
Abstract
Background
Chemical signaling between a mammalian host and intestinal microbes is health and maintenance of ‘healthy’ intestinal microbiota. Escherichia coli O157:H7 can hijack host- and microbiota-produced chemical signals for survival in a harsh and nutritionally competitive gastrointestinal environment and for intestinal colonization. Norepinephrine (NE) produced by sympathetic neurons of the enteric nervous system has been shown in vitro to induce expression of genes controlling E. coli O157:H7 swimming motility, acid resistance, and adherence to epithelial cells. A previous study used a microarray approach to identify differentially expressed genes in E. coli O157:H7 strain EDL933 in response to NE. To elucidate a comprehensive transcriptional response to NE, we performed RNA-Seq on rRNA-depleted RNA of E. coli O157:H7 strain NADC 6564, an isolate of a foodborne E. coli O157:H7 strain 86–24. The reads generated by RNA-Seq were mapped to NADC 6564 genome using HiSat2. The mapped reads were quantified by htseq-count against the genome of strain NADC 6564. The differentially expressed genes were identified by analyzing quantified reads by DESeq2.
Results
Of the 585 differentially expressed genes (≥ 2.0-fold; p < 0.05), many encoded pathways promoting ability of E. coli O157:H7 strain NADC 6564 to colonize intestines of carrier animals and to produce disease in an incidental human host through increased adherence to epithelial cells and production of Shiga toxins. In addition, NE exposure also induced the expression of genes encoding pathways conferring prolonged survival at extreme acidity, controlling influx/efflux of specific nutrients/metabolites, and modulating tolerance to various stressors. A correlation was also observed between the EvgS/EvgA signal transduction system and the ability of bacterial cells to survive exposure to high acidity for several hours. Many genes involved in nitrogen, sulfur, and amino acid uptake were upregulated while genes linked to iron (Fe3+) acquisition and transport were downregulated.
Conclusion
The availability of physiological levels of NE in gastrointestinal tract could serve as an important cue for E. coli O157:H7 to engineer its virulence, stress, and metabolic pathways for colonization in reservoir animals, such as cattle, causing illness in humans, and surviving outside of a host.
Similar content being viewed by others

Background
Escherichia coli O157:H7 (O157) infections in healthy human adults usually result in an asymptomatic and self-resolvable watery diarrhea [1]. However, in children and elderly individuals, O157 infections can lead to development of more serious symptoms such as abdominal pain, bloody diarrhea or hemorrhagic colitis, and hemolytic uremic syndrome [2, 3]. Besides producing Shiga toxins, which are a major cause of kidney failure and even death in infected humans [4, 5], O157 strains encode virulence factors that promote their ability to colonize the large intestine of incidental human hosts and carrier animals, such as cattle [6]. These adherence-promoting virulence factors are secreted through a type-3 secretion system, genes for which are encoded by a pathogenicity island, called the locus of enterocyte effacement (LEE) [6,7,8,9]. LEE is composed of five major operons and three of these five operons (LEE1 – LEE3) are involved in formation of the type three system which secrets adhesin intimin, intimin receptor called translocated intimin receptor (Tir), and many other proteins involved in the formation of attaching and effacing lesions on intestinal mucosa [10, 11]. Although LEE expression is positively regulated by Ler [11], several positive and negative transcriptional regulators, some LEE- and some non-LEE-encoded, and a variety of bacterial, host, and environmental signals control Ler expression to ensure optimal LEE expression occurs in the appropriate intestinal compartment [12, 13].
Since the preferred site for O157 colonization in ruminants, such as cattle, is the terminal colon, specifically the rectoanal junction (RAJ) [7, 8], O157 first traverses the highly acidic environment of the abomasum before reaching RAJ. Several studies have shown that the ability to sense a variety of chemical signals and metabolites produced by the host and intestinal microbiota play an important role in the survival of O157 at the extreme acidic pH of the stomach and subsequent O157 colonization at the RAJ. For example, in cattle, which are the primary reservoir for O157 and source of O157 infections in humans, bacterial members of the rumen microbiota produce acyl-homoserine lactones that are perceived as a quorum-sensing (QS) signal by a LuxR homolog SdiA to induce expression of glutamate-dependent acid resistance pathway 2 (ARP2) [14, 15]. ARP2 ensures survival of O157 at a very low pH (pH 2) and thus accounts for the low infectious dose of O157 in human infections [14, 16,17,18]. The expression of ARP2, which uses glutamate decarboxylase A (GadA) and GadB, and an antiporter (GadC) to confer acid resistance, is regulated by GadE [19]. GadE expression is controlled by transcriptional regulators GadX, GadW, and a two-component signal transduction system EvgS/EvgA [20,21,22]. EvgS/EvgA responds to low pH and alkali metals to regulate acid resistance and multidrug resistance efflux pumps in E. coli [23]. The response regulator EvgA has been shown to induce gadE transcription either through the induction of YdeO, which interacts with the gadE promoter, or through direct interaction of EvgA at an undefined site in the gadE promoter [22, 24].
While the sensing of acyl-homoserine lactones enhances expression of ARP2 by GadE, LEE expression is repressed by GadE since LEE functions are not needed in the rumen. To colonize the RAJ, O157 uses different QS pathways to sense signals, such as autoinducer-3 (AI-3) produced by many bacterial species of intestinal microbiota [25,26,27,28], and host-produced stress hormones norepinephrine/epinephrine [29,30,31]. About half of norepinephrine (NE) is synthesized and utilized locally within the enteric nervous system by adrenergic neurons in the basal-lateral layer of the gut, epinephrine (E), on the other hand, is mostly synthesized in the adrenal medulla and reaches the small intestine via blood [30, 32, 33]. According to many studies, NE not only enhances growth, iron acquisition, motility and Shiga toxin expression, but also induces acid resistance and promotes adherence of O157 to epithelial cells [34,35,36,37,38,39,40]. The mechanism for NE-mediated growth and virulence enhancement of O157 is linked to the release of iron (Fe3+) from transferrin and lactoferrin, which are important innate immune defense proteins in mammalian hosts [35, 41, 42]. NE and NE-Fe3+ complexes reach the periplasm through the outer membrane embedded ferric iron transport system, and outer membrane proteins OmpA, and OmpC [35, 42]. Once in the periplasm, NE is sensed by the inner membrane-embedded quorum-sensing signal transduction systems (QseBC and QseEF) that through a phospho-relay cycle leads to the activation of a cascade of response regulators, which induce expression of motility, LEE, and Shiga toxins [30]. In addition to impacting gene expression directly, NE is also metabolized by commensal E. coli to 3,4-dihydroxymandelic acid (DHMA), which serves as a bacterial chemoattractant, induces the expression of virulence genes, and enhances attachment of O157 to intestinal epithelial cells in a QseC-dependent manner [27].
A recent study that used a probe set of 610 genes in a microarray-based transcriptional profiling of O157 strain EDL933 demonstrated that NE enhanced expression of genes involved in tissue adherence, Shiga toxin production, motility, ARP2, and reduced expression of genes for iron acquisition [34]. In the current study, we describe the use of RNA-Seq to determine the differential gene expression profile of O157 strain NADC 6564 [43] when grown in the presence of NE. Like the previous microarray-based gene expression profiling [34], we found that NE-mediated signaling resulted in the differential expression of genes encoding pathways for survival at a very low pH and for intestinal colonization. We also showed that NE-mediated induction of the EvgS/EvgA signaling system is directly linked to the expression of acid resistance phenotype. In addition, we demonstrated that exposure of strain NADC 6564 to NE resulted in the repression of enterobactin siderophore biosynthesis used for iron (Fe3+) acquisition but enhanced the expression of ferrous uptake pathway that is more active under low pH and anaerobic conditions. The differential regulation of numerous other pathways, such as those controlling transport of amino acids and peptides, salvage of pyrimidines, storage and utilization of carbohydrate substrates, nitrogen and sulfur metabolism, and sensing of various stressors indicated that strain NADC 6564 uses NE to alter its cellular physiology and cell membrane functions that in all likelihood are advantageous for O157 survival, growth, and colonization of specific sites in the large intestinal compartment of its carrier animal and the incidental human host.
