have various potential functions in different organs (Fig. 9). In line with this, it has been noted in previous research that distinct gene expression profiles in different organs can provide insights into gene functions, especially in relation to plant development and morphology [24].

Bar plots showing the expression of MsYTH genes in various alfalfa organs. The x-axis represents the different organs, while the y-axis shows the relative gene expression calculated using the 2−∆∆Ct method. Each bar represents the mean ± standard deviation (SD) of three biological replicates. Lowercase letters above the bars indicate significant differences (p < 0.05) according to one-way ANOVA
YTH-containing genes in many species can transcriptionally respond to abiotic stress, such as excessive salinity, drought, heat, cold, and polyethylene glycol stress [24, 58]. In this study, several stress-related cis-elements have been discovered in the upstream regions of the promoters of YTH members, including heat stress-responsive elements, drought-responsive elements, defense- and stress-responsive elements (TC-rich repeats), anaerobic induction elements, and low temperature-responsive elements, all of which respond to external environmental stresses [24].
The qRT-PCR results revealed distinct expression patterns for various genes in response to both cold and salt treatments. For example, MsYTH2 and MsYTH14 exhibited significant up-regulation under both stress conditions, implying a potential shared regulatory mechanism in addressing these environmental challenges. In contrast, both MsYTH33 and MsYTH35 consistently displayed down-regulation in response to cold and salt stress. MsYTH9 showed no significant difference in gene expression under cold stress but demonstrated up-regulation in response to salt stress. Notably, AtYHT3 encoding AtCPSF30, which is the homologous to MsYTH9, has been proven to enhance tolerance to oxidative stress by influencing poly(A) site selection and mRNA profiles [24]. This suggests the possibility of similar stress tolerance mechanisms among YTH genes in alfalfa.
Conclusion
In conclusion, our study identified and characterized 53 putative YTH domain-containing genes in alfalfa, highlighting their potential roles in the regulating various physiological processes, particularly in response to abiotic stress conditions. Expression profile analysis revealed that specific genes, such as MsYTH2, exhibited enhanced expression in response to salt and cold treatments, suggesting their possible involvement in managing abiotic stress responses. Additionally, the distinct motif distributions and domain architectures within the YTH gene family were observed. Our findings shed light on the biological functions of YTH genes in alfalfa and provide valuable insights for future genetic enhancement strategies aimed at improving salt and cold tolerance in this important forage crop.
Availability of data and materials
The whole alfalfa genome sequence information was obtained from the database (https://figshare.com/projects/whole_genome_sequencing_and_assembly_of_Medicago_sativa/66380). Alfalfa seeds used for experimental is given by Dr. Dong from Guizhou University. The datasets supporting the conclusions of this article are included in the article and its Additional files.
References
Ambrosone A, Costa A, Leone A, Grillo S. Beyond transcription: RNA-binding proteins as emerging regulators of plant response to environmental constraints. Plant Sci. 2012;182:12–8.
Anderson SJ, Kramer MC, Gosai SJ, Yu X, Vandivier LE, Nelson AD, Gregory BD. N6-methyladenosine inhibits local ribonucleolytic cleavage to stabilize mRNAs in Arabidopsis. Cell reports. 2018;25(5):1146–57.
Arribas-Hernández L, Bressendorff S, Hansen MH, Poulsen C, Erdmann S, Brodersen P. An m6A-YTH module controls developmental timing and morphogenesis in Arabidopsis. Plant Cell. 2018;30(5):952–67. https://doi.org/10.1105/tpc.17.00833.
Arribas-Hernández L, Simonini S, Hansen MH, Paredes EB, Bressendorf S, Dong Y, Østergaard L, Brodersen P. Recurrent requirement for the m6 A-ECT2/ECT3/ECT4 axis in the control of cell proliferation during plant organogenesis. Development. 2020;147(14):dev189134. https://doi.org/10.1242/dev.189134.
Bailey TL, Johnson J, Grant CE, Noble WS. The MEME suite. Nucleic Acids Res. 2015;43(W1):W39-49.
Bodi Z, Zhong S, Mehra S, Song J, Graham N, Li H, May S, Fray RG. Adenosine methylation in Arabidopsis mRNA is associated with the 3’ end and reduced levels cause developmental defects. Front Plant Sci. 2012;3:48. https://doi.org/10.3389/fpls.2012.00048.
Cannon SB, Mitra A, Baumgarten A, Young ND, May G. The roles of segmental and tandem gene duplication in the evolution of large gene families in Arabidopsis thaliana. BMC Plant Biol. 2004;4(1):1–21.