Results
Large numbers of genes were upregulated in response to norepinephrine
The results described below are based on a genome-scale transcriptomic analysis of E. coli O157H:H7 (O157) strain NADC 6564 [43] in response to NE that overcomes the limitations of a previously reported study involving only a 610 gene array to determine the differential response of E. coli O157:H7 strain EDL933 to norepinephrine (NE). Although some of the major findings of the current study were similar to the 610 gene array-based study, we identified many other differentially expressed genes regulating a variety of pathways in O157 in response to NE. We used an FDR-adjusted p-value of 0.05 and ≥ 2.0-fold change in gene expression as a threshold for considering a gene being differentially expressed (DE) in NE-treated relative to untreated bacterial cultures. Many of the genes that we identified as DE at ≥2.0-fold, p ≤ 0.05 in response to NE (this report) were also identified as DE in a previously reported microarray-based study that used a threshold of 1.5-fold for a gene to be considered DE [34]. RNA-Seq analysis of the genome-scale transcriptome of the norepinephrine (NE)-treated and untreated cultures of strain NADC 6564 allowed us to determine the proportion of DE genes. Overall, 5509 genes (Fig. 1A, Table S1), representing 98.76% of 5578 total genes predicted in the chromosomal sequence of NADC 6564 [43], generated reads mapping to the reference genome. Using the ≥2.0-fold threshold, 585 genes, representing 10.6% of 5509 genes, were DE (p > 0.05) in response to NE (Fig. 1A). Of these 585 DE genes, 321 genes (about 5.82%) were upregulated and 264 genes (about 4.79%) were downregulated in NE-treated cultures (p < 0.05) (Fig. 1A and Inset Table in Fig. 1; Table S2 and Table S3). About 31.8% (102 of 321 genes) of the upregulated genes and about 14.4% (38 of 264 genes) of the downregulated genes were predicted to encode hypothetical proteins with unknown functions (Fig. 1B and Inset Table Fig. 1). The DE genes were not localized to any specific region of the chromosome, rather both up- and downregulated genes were distributed randomly throughout the whole chromosome of strain NADC 6564 (Fig. 1C). However, the region of the chromosome (located between 2000 kbp – 2300 kbp) containing a bacteriophage labeled P6 and a genomic island G19 contained a cluster of several upregulated and downregulated genes, respectively, although majority of these genes encoded hypothetical proteins of unknown functions (Fig. 1C). Therefore, the biological significance of the differential expression of genes in this cluster is not clear and will depend on the functional characterization of these genes/gene products and identification of regulatory networks controlling these genes.

Graphical representation of differentially expressed gene by E. coli O157:H7 strain NADC 6564 in response to norepinephrine (A) A pie chart showing proportion of significantly upregulated (dark blue slice), downregulated (light blue slice), and unaffected genes (red slice) in total of 5509 chromosomal genes analyzed by RNA-Seq, (B) A bar graph showing number of upregulated genes with known function (dark blue bar), upregulated genes assigned hypothetical function (light blue bar), downregulated genes (dark green bar), and downregulated genes assigned hypothetical function (light green bar). Inset Table in Fig. 1 shows up- and downregulated genes of known and hypothetical functions and their fold change in expression, and (C) BRIG plot showing distribution of upregulated genes (green), downregulated genes (red), Genomic islands 1–53 (blue) and bacteriophages P1 – P19 (purple) on the chromosome of strain NADC 6564. Chromosomal size (5,466,770 bp) of strain NADC 6564 is listed in the center of the inner circle, which is marked on the inside using a 500 kbp (kilo base pairs) scale
Norepinephrine-treated cultures showed enhanced expression of virulence genes
Among the virulence genes, majority of LEE-encoded genes and the stx2 gene (encodes for Stx2 subunits A and B) were significantly upregulated (≥ 2.0-fold, p < 0.05) in NE-treated cultures (Table 1; Table S2). Since LEE expression is activated by LEE-encoded ler, and ler expression in turn is modulated by a network of transcriptional factors, we analyzed the RNA-Seq data to determine if NE exposure resulted in enhanced expression of ler and differential expression of specific LEE- and non-LEE-encoded transcriptional factors. This was done to gain insight into the mechanism of regulation of LEE by these transcriptional factors. We did not detect any change in the expression of ler, which encodes the transcriptional factor Ler for activating LEE expression (Table S2), However, two of the four copies of the perC gene, which encodes transcriptional factor PerC, were upregulated by ≥2-fold (p < 0.05) in NE-treated cultures (Table 1; Table S2). The four copies of perC are located at different chromosomal locations.
Although LEE encoded proteins are critical for O157 adherence to epithelial cells, many other adhesins, particularly those represented by fimbriae also play an important role in adherence of O157 to mammalian tissues and to abiotic matrices to produce biofilms. RNA-Seq analyses revealed significantly higher expression of several genes belonging to Lpf1 (lpfB and lpfD), Ygp, and curli (csgB) fimbrial groups (Table 1 and Table S2) in response to NE. The csgB gene, which is located in the csgBAC operon [44] and encodes CsgB for nucleating CsgA subunits into curli fimbriae, showed the highest increase (+ 7.8-fold, p < 0.05) in its expression (Table 1), but no other genes involved in curli biogenesis were differentially expressed. A cdGMP encoding dosC gene was also upregulated (+ 2.86-fold, p < 0.05) (Table 2, Table S2) in response to NE and increased expression of dosC has been shown to enhance csgB expression and biofilm formation in E. coli [45].
Norepinephrine enhanced expression of genes encoding acid resistance and signaling system EvgS/EvgA
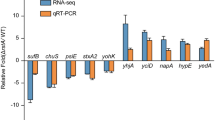
O157 can survive for several hours in highly acidic environments (pH 2.0 to 2.5) resembling those encountered in a mammalian stomach [46]. Similar to the results reported in a previous microarray-based transcriptional study [34], we also observed that NE induced significantly higher (≥ 2.0-fold, p < 0.05) expression of acid resistance pathway (ARP2) genes involved in O157 strains’ extreme acid tolerance (Table 3 and Table S2). The highly (p < 0.05) induced ARP2 genes included gadA (+ 4.21-fold), gadB (+ 4.36-fold), and gadC (+ 5.02-fold), which encode enzymes for reducing cytoplasmic H+ ion concentration when external pH is very acidic. Similarly, expression of hdeD, hdeB, and hdeA, which are present on an acid fitness island and encode proteins that serve as chaperons during acid stress [47], was upregulated by + 4.76, + 3.46, and + 3.94 -fold, respectively (Table 3, Table S2). The gadE gene, a LuxR-like family of proteins and a master regulator of ARP2 genes [48], was significantly upregulated (+ 3.72-fold, p < 0.05) in NE-treated cultures. The expression of gadX (+ 3.27-fold) and gadW (+ 2.37-fold), which encode AraC-family of transcriptional regulators GadX and GadW [20], respectively, and control gadA/BC and gadE expression, was significantly upregulated in NE-treated cultures. We also used RT-qPCR to determine the relative expression of some of the genes (gadB, hdeA, gadE, and gadX) of ARP2 that showed elevated expression by RNA-Seq. Although RNA-Seq analysis showed an increase of ≥2.0 -fold in the expression of gadB, hdeA, gadE, and gadX in NE-treated cultures, RT-qPCR analysis showed significant increases in the expression of these genes but the fold increase in their expression was < 2-fold (Fig. 2) (Table 3; Table S2). The most important reason for the observed differences in gene expression by these two approaches could be that RNA-Seq libraries were normalized and gene expression values were calculated relative to the mRNA pool. In RT-qPCR, the amount of RNA used for cDNA synthesis was based on total RNA levels, which could lead to variability in RNA available for cDNA synthesis necessitating the use of an internal reference for normalization. But despite these underlying technical differences, the trend showing upregulation of four ARP2 genes was similar between the two methods. Similar findings were apparent in a study where use of a RT-qPCR as validation approach produced lower fold changes in the expression of genes compared to that detected for the same genes by microarray-based transcriptional analysis [34].

Determination of expression of genes encoding acid resistance pathway 2. Relative expression of gadB, hdeA, gadE and gadX genes was determined by using total RNA prepared from strain NADC 6564 grown without (green bars) or with norepinephrine (red bars). Error bars represent standard deviation of the mean of three independent assays. *** p = 0.00043, **** p = 0.000086; ** = p = 0.0037; **** p 0.000064
In addition to the upregulation of gadE, gadX, and gadW, NE enhanced expression of evgS (+ 2.1-fold; p < 0.05) and evgA (+ 2.93-fold; p < 0.05) (Table 3; Table S2). The EvgS/EvgA signaling system is involved in the regulation of ARP2 in E. coli through YdeO, the transcriptional factor that activates gadE [24]. However, ydeO expression was not enhanced in NE-treated cultures suggesting that EvgS/EvgA might activate gadE directly without the intermediate of YdeO. To confirm a direct requirement of EvgS/EvgA in ARP2 expression and NE signaling, we constructed an evgS/evgA deletion mutant of strain NADC 6564 and compared the evgS/evgA mutant and the same mutant complemented with an evgS/evgA recombinant plasmid to the parental strain NADC 6564 in their ability to survive exposure to highly acidic (pH 2.5) conditions before or after exposure to NE. As shown in Fig. 3, the evgS/evgA mutant grown overnight without NE and then incubated for 3 h in phosphate-citrate minimal medium (pH 2.5) was recovered at significantly lower numbers (0.47%, p < 0.05) compared to 5.16 and 9.3% recovery of viable cells of parental strain NADC 6564 and the evgS/evgA mutant complemented with an evgS/evgA-recombinant plasmid, respectively. On the other hand, the evgS/evgA mutant grown overnight in the presence of NE and then exposed to an acidified medium for 3 h resulted in the recovery of significantly higher numbers of viable cells (6.74%, p < 0.05) compared to the mutant strain (0.47%) grown overnight in the absence of NE (Fig. 3). However, recovered numbers of viable cells for the evgS/evgA mutant even after an overnight growth in NE were still lower (6.74%, p < 0.05) compared to the similarly grown cultures of parental strain (9.17%) and the complemented evgS/evgA mutant strain (10.55%) (Fig. 3). These results indicated that evgS/evgA genes are involved in ARP2-mediated acid resistance of strain NADC 6564 as mutants lacking these genes were highly sensitive to very low pH conditions. However, the evgS/evgA mutant was still able to respond to NE signaling but at levels that were only slightly lower in terms of recovery of viable cells (6.74%, p = 0.03) after 3 h exposure to acidic medium than the similarly grown parental strain (9.17%) (Fig. 3).

Determination of the requirement of evgS/evgA genes in survival of E. coli O157:H7 strain NADC6564 in highly acidic medium and in NE signaling. The overnight bacterial cultures of parental strain NADC 6564 carrying the cloning vector pACYC177 (NADC 6564/pACYC177), evgS/evgA deletion mutant of NADC 6564 carrying pACYC177 (strain NADC 6662/pACYC177), evgS/evgA deletion mutant complemented with evgS/evgA recombinant plasmid pSM779 (strain NADC 6662/pSM779), and an acid sensitive strain of E. coli O157:H7 carrying pACYC177 (strain NADC 6565/pACYC177) were grown in DMEM medium lacking (green bars) or containing norepinephrine (red bars) were diluted in a phosphate-citrate minimal medium acidified to pH 2.5. After 3 h of incubation, cultures were plated for viable cell count determination (as described in materials and methods) Error bars represent standard deviation of the mean of three independent assays. **** p < 0.0001, * p = 0.033; ** = p = 0.0086
Norepinephrine enhanced expression of genes encoding various stress response, cell division, and biofilm formation pathways
In addition to enhancing expression of ARP2 genes, the presence of NE resulted in the differential expression of genes linked to various stress responses. Prominent among these genes were those that enable E. coli strains to survive in the stationary phase of growth. As listed in Table 2 and Table S2, stationary phase genes that were significantly (≥ 2.0-fold, p < 0.05) upregulated in NE-treated cultures was the DNA starvation/stationary phase protection gene dps (+ 2.88-fold) [49] and genes uspA, uspB, uspE, uspF, and uspG, which encode universal stress proteins [50]. The expression of usp genes was upregulated in the range + 2.01-fold through + 3.07-fold in response to NE. Another set of genes that was upregulated in response to NE is an important component of heat shock response in E. coli [51]. Some of these genes included hspQ (+ 2.2-fold) and ibpA (+ 2.15-fold) (Table 2, Table S2). The heat shock response is initiated in response to a sudden increase in the growth temperature while the cold shock proteins (Csp) are produced in response to rapid temperature downshifts [52]. As shown in Table 2 and Table S2, cspG expression was significantly upregulated (+ 2.99-fold) in response to NE. Also, upregulated was clpB (+ 2.72-fold) encoding a protease produced during stress [52]. The sodC gene that encodes a periplasmic superoxide dismutase C in NE-treated cultures (Table 2) and serves as an important antioxidant in protecting bacterial cells from oxidative stress [53,54,55,56] was also upregulated by + 2.81-fold in response to NE.
Since unfavorable growth conditions promote biofilm formation that requires the induction of many stationary phase-dependent genes [57, 58], NE-treatment induced significantly (p < 0.05) higher expression of some of the genes linked to biofilm formation. Prominent among these genes was csgB (+ 7.85-fold) (Table 1 and Table S2) that encodes the CsgB protein essential for the formation of curli fimbriae. Curli fimbriae are essential for the initial, reversible bacterial adherence to abiotic/biotic surfaces and in subsequent stages of biofilm formation [59]. The expression of genes (wcaA and wcaB) encoding enzymes for the biosynthesis of extracellular polysaccharide colanic acid was significantly downregulated (≥ − 2.0-fold, p < 0.05) in response to NE (Table S3). Other genes that were upregulated in response to NE have been implicated in the control of biofilm formation including bolA (+ 3.2-fold), bssR (+ 2.76-fold), and bssS (+ 7.75-fold) (Table 2 and Table S2). The bolA gene confers round cell morphology to E. coli cells, is expressed in the stationary phase of growth in a RpoS-dependent manner, and controls biofilm formation in E. coli [60, 61]. The bssR (yliH) and bssS (yceP) genes encode transcriptional regulators involved in the regulation of biofilm formation through autoinducer-2 secretion in E. coli K12 [62].