Chen H, Zeng Y, Yang Y, Huang L, Tang B, Zhang H, Hao F, Liu W, Li Y, Liu Y, Zhang X. Allele-aware chromosome-level genome assembly and efficient transgene-free genome editing for the autotetraploid cultivated alfalfa. Nat Commun. 2020;11(1):2494.
Chen J, Cao H, Chen D, Kuang L, Wu D. Transcriptome-wide analysis of m6A methylation reveals genetic responses to cadmium stress at germination stage in rice. Environ Exp Bot. 2023;205:105130.
Chou KC, Shen HB. Cell-PLoc: a package of Web servers for predicting subcellular localization of proteins in various organisms. Nat Protoc. 2008;3(2):153–62.
Crooks GE, Hon G, Chandonia JM, Brenner SE. WebLogo: a sequence logo generator. Genome Res. 2004;14(6):1188–90.
Dai X, Zhuang Z, Boschiero C, Dong Y, Zhao PX. LegumeIP V3: from models to crops—an integrative gene discovery platform for translational genomics in legumes. Nucleic Acids Res. 2021;49(D1):D1472–9.
Floris M, Mahgoub H, Lanet E, Robaglia C, Menand B. Post-transcriptional regulation of gene expression in plants during abiotic stress. Int J Mol Sci. 2009;10(7):3168–85. https://doi.org/10.3390/ijms10073168.
Forman-Kay JD, Mittag T. From sequence and forces to structure, function, and evolution of intrinsically disordered proteins. Structure. 2013;21(9):1492–9.
Gasteiger E, Hoogland C, Gattiker A, Duvaud SE, Wilkins MR, Appel RD, Bairoch A. Protein identification and analysis tools on the ExPASy server. In: The Proteomics Protocols Handbook, J.M. Walker, editors. Humana Press. pp. 571–607
Govindan G, et al. mRNA N6 -methyladenosine is critical for cold tolerance in Arabidopsis. Plant J. 2022;111(4):1052–68.
Hazra D, Chapat C, Graille M. m6A mRNA Destiny: Chained to the rhYTHm by the YTH-Containing Proteins. Genes. 2019;10(1):49.
Jang JC. Arginine-rich motif-tandem CCCH zinc finger proteins in plant stress responses and post-transcriptional regulation of gene expression. Plant Sci. 2016;252:118–24.
Jia G, Fu Y, Zhao X, et al. N6-Methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nat Chem Biol. 2011;7:885–7. https://doi.org/10.1038/nchembio.687.
Krepl M, Damberger FF, von Schroetter C, Theler D, Pokorná P, Allain FH-T, Šponer J. Recognition of N6-methyladenosine by the YTHDC1 YTH domain studied by molecular dynamics and NMR spectroscopy: the role of hydration. J Phys Chem B. 2021;125(28):7691–705. https://doi.org/10.1021/acs.jpcb.1c03541.
Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Higgins DG. Clustal W and Clustal X version 2.0. Bioinform. 2007;23(21):2947–48.
Lescot M, Déhais P, Thijs G, Marchal K, Moreau Y, Van de Peer Y, Rouzé P, Rombauts S. PlantCARE, a database of plant cis-acting regulatory elements and a portal to tools for in silico analysis of promoter sequences. Nucleic Acids Res. 2002;30(1):325–7.
Letunic I, Bork P. Interactive Tree Of Life (iTOL) v5: an online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 2021;49(W1):W293–6.
Li D, Zhang H, Hong Y, Huang L, Li X, Zhang Y, Ouyang Z, Song F. Genome-wide identification, biochemical characterization, and expression analyses of the YTH domain-containing RNA-binding protein family in Arabidopsis and rice. Plant Mol Biol Report. 2014;32(6):1169–86.
Liao S, Sun H, Xu C. YTH domain: a family of N6-methyladenosine (m6A) readers. Genomics Proteomics Bioinformatics. 2018;16(2):99–107. https://doi.org/10.1016/j.gpb.2018.04.002.
Librado P, Rozas J. DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics. 2009;25(11):1451–2. https://doi.org/10.1093/bioinformatics/btp187. Epub 2009 Apr 3 PMID: 19346325.
Licatalosi DD. Roles of RNA-binding proteins and post-transcriptional regulation in driving male germ cell development in the mouse. RNA Processing: Disease and Genome-wide Probing; 2016. p. 123–51.