NE-treatment resulted in differential expression of genes encoding metabolic pathways
Table 4 lists the numbers of genes differentially expressed in various metabolic pathways in response to NE. Tables S2 and S3 list names of differentially expressed genes (≥ 2.0-fold, p < 0.05), pathways represented by these genes, and fold-change in the expression of these genes. Among the metabolic pathways that had their representative genes significantly upregulated were ABC transporter systems for uptake of amino acids, glutamine, and sulfate/sulfite (Table 4; Table S2). Also upregulated were genes encoding phosphofructokinase fruK (+ 3.12-fold, p < 0.05) and PTS fructose transporter subunit IIBC (+ 4.32, p < 0.05) (Table 4; Table S2), suggesting that gluconeogenic activity might be enhanced in response to NE. The genes mglA (− 4.26-fold, p < 0.05) and mglC (− 2.66-fold, p < 0.05) encoding galactose/methyl galactoside ABC transporter ATP-binding protein and galactoside ABC transporter permease, respectively, were significantly downregulated suggesting that the transport of readily metabolizable sugars was reduced in NE-treated cultures (Table 4 and Table S3). The other downregulated (≥ 2.0-fold, p < 0.05) genes included livG and livM encoding a high-affinity branched-chain amino acid ABC transporter ATP-binding protein LivG and high-affinity branched-chain amino acid ABC transporter permease LivM, respectively; dppB, dppC, dppD and dppF encoding dipeptide ABC transport system; cstA encoding an inner membrane peptide transporter [63]; and genes pstA, pstB, pstC and pstS encoding uptake system for inorganic phosphate, which is required for phosphorylation of cellular proteins, lipids, and carbohydrates (Table S3). The other upregulated (≥ 2.0-fold, p < 0.05) genes were those that encoded the pyrimidine utilization pathway for assimilating pyrimidine as a sole nitrogen source [64]. Also significantly upregulated were genes encoding nitrate/nitrite transport and nitrite/nitrate reductases that oxidize nitrite and nitrate, respectively, as terminal electron acceptors during anaerobic metabolism [65] (Table 4; Table S2). With respect to iron transport, the expression of feoABC genes, which are involved in ferrous iron uptake system, was significantly upregulated, but the expression of genes for ferric iron uptake system (fepC and fepG) and genes (entA, entC, entE) for the biosynthesis of siderophore enterobactin were downregulated by ≥2.0-fold (Table 4 and Table S2 and Table S3). On the other hand, genes cirA and fiu encoding catecholate siderophore (enterobactin) receptor proteins were significantly downregulated. The expression of many genes representing pathways for transcription, translation, and energy production was also significantly downregulated in response to NE (Table 4 and Table S3). These downregulated genes included rpoA and rpoB, which encode DNA-directed RNA polymerase subunit α and β, respectively; 11 and 15 genes encoding 30S and 50S ribosomal proteins essential for protein synthesis; two genes (trmG and tgt) involved in tRNA modification; and genes fus, tsf, tuf encoding translational elongation factors G, Ts, and Tu. For the energy generating pathways, the genes encoding for cytochrome b, c, and o; electron transport complex subunit RsxE and RsxG; and subunits α, β, γ, δ and ε of F0F1 ATP synthase required for the synthesis of ATP [66] were also significantly downregulated (Table 4 and Table S3). In addition, expression of many genes encoding enzymes involved in carbohydrate, amino acid, and fatty acid metabolism was also significantly downregulated in NE-treated cultures (Table 4; Table S3). The representative genes of these pathways were glpT, which encodes a transporter for the uptake of glycerol-3-phosphate used as a substrate in glycolysis and phospholipid biosynthesis [67]; sucA encoding 2-oxoglutarate dehydrogenase E1 component for converting 2-oxoglutarate to succinyl-CoA and CO2 in the TCA cycle; sdhCDAB encoding a succinate dehydrogenase enzyme complex for synthesizing fumarate from succinate; genes encoding NADH-quinone oxidoreductase that serve as a main entry point for electron transfer to the electron transport chain to generate ATP [68]; purCDFLMNT catalyzing de novo purine biosynthesis; and fadB that encodes a multifunctional fatty acid oxidation complex subunit alpha for aerobic and anaerobic degradation of long-chain fatty acids. Also downregulated was the expression of genes tnaA (+ 2.87-fold) and tnaL (+ 6.99-fold) required for the hydrolysis of tryptophan to produce indole that plays an important role in the regulation of biofilm formation [69] (Table 4 and Table S3). Besides inducing differential expression of genes encoding various metabolic pathways, NE also impacted expression of genes involved in bacterial cell division. Some of these genes, such as cpoB, which coordinates cell wall production and the movement of the outer membrane during cell division [70], and mukB, which is essential for the correct partitioning of replicated chromosomes during cell division so that both daughter cells inherit a copy of the replicated chromosome [71], were significantly downregulated (≥ − 2.00-fold, p < 0.05) (Table 2 and Table S3). On the other hand, the expression of fic-1, whose function is not fully understood but impacts cell division by leading to cell filamentation [72], was significantly upregulated (+ 2.95-fold) in response to NE (Table 2 and Table S2). Strain NADC 6564, like other E. coli O157:H7 strains, harbors a cluster of genes (terABCWZ and tehB) encoding resistance to tellurium. The expression of tellurium resistance genes that might be involved in bacterial resistance to host cellular defenses [73] was also downregulated (≥ − 2.0-fold, p < 0.05) in response to NE (Table 4 and Table S3).
NE had no effect on growth rate but enhanced viability
Although the bacterial growth curves generated over a 24 h of growth in the presence or absence of NE were almost identical (Fig. S1A), viable counts recovery was about 2-fold higher in NE-treated cultures compared to the culture grown without NE (Fig. S1B).
Discussion
The RNA-Seq-based transcriptional profiling of E. coli O157:H7 strain NADC 6564 [43] grown in the presence of norepinephrine (NE) to an early stationary phase showed differential expression (DE) of large number of genes that are usually turned on in the stationary phase plus genes involved in bacterial virulence, stress response, and various metabolic pathways. The pattern of differential expression for many of these genes was highly similar to that reported for these genes by a microarray-based approach in E. coli O157:H7 (O157) strain EDL933 [34]. Despite differential expression of many related genes in response to NE between EDL933 and NADC 6564 strains, which are classified as lineage I strains [74], these two strains differ in having distinct regulatory systems controlling expression of virulence genes. The RNA-Seq-based approach identified many differentially expressed genes in strain NADC6564 that were not represented in a microarray-based transcriptional profile of strain EDL933, since that study only probed 610 genes for DE in response to NE [34]. For example, NE exposure not only enhanced the expression of genes encoding (gadABC) and controlling (gadE, gadX, and gadW) glutamate-dependent resistance pathway 2 (ARP2) [15, 19, 20] similar to that was observed for strain EDL933 [34], but also upregulated genes encoding the EvgS/EvgA signaling system implicated in the regulation of ARP2 in E. coli [22, 75]. We validated this finding for the first time by demonstrating that deleting evgS/evgA genes reduced resistance of strain NADC 6564 to highly acidic environment but the NE signaling was not dependent on EvgS/EvgA.
Similar to previous findings using EDL933 strain [34], the expression of several LEE genes was significantly increased in strain NADC 6564 in response to NE, but the expression of ler, which encodes Ler for activating LEE expression, was not significantly altered with or without exposure to NE. We have shown previously that the basal level of ler transcription is very high in strain NADC 6564 [76], and in the current study we found that the growth of this strain in the presence of NE had no significant effect on ler expression (− 1.40-fold, p = 0.311, Table S1). Both LEE- and non-LEE-encoded transcriptional factors, such as RpoS, QseA, PerC, Hha, IHF, SdiA, H-NS, Fis, GrlA, GrlR, GadE, GadX, RcsB, and Hfq control ler expression by their direct interactions with ler promoter region [10, 77]. For example, sRNA chaperone Hfq acts as a negative regulator of LEE in EDL933 but in strain NADC 6564 LEE expression is positively regulated by Hfq [78]. Similarly, RpoS, the stationary sigma factor, directly or in conjunction with other regulators, can have positive or negative regulatory effect on LEE expression [79, 80]. However, NE exposure had no significant effect on Hfq and RpoS expression in strain NADC 6564 suggesting that LEE activation in response to NE occurs via a different regulatory mechanism. The transcriptional factors that were upregulated in response to NE in strain NADC 6564 were GadE, GadX, and 2 of the 4 copies of perC, which encode transcriptional factor PerC. Despite the upregulation of gadE and gadX, which have been shown to repress LEE gene expression [10, 77], LEE expression was upregulated in response to NE. Thus, it is possible that upregulated PerC family of proteins, which have been shown to increase LEE expression by interacting with the ler promoter [81], could account for the upregulation of LEE gene expression by inhibiting GadE- and GadX-mediated repression of ler. Besides LEE-encoded virulence adherence proteins, E. coli O157:H7 strains can express different types of fimbriae in response to a variety of intestinal metabolites, such as ethanolamine, choline, and serine [82]. In the RNA-Seq data, we also observed significantly increased expression of fimbrial genes belonging to Lpf1 and Ybp fimbrial groups in response to NE. The Lpf1 fimbriae have been shown to promote bacterial adherence to epithelial cells and mutants lacking genes encoding Lpf1 fimbriae show poor colonization in animal models [83, 84]. Since expression of these fimbriae is modulated in vitro both by environmental factors and bacterial- and host-produced metabolites [82, 84], presence of NE in mammalian intestine at concentrations sufficient to induce differential gene expression could promote expression of the above listed fimbriae leading to increased adherence of E. coli O157:H7 to host tissues.
Transcriptional profiling in response to NE revealed differential expression of many stress response genes that are usually turned on as bacterial cells enter a stationary growth phase or encounter conditions less optimal for growth. Some of these genes, which were also differentially expressed in EDL933 using the microarray approach [34], were represented by usp genes that encode Usp superfamily of proteins performing a diverse array of functions related to oxidative stress, iron homeostasis, motility/adhesion, which could impact pathogenesis of O157 strains [50, 85, 86]. Similarly, the significant induction of heat and cold shock response genes in NE-treated cultures would be important in maintaining protein homeostasis by assisting in the folding of newly synthesized proteins, preventing protein aggregation, rescuing partially or completely un-folded proteins formed under stress, and preventing the formation of secondary structures in mRNA at low temperatures to allow the initiation of translation [51, 52]. In addition, we also observed upregulation of genes dps and clpB that are important in preventing oxidative damage to DNA from hydrogen peroxide produced during specific metabolic activities and removal of damaged polypeptides from stressed bacterial cells, respectively [49, 52].
Since biofilm formation is induced under conditions unfavorable for growth, such as when bacterial cells enter the stationary phase, experience nutritional stress, or low temperatures [59], several genes linked directly or indirectly to biofilm formation were differentially expressed by NE. Among the genes that are directly linked to biofilm formation, csgB was highly upregulated in response to NE. The CsgB protein facilitates assembly of CsgA, a major curli subunit into mature curli fimbriae, which are essential for the initial bacterial adherence to abiotic/biotic surfaces during biofilm formation [57, 59, 87, 88]. The genes csgB and csgA constitute, along with csgC, the csgBAC operon transcribed divergently from the csgDEFG operon [44]. The expression of genes in csgBAC and csgDEFG opeons is positively regulated by the global transcriptional factor CsgD encoded by the csgD gene of csgDEFG operon [89, 90]. In addition, the csgEFG gene products are essential in the secretion and assembly of CsgA in to curli fimbriae [59, 91] and CsgD regulates expression of other genes, such as those encoding bacterial cellulose that are essential for biofilm formation [92]. Despite the upregulation of csgB and presumably other genes in this operon, we did not detect any change in the expression of csgD or the csgEFG genes when cultured with or without NE. These results were corroborated by the inability of strain NADC 6564 to produce biofilms when grown with or without NE for 72 h (data not shown) according to a previously described biofilm detection procedure [93]. The apparent lack of any increase in csgD expression could be attributed to the inability of NE to cause any changes in the differential expression of rpoS, rcsB, fis and hha genes, which have been shown to play important role in biofilm formation by O157 by affecting csgD expression [93,94,95]. Since increased expression of perC homologs has been shown to repress csgD expression and biofilm formation [96], it is also possible that increased expression of two of the four copies of perC homologs in response to NE could have resulted in csgD repression leading to no increases in biofilm formation by strain NADC 6564.
Besides upregulation of pathways impacting bacterial virulence and response to various stressors, large number of genes encoding a variety of metabolic pathways were differentially impacted in their expression by NE. The majority of upregulated genes were those that enabled E. coli O157:H7 to utilize alternative sources of carbon and nitrogen, such as amino acids and pyrimidines, rather than the readily utilizable sugars that probably are scarce in the stationary phase-like growth conditions and in the host intestinal environment. There was also significant upregulation of nitrate/nitrite transport and nitrite/nitrate reductases that oxidize nitrite and nitrate as terminal electron acceptors in anaerobic metabolism [65]. Additional support that the metabolism of NE-treated cultures become less aerobic is garnered by the increased expression of fumarate reductase, which is a terminal electron receptor in fermentative metabolism of carbon substrates [97]. Thus, this shift to less aerobic metabolic activity in response to NE may account for the downregulation of other prominent metabolic pathways such as glycolysis, TCA cycle, electron transport system for producing ATP, fatty acid oxidation, gene transcription, and protein synthesis. This altered metabolic physiology and differential upregulation of genes promoting adherence to tissues and resistance to various stressors might also be correlated to differential expression of genes that inhibit cell division (cpoB, mukB, and fic-1) and alter cellular morphology (bolA). It has been suggested that altered cellular morphology during stationary phase might be a strategy to tolerate variety of stresses and nutritional starvation [98]. Although, some studies have reported that the exposure of E. coli O157:H7 strains to NE for 4 to 6 h can increase growth rate by a 1/100 of an A600 [34], we didn’t detect such a small change in growth of strain NADC 6564 grown in minimal medium containing NE relative to that grown without NE. However, a higher number of viable cells were recovered from cultures grown in the presence of NE, suggesting that altered metabolic profile, and differential expression of many stress-related, and stationary phase-dependent pathways might enhance survival and host colonization potential of O157 strains when exposed to NE during the stationary phase-like growth conditions [27, 98,99,100].
Conclusions
Based on the whole genome transcriptional profiling of E. coli O157:H7 strain NADC 6564 grown in the presence of NE to an early stationary growth phase, we observed that NE exposure had a major impact on the expression of genes attributable to bacterial survival under suboptimal growth conditions, such as those encountered during stationary phase of growth, during colonization of the host intestinal mucosa, and during bacterial persistence in the environment outside of the host animal. Thus, the availability of NE and other host-produced metabolites could serve as signals and/or nutrients to not only alter the global gene expression profile but also skew the gene expression profile to the benefit of E. coli O157:H7 by enhancing its ability to colonize the carrier host animal, produce disease in the susceptible human host, and survival outside the host animal.
Materials and methods
Bacterial strains and growth conditions
Bacterial strains used in this study are listed in Table 5. Escherichia coli O157:H7 strain NADC 6564 served as the parental strain and all other strains were derivatives of this strain, either described previously or in the current study. E. coli TOP10 was used as a host for the propagation of recombinant plasmids. Bacterial strains were propagated in Luria-Bertani broth (LB) or LB containing 1.5% agar (LB-agar). Antibiotics were added to liquid or solid media as needed (streptomycin 100 mg per liter; carbenicillin 100 mg per liter; kanamycin 50 mg per liter).
Transcriptional profiling
For RNA isolation, an overnight bacterial culture grown at 37 °C in LB broth was diluted 1:100 (A600 = 0.10) into a low-glucose Dulbecco’s Minimal Eagles Medium (DMEM) lacking or containing 50 μM norepinephrine, the amount considered to be reached locally in various areas of GIT [101]. After about 5.5 h of incubation at 37 °C with shaking (250 rpm) to allow cultures to attain A600 ≈ 1.2, (the incubation period which we and others have shown in a previous study to be long enough to allow bacterial cultures to reach the early stationary phase of growth [34, 76]), total RNA was isolated using RNeasy isolation kit according to the manufacturer’s instructions (Qiagen, Valencia, CA). RNA was treated with DNase (TURBO DNA-free kit; ThermoFisher Scientific, Grand Island, NY). The DNase-treated RNA was used for RT-qPCR or treated with Ribo-Zero rRNA kit reagents according to the manufacturer’s instructions (Gram-negative bacteria; Illumina, Inc., San Diego, CA) to remove rRNA. The strand-specific RNA-Seq libraries were prepared from the rRNA-free RNA and sequenced with Illumina HiSeq (Iowa State University, Ames, Iowa). The trimmed, single-end reads were mapped to the reference genome (strain NADC 6564) using HiSat2 v2.05 to generate SAM files that were fed into htseq-count v0.11.2 along with the reference genome file for unnormalized read quantification. DESeq2 was used to determine differential gene expression by analyzing quantified htseq-counts. The set of differentially expressed genes for each comparison were sorted by the adjusted p-value of less than 0.05. A total of three biological replicates of bacterial cultures grown independently were used for RNA-Seq analysis.