Liu G, Wang J, Hou X. Transcriptome-wide N6-methyladenosine (m6A) methylome profiling of heat stress in pak-choi (Brassica rapa ssp. chinensis). Plants. 2020;9(9):1080.
Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods. 2001;25:402–8.
Ma C, Liao S, Zhu Z. Crystal structure of human YTHDC2 YTH domain. Biochem Biophys Res Commun. 2019;518:678–84.
Ma W, Cui S, Lu Z, Yan X, Cai L, Lu Y, Cai K, Zhou H, Ma R, Zhou S, et al. YTH domain proteins play an essential role in rice growth and stress response. Plants. 2022;11:2206. https://doi.org/10.3390/plants11172206.
Mao X, Hou N, Liu Z, He J. Profiling of N6-methyladenosine (m6A) modification landscape in response to drought stress in apple (Malus prunifolia (Willd.) Borkh). Plants. 2022;11(1):103.
Martínez-Pérez M, Aparicio F, López-Gresa MP, Bellés JM, Sánchez-Navarro JA, Pallás V. Arabidopsis m6A demethylase activity modulates viral infection of a plant virus and the m6A abundance in its genomic RNAs. Proc Natl Acad Sci USA. 2017;114(40):10755–60. https://doi.org/10.1073/pnas.1703139114.
Mirdita M, Schütze K, Moriwaki Y, Heo L, Ovchinnikov S, Steinegger M. ColabFold: making protein folding accessible to all. Nat Methods. 2022;19(6):679–82. https://doi.org/10.1038/s41592-022-01488-1.
Ning W, Wei Y, Gao L, Han C, Gou Y, Fu S, Liu D, Zhang C, Huang X, Wu S, Peng D. HemI 2.0: an online service for heatmap illustration. Nucleic Acids Res. 2022;50(W1):W405–11.
Nishizawa Y, Konno M, Asai A, Koseki J, Kawamoto K, Miyoshi N, Takahashi H, Nishida N, Haraguchi N, Sakai D, Kudo T. Oncogene c-Myc promotes epitranscriptome m6A reader YTHDF1 expression in colorectal cancer. Oncotarget. 2018;9(7):7476.
Ouyang Z, Duan H, Mi L, Hu W, Chen J, Li X, Zhong B. Genome-wide identification and expression analysis of the YTH domain-containing RNA-binding protein family in citrus sinensis. J Am Soc Hortic Sci. 2019;144(2):79–91.
Patil DP, Pickering BF, Jaffrey SR. Reading m6A in the transcriptome: m6A-binding proteins. Trends Cell Biol. 2018;28(2):113–27. https://doi.org/10.1016/j.tcb.2017.10.001.
Paysan-Lafosse T, Blum M, Chuguransky S, Grego T, Pinto BL, Salazar GA, Bileschi ML, Bork P, Bridge A, Colwell L, Gough J. InterPro in 2022. Nucleic Acids Res. 2023;51(D1):D418–27.
Pincus MR. Physiological structure and function of proteins. In: Cell physiology source book. Amsterdam: Elsevier; 2001. p. 19–42.
Reichel M, Köster T, Staiger D. Marking RNA: m6A writers, readers, and functions in Arabidopsis. J Mol Cell Biol. 2019;11(10):899–910. https://doi.org/10.1093/jmcb/mjz085.
Růžička K, Zhang M, Campilho A, Bodi Z, Kashif M, Saleh M, Eeckhout D, El-Showk S, Li H, Zhong S, De Jaeger G, Mongan NP, Hejátko J, Helariutta Y, Fray RG. Identification of factors required for m6A mRNA methylation in Arabidopsis reveals a role for the conserved E3 ubiquitin ligase HAKAI. New Phytol. 2017;215(1):157–72. https://doi.org/10.1111/nph.14586.
Scutenaire J, Deragon JM, Jean V, Benhamed M, Raynaud C, Favory JJ, Merret R, Bousquet-Antonelli C. The YTH domain protein ECT2 is an m6A reader required for normal trichome branching in Arabidopsis. Plant Cell. 2018;30(5):986–1005. https://doi.org/10.1105/tpc.17.00854.
Shi R, Ying S, Li Y, et al. Linking the YTH domain to cancer: the importance of YTH family proteins in epigenetics. Cell Death Dis. 2021;12:346. https://doi.org/10.1038/s41419-021-03625-8.
Sun J, Bie XM, Wang N, Zhang XS, Gao XQ. Genome-wide identification and expression analysis of YTH domain-containing RNA-binding protein family in common wheat. BMC Plant Biol. 2020;20(1):1–4.