Read QC and mapping
The trimmed reads acquired from sequencing were first run through FastQC v0.11.5 to check for any glaring issues in the quality of reads. After it had been determined that there weren’t any major discrepancies associated with the reads, the single-end reads were mapped to the reference genome using HiSat2 v2.05. HiSat2 utilizes a novel indexing scheme termed Hierarchical Graph FM index which improves the efficiency of pattern recognition [102]. First, HiSat2 was provided with the reference FASTA file for E. coli O157:H7 strain NADC 6564 acquired from the NCBI database [43, 103]. The reference FASTA file was indexed by HiSat2 to make mapping possible using HiSat2 algorithms. Once the HiSat2 index for NADC 6564 had been built, the trimmed FASTQ files for all NADC 6564 control and norepinephrine-treated replicates were passed as input to HiSat2. Other than multiple threads being used to speed up the processing time, default HiSat2 parameters were used to conduct the mapping. The output SAM file for each replicate was used as an input for read quantification.
Read quantification
The mapped reads were fed into htseq-count v0.11.2 along with the reference GFF file for unnormalized read quantification [103]. The GFF annotation file was acquired from the same NCBI accession for E. coli O157:H7 strain NADC 6564 as mentioned previously. The stranded option was specified as “no” because a strand-specific sequencing protocol was not used. The default “union” overlap resolution mode was used in order to avoid discriminating valid reads [103]. The “CDS” tag was used as the feature type to quantify. The output text file was formatted for the next step by removing the quantification statistics at the end and adding column names.
Differential gene expression analysis
DESeq2 was used to identify differentially expressed genes in the data set [104]. To do so, DESeq2 was launched in an R environment and the quantified reads were imported as a tab-delimited table. An experimental design table specifying control and experimental groups was created according to the format specified by the DESeq2 vignette. Once the quantified read data and experimental design was provided, DESeq2 was run with the default false detection rate set to 0.1. The genes were tested using the default null hypothesis of not being different. Gene expression was compared between the control and treated strain. The set of differentially expressed genes were sorted by the adjusted p-value of less than 0.05.
Pathway analysis
The resultant set of differentially expressed genes were mapped to genes in E. coli O157:H7 str. EDL933. The corresponding differentially expressed genes in EDL933 were used as input to STRING-DB. STRING-DB is a database of known and predicted protein-protein interactions that was used to search for correlations between the gene products that were found to be differentially expressed [105]. Images characterizing the network of protein-protein interactions between the differentially expressed products were created by STRING-DB. In addition, KEGG annotation for the differentially expressed genes are also provided by STRING-DB.
Recombinant DNA procedures
The evgS/evgA deletion mutant of E. coli O157:H7 strain NADC 6564 was constructed by using a phage lambda-derived Red recombination system [106]. Briefly, a 1.5 kb fragment containing the gene encoding kanamycin (kan) resistance, which is flanked at its 5′ and 3′ ends by a FRT sequence for enabling a FLP catalyzed deletion of the kan resistance gene, was isolated from the pKD4 plasmid (Table 5) [106] by PCR using a primer pair evgSAF (forward deletion primer) evgSAR (reverse deletion primer) with their nucleotide sequences listed in Table 6. The underlined nucleotides in these primers are complementary to nucleotides at 5′ and 3′ ends, respectively, of evgS and evgA and nucleotides not underlined are complementary to 5′ and 3′ ends, respectively, of the kan FRT fragment (Table 6). The procedures for PCR amplification, purification of the amplified DNA fragments, electroporation of the purified DNA fragments in to arabinose-induced competent bacterial cells (strain NADC 6564 in the current study) containing the pKD46 plasmid, selection of kan-resistant isolates, removal of the kan gene, and confirmation of evgS/evgA gene deletion have been described previously [93]. The deletion of evgS/evgA genes was confirmed by PCR amplification of genomic DNA of kanamycin-sensitive isolates using an evgSAF (evgSA operon isolation forward primer) and evgSAR (evgSA operon isolation reverse primer) primer pair as listed in Table 6. These primers were complementary to a short nucleotide sequence located upstream of evgS and a short nucleotide sequence located downstream of evgA, respectively. The PCR amplified DNA was analyzed by a standard agarose gel electrophoresis to determine the size of the amplified fragments [93].
The plasmid for complementing an evgS/evgA deletion mutation (as constructed above) in strain NADC 6564 was generated by cloning a 4.17 kb DNA fragment containing the evgS/evgA operon at the SmaI site located in the kanamycin gene of a low copy vector pACYC177 (Table 5; New England Biolabs Inc., Ipswich, MA). The 4.17 kb DNA fragment was isolated by PCR amplification of DNA purified from strain NADC 6564 using primers evgSAF (evgSA operon isolation forward primer) and evgSAR (evgSA operon isolation reverse primer) as listed in Table 6. Procedures for PCR DNA amplification, purification of amplified DNA fragments, ligating the SmaI-linearized 4.17 kb fragment in SmaI-linearized pACYC177, transformation of ligated DNA fragments into E. coli TOP 10 electrocompetent cells, and confirming the presence of a cloned 4.17 kb fragment in pACYC177 have been described previously [93].
Bacterial growth curves
The overnight bacterial cultures grown in LB-broth at 37 °C with shaking (200 rpm) were diluted 1:100 in DMEM containing 100 μg per ml of streptomycin. Aliquots (300 μl) of diluted cultures were added to wells of a 100-well Honeycomb 2 plate (Growth Curves USA, Piscataway, NJ). The plate was incubated at 37 °C in an automated growth curve reader for recording optical density at 600 nm (Growth Curves USA, Piscataway, NJ). The growth curve data was collected by analyzing three independently grown bacterial cultures and each culture being assayed in triplicate wells.
Acid resistance assays and detection of biofilm formation
For determining relative survival of bacterial strains at pH 2.5, three independently grown overnight cultures of each bacterial strain were diluted at 1:1000 in a phosphate-citrate minimal medium (pH 2.5) containing 0.4% glucose and 1.5% sodium glutamate [107, 108]. After 3 h of incubation at 37 °C, the viable bacterial cell counts were determined by plating 10-fold serial dilutions on LB agar medium containing carbenicillin (100 μg per ml). Bacterial survival was calculated by dividing the viable counts at 3 h with the viable counts of the same strain at 0 min. Bacterial survival was plotted as a percent survival.
Quantitative RT-qPCR
Total DNA-free RNA was prepared from three biological replicates of control and NE-treated bacterial strain NADC 6564 as described above in the section ‘Transcriptional Profiling’. The expression of acid resistance pathway 2 (ARP2) encoding genes was determined by transcribing DNA-free RNA into cDNA and amplifying the cDNA using the iTaq Universal One-Step RT-qPCR Kit in CFX96 PCR system according to the manufacturer’s instructions (Bio-Rad, Hercules, CA). The fold change in gene expression was determined using the software and according to the instructions of the manufacturer (Bio-Rad, Hercules, CA). The expression data were normalized to endogenous levels of rpoA in order to account for any minor variations in the amounts of RNA across samples [93]. Biofilm formation was detected by crystal violet staining [93, 94] of any potential biofilm produced by strain NADC 6564 grown for 72 h in a biofilm formation-supporting medium containing or lacking NE [93, 94].
Statistical analyses
Student’s t-test was used to determine the significance of differences in the acid resistance of evgS/evgA mutant or evgS/evgA mutant complemented with the evgS/evgA recombinant plasmid to the parental strain 6564. The difference in growth rate of strain NADC 6564 in the presence or absence of norepinephrine was evaluated by the t-test as described above. Data were analyzed with GraphPad Prism8 (GraphPad Software, La Jolla, CA). The difference was considered significant at p < 0.05.
Availability of data and materials
The complete chromosomal sequence of NADC 6564 is available at the GenBank under the assigned accession number CP017251. E. coli O157:H7 strain NADC 6564 will be provided pending that the requestor would fulfill requirements for shipment of RG2 bacterial agents.
The RNA-Seq raw data is available in the NCBI SRA database under Study SRP091887 comprising accessions SRR16601911 - SRR16601916, which is linked to BioProject PRJNA341860 and BioSamples SAMN22608725 and SAMN22608726.
Abbreviations
- NE:
-
Norepinephrine
- RNA:
-
Ribonucleic acid
- rRNA:
-
Ribosomal ribonucleic acid
- DE:
-
Differentially expressed genes
- Fe3+ :
-
Ferric ion
- STEC:
-
Shiga toxigenic Escherichia coli
- HUS:
-
Hemolytic uremic syndrome
- T3SS:
-
type III secretion system
- Ler:
-
Locus of enterocyte effacement regulator
- GIT:
-
Gastrointestinal tract
- QS:
-
Quorum sensing
- AHL:
-
acyl-homoserine lactones
- ARP2:
-
Acid resistance pathway 2
- AI-3:
-
Autoinducer-3
- ENS:
-
Enteric nervous system
- E:
-
Epinephrine
- DHMA:
-
dihydroxymandelic acid
- stx :
-
Shiga toxin encoding genes
- NADC:
-
National Animal Disease Center
- DMEM:
-
Dulbecco’s minimal Eagles medium
- TCSS:
-
Two component signal transduction system
- cdGMP:
-
cyclic diguanylate monophosphate
- Usp:
-
Universal stress proteins
- PTS:
-
phosphotransferase system
- ATP:
-
Adenosine triphosphate
- LB:
-
Luria Bertani broth
- mg:
-
milligram
- A600 :
-
Absorbance at 600 nm
- nm:
-
nanometers
- kan :
-
Kanamycin
- μg:
-
microgram
- μl:
-
microliter
- cDNA:
-
complementary deoxyribonucleic acid
- RT-qPCR:
-
Reverse transcriptase-quantitative polymerase chain reaction
- Cfu:
-
Colony forming units
References
Nataro JP, Kaper JB. Diarrheagenic Escherichia coli. Clin Microbiol Rev. 1998;11(1):142–201.
Griffin PM, Tauxe RV. The epidemiology of infections caused by Escherichia coli O157:H7, other enterohemorrhagic E. coli, and the associated hemolytic uremic syndrome. Epidemiol Rev. 1991;13:60–98.
Wachsmuth IK, Griffin PM, Wells JG. Escherichia coli O157:H7, a cause of hemorrhagic colitis and hemolytic uremic syndrome. Acta Paediatr Jpn. 1991;33(5):603–12.
Obrig TG, Karpman D. Shiga toxin pathogenesis: kidney complications and renal failure. Curr Top Microbiol Immunol. 2012;357:105–36.
Strockbine NA, Marques LR, Newland JW, Smith HW, Holmes RK, O'Brien AD. Two toxin-converting phages from Escherichia coli O157:H7 strain 933 encode antigenically distinct toxins with similar biologic activities. Infect Immun. 1986;53(1):135–40.
Jarvis KG, Kaper JB. Secretion of extracellular proteins by enterohemorrhagic Escherichia coli via a putative type III secretion system. Infect Immun. 1996;64(11):4826–9.
Naylor SW, Low JC, Besser TE, Mahajan A, Gunn GJ, Pearce MC, et al. Lymphoid follicle-dense mucosa at the terminal rectum is the principal site of colonization of enterohemorrhagic Escherichia coli O157:H7 in the bovine host. Infect Immun. 2003;71(3):1505–12.
Naylor SW, Roe AJ, Nart P, Spears K, Smith DGE, Low JC, Gally DL: Escherichia coli O157 : H7 forms attaching and effacing lesions at the terminal rectum of cattle and colonization requires the LEE4 operon. Microbiology (Reading) 2005, 151(Pt 8):2773–2781.
McDaniel TK, Jarvis KG, Donnenberg MS, Kaper JB. A genetic locus of enterocyte effacement conserved among diverse enterobacterial pathogens. Proc Natl Acad Sci U S A. 1995;92(5):1664–8.
Franzin FM, Sircili MP. Locus of enterocyte effacement: a pathogenicity island involved in the virulence of enteropathogenic and enterohemorragic Escherichia coli subjected to a complex network of gene regulation. Biomed Res Int. 2015;2015:534738.
Elliott SJ, Sperandio V, Giron JA, Shin S, Mellies JL, Wainwright L, et al. The locus of enterocyte effacement (LEE)-encoded regulator controls expression of both LEE- and non-LEE-encoded virulence factors in enteropathogenic and enterohemorrhagic Escherichia coli. Infect Immun. 2000;68(11):6115–26.
Furniss RCD, Clements A. Regulation of the locus of enterocyte effacement in attaching and effacing pathogens. J Bacteriol. 2018;200(2).
Walters M, Sperandio V. Autoinducer 3 and epinephrine signaling in the kinetics of locus of enterocyte effacement gene expression in enterohemorrhagic Escherichia coli. Infect Immun. 2006;74(10):5445–55.
Hughes DT, Terekhova DA, Liou L, Hovde CJ, Sahl JW, Patankar AV, et al. Chemical sensing in mammalian host-bacterial commensal associations. Proc Natl Acad Sci U S A. 2010;107(21):9831–6.
Lin J, Smith MP, Chapin KC, Baik HS, Bennett GN, Foster JW. Mechanisms of acid resistance in enterohemorrhagic Escherichia coli. Appl Environ Microbiol. 1996;62(9):3094–100.
Bergholz TM, Whittam TS. Variation in acid resistance among enterohaemorrhagic Escherichia coli in a simulated gastric environment. J Appl Microbiol. 2007;102(2):352–62.
Price SB, Wright JC, DeGraves FJ, Castanie-Cornet MP, Foster JW. Acid resistance systems required for survival of Escherichia coli O157:H7 in the bovine gastrointestinal tract and in apple cider are different. Appl Environ Microbiol. 2004;70(8):4792–9.
Tuttle J, Gomez T, Doyle MP, Wells JG, Zhao T, Tauxe RV, et al. Lessons from a large outbreak of Escherichia coli O157:H7 infections: insights into the infectious dose and method of widespread contamination of hamburger patties. Epidemiol Infect. 1999;122(2):185–92.
Foster JW. Escherichia coli acid resistance: tales of an amateur acidophile. Nat Rev Microbiol. 2004;2(11):898–907.
Sayed AK, Odom C, Foster JW. The Escherichia coli AraC-family regulators GadX and GadW activate gadE, the central activator of glutamate-dependent acid resistance. Microbiology (Reading). 2007;153(Pt 8):2584–92.
Tucker DL, Tucker N, Ma Z, Foster JW, Miranda RL, Cohen PS, et al. Genes of the GadX-GadW regulon in Escherichia coli. J Bacteriol. 2003;185(10):3190–201.
Itou J, Eguchi Y, Utsumi R. Molecular mechanism of transcriptional cascade initiated by the EvgS/EvgA system in Escherichia coli K-12. Biosci Biotechnol Biochem. 2009;73(4):870–8.
Eguchi Y, Utsumi R. Alkali metals in addition to acidic pH activate the EvgS histidine kinase sensor in Escherichia coli. J Bacteriol. 2014;196(17):3140–9.
Ma Z, Masuda N, Foster JW. Characterization of EvgAS-YdeO-GadE branched regulatory circuit governing glutamate-dependent acid resistance in Escherichia coli. J Bacteriol. 2004;186(21):7378–89.
Sperandio V, Torres AG, Jarvis B, Nataro JP, Kaper JB. Bacteria-host communication: the language of hormones. Proc Natl Acad Sci U S A. 2003;100(15):8951–6.
Walters M, Sircili MP, Sperandio V. AI-3 synthesis is not dependent on luxS in Escherichia coli. J Bacteriol. 2006;188(16):5668–81.
Sule N, Pasupuleti S, Kohli N, Menon R, Dangott LJ, Manson MD, et al. The norepinephrine metabolite 3,4-Dihydroxymandelic acid is produced by the commensal microbiota and promotes Chemotaxis and virulence gene expression in Enterohemorrhagic Escherichia coli. Infect Immun. 2017;85(10).
Kim CS, Gatsios A, Cuesta S, Lam YC, Wei Z, Chen H, Russell RM, Shine EE, Wang R, Wyche TP et al: Characterization of Autoinducer-3 Structure and Biosynthesis in E. coli. ACS Cent Sci 2020, 6(2):197–206.
Chen Y, Arsenault R, Napper S, Griebel P. Models and methods to investigate acute stress responses in cattle. Animals (Basel). 2015;5(4):1268–95.
Moreira CG, Russell R, Mishra AA, Narayanan S, Ritchie JM, Waldor MK, et al. Bacterial adrenergic sensors regulate virulence of enteric pathogens in the gut. MBio. 2016;7(3).
Stein RA, Katz DE. Escherichia coli, cattle and the propagation of disease. FEMS Microbiol Lett. 2017;364(6).
Costa M, Brookes SJ, Hennig GW: Anatomy and physiology of the enteric nervous system. Gut 2000, 47 Suppl 4:iv15-19; discussion iv26.
Lyte M. Microbial endocrinology and infectious disease in the 21st century. Trends Microbiol. 2004;12(1):14–20.
Dowd SE. Escherichia coli O157:H7 gene expression in the presence of catecholamine norepinephrine. FEMS Microbiol Lett. 2007;273(2):214–23.
Freestone PP, Haigh RD, Williams PH, Lyte M. Involvement of enterobactin in norepinephrine-mediated iron supply from transferrin to enterohaemorrhagic Escherichia coli. FEMS Microbiol Lett. 2003;222(1):39–43.
Lyte M, Arulanandam B, Nguyen K, Frank C, Erickson A, Francis D. Norepinephrine induced growth and expression of virulence associated factors in enterotoxigenic and enterohemorrhagic strains of Escherichia coli. Adv Exp Med Biol. 1997;412:331–9.
Lyte M, Arulanandam BP, Frank CD. Production of Shiga-like toxins by Escherichia coli O157:H7 can be influenced by the neuroendocrine hormone norepinephrine. J Lab Clin Med. 1996;128(4):392–8.
Lyte M, Ernst S. Catecholamine induced growth of gram negative bacteria. Life Sci. 1992;50(3):203–12.
Lyte M, Nguyen KT. Alteration of Escherichia coli O157:H7 growth and molecular fingerprint by the neuroendocrine hormone noradrenaline. Microbios. 1997;89(360–361):197–213.
Vlisidou I, Lyte M, van Diemen PM, Hawes P, Monaghan P, Wallis TS, et al. The neuroendocrine stress hormone norepinephrine augments Escherichia coli O157:H7-induced enteritis and adherence in a bovine ligated ileal loop model of infection. Infect Immun. 2004;72(9):5446–51.
Freestone PP, Lyte M, Neal CP, Maggs AF, Haigh RD, Williams PH. The mammalian neuroendocrine hormone norepinephrine supplies iron for bacterial growth in the presence of transferrin or lactoferrin. J Bacteriol. 2000;182(21):6091–8.
Sandrini SM, Shergill R, Woodward J, Muralikuttan R, Haigh RD, Lyte M, et al. Elucidation of the mechanism by which catecholamine stress hormones liberate iron from the innate immune defense proteins transferrin and lactoferrin. J Bacteriol. 2010;192(2):587–94.
Sharma VK, Bayles DO, Alt DP, Looft T. Complete genome sequences of Curli-negative and Curli-positive isolates of foodborne Escherichia coli O157:H7 strain 86-24. Genome Announc. 2016;4(6).
Hammar M, Arnqvist A, Bian Z, Olsen A, Normark S. Expression of two csg operons is required for production of fibronectin- and Congo red-binding curli polymers in Escherichia coli K-12. Mol Microbiol. 1995;18(4):661–70.
Tuckerman JR, Gonzalez G, Sousa EH, Wan X, Saito JA, Alam M, et al. An oxygen-sensing diguanylate cyclase and phosphodiesterase couple for c-di-GMP control. Biochemistry. 2009;48(41):9764–74.
Benjamin MM, Datta AR. Acid tolerance of enterohemorrhagic Escherichia coli. Appl Environ Microbiol. 1995;61(4):1669–72.
Mates AK, Sayed AK, Foster JW. Products of the Escherichia coli acid fitness island attenuate metabolite stress at extremely low pH and mediate a cell density-dependent acid resistance. J Bacteriol. 2007;189(7):2759–68.
Ma Z, Gong S, Richard H, Tucker DL, Conway T, Foster JW. GadE (YhiE) activates glutamate decarboxylase-dependent acid resistance in Escherichia coli K-12. Mol Microbiol. 2003;49(5):1309–20.
Almiron M, Link AJ, Furlong D, Kolter R. A novel DNA-binding protein with regulatory and protective roles in starved Escherichia coli. Genes Dev. 1992;6(12B):2646–54.
Kvint K, Nachin L, Diez A, Nystrom T. The bacterial universal stress protein: function and regulation. Curr Opin Microbiol. 2003;6(2):140–5.
Roncarati D, Scarlato V. Regulation of heat-shock genes in bacteria: from signal sensing to gene expression output. FEMS Microbiol Rev. 2017;41(4):549–74.
Keto-Timonen R, Hietala N, Palonen E, Hakakorpi A, Lindstrom M, Korkeala H. Cold shock proteins: a Minireview with special emphasis on Csp-family of Enteropathogenic Yersinia. Front Microbiol. 2016;7:1151.
Gort AS, Ferber DM, Imlay JA. The regulation and role of the periplasmic copper, zinc superoxide dismutase of Escherichia coli. Mol Microbiol. 1999;32(1):179–91.
Kim YH, Lee Y, Kim S, Yeom J, Yeom S, Seok Kim B, et al. The role of periplasmic antioxidant enzymes (superoxide dismutase and thiol peroxidase) of the Shiga toxin-producing Escherichia coli O157:H7 in the formation of biofilms. Proteomics. 2006;6(23):6181–93.
Scotti R, Nicolini L, Gabbianelli R. sodC genes expression in Escherichia coli O157:H7 strains. Ann Ist Super Sanita. 2016;52(2):309–12.
Scotti R, Nicolini L, Stringaro A, Gabbianelli R. A study on prophagic and chromosomal sodC genes involvement in Escherichia coli O157:H7 biofilm formation and biofilm resistance to H2O2. Ann Ist Super Sanita. 2015;51(1):62–6.
Beloin C, Roux A, Ghigo JM. Escherichia coli biofilms. Curr Top Microbiol Immunol. 2008;322:249–89.
Ren D, Bedzyk LA, Thomas SM, Ye RW, Wood TK. Gene expression in Escherichia coli biofilms. Appl Microbiol Biotechnol. 2004;64(4):515–24.
Barnhart MM, Chapman MR. Curli biogenesis and function. Annu Rev Microbiol. 2006;60:131–47.
Dressaire C, Moreira RN, Barahona S, Alves de Matos AP, Arraiano CM. BolA is a transcriptional switch that turns off motility and turns on biofilm development. mBio. 2015;6(1):e02352–14.
Vieira HL, Freire P, Arraiano CM. Effect of Escherichia coli morphogene bolA on biofilms. Appl Environ Microbiol. 2004;70(9):5682–4.
Domka J, Lee J, Wood TK. YliH (BssR) and YceP (BssS) regulate Escherichia coli K-12 biofilm formation by influencing cell signaling. Appl Environ Microbiol. 2006;72(4):2449–59.
Schultz JE, Matin A. Molecular and functional characterization of a carbon starvation gene of Escherichia coli. J Mol