Tompa P. Multisteric regulation by structural disorder in modular signaling proteins: an extension of the concept of allostery. Chem Rev. 2014;114(13):6715–32.
Wang J, Chitsaz F, Derbyshire MK, Gonzales NR, Gwadz M, Lu S, Marchler GH, Song JS, Thanki N, Yamashita RA, Yang M, Zhang D, Zheng C, Lanczycki CJ, Marchler-Bauer A. The conserved domain database in 2023. Nucleic Acids Res. 2022. https://doi.org/10.1093/nar/gkac1096.
Wang Y, Tang H, Debarry JD, Tan X, Li J, Wang X, Lee TH, Jin H, Marler B, Guo H, Kissinger JC. MCScanX: a toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 2012;40(7):e49.
Wei LH, Song P, Wang Y, Lu Z, Tang Q, Yu Q, Xiao Y, Zhang X, Duan HC, Jia G. The m6A reader ECT2 controls trichome morphology by affecting mRNA stability in Arabidopsis. Plant Cell. 2018;30(5):968–85. https://doi.org/10.1105/tpc.17.00934.
Xiao W, Adhikari S, Dahal U, Chen YS, Hao YJ, Sun BF, Yang YG. Nuclear m6A reader YTHDC1 regulates mRNA splicing. Mol Cell. 2016;61(4):507–19.
Yang D, Xu H, Liu Y, Li M, Ali M, Xu X, Lu G. RNA N6-methyladenosine responds to low-temperature stress in tomato anthers. Front Plant Sci. 2021;12:687826.
Zaccara S, Ries RJ, Jaffrey SR. Reading, writing and erasing mRNA methylation. Nat Rev Mol Cell Biol. 2019;20(10):608–24. https://doi.org/10.1038/s41580-019-0168-5.
Zhang Z, Theler D, Kaminska KH, Hiller M, de la Grange P, Pudimat R, Rafalska I, Heinrich B, Bujnicki JM, Allain FH, Stamm S. The YTH domain is a novel RNA binding domain. J Biol Chem. 2010;285(19):14701–10. https://doi.org/10.1074/jbc.M110.104711.
Zhao BS, Roundtree IA, He C. Post-transcriptional gene regulation by mRNA modifications. Nat Rev Mol Cell Biol. 2017;18(1):31–42.
Zheng G, Dahl JA, Niu Y, Fedorcsak P, Huang CM, Li CJ, Vågbø CB, Shi Y, Wang WL, Song SH, Lu Z. ALKBH5 is a mammalian RNA demethylase that impacts RNA metabolism and mouse fertility. Mol Cell. 2013;49(1):18–29.
Zheng H, Gao Y, Dang Y, Wu F, Wang X, Zhang F, Sui N. Characterization of the m6A gene family in sorghum and its function in growth, development and stress resistance. Ind Crops Prod. 2023;198:116625.
Zhong S, Li H, Bodi Z, Button J, Vespa L, Herzog M, Fray RG. MTA is an Arabidopsis messenger RNA adenosine methylase and interacts with a homolog of a sex-specific splicing factor. Plant Cell. 2008;20(5):1278–88.
Zhou Y, Hu L, Jiang L, Liu S. Genome-wide identification and expression analysis of YTH domain-containing RNA-binding protein family in cucumber (Cucumis sativus). Genes Genomics. 2018;40(6):579–89.
Funding
This research was funded by Key Program of the National Natural Science Foundation of China (42320104006), the Key R&D Program of Shandong Province, China (2022TZXD0045) and National Natural Science Foundation of China (3190139).
Author information
Authors and Affiliations
Contributions
S.F. and Y.Z. contributed to conception and design of the study. X.X. and J.C. performed the statistical analysis. S.F. and Y.Y. wrote the first draft of the manuscript. All authors contributed to manuscript revision, read and approved the submitted version.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
We guarantee that the collection of plant material and experimental research on plants comply with relevant institutional, national, and international guidelines and legislation.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Table S1.
Sequences of all the MsYTH protein.
Additional file 2: Table S2.
List of Primers used for qRT-PCR genes.
Additional file 3: Table S3.
Synonymous and Non-Synonymous substitution rates (Ka/Ks) for corresponding gene pairs within the alfalfa YTH gene family.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Fan, S., Xu, X., Chen, J. et al. Genome-wide identification, characterization, and expression analysis of m6A readers-YTH domain-containing genes in alfalfa. BMC Genomics 25, 18 (2024). https://doi.org/10.1186/s12864-023-09926-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-023-09926-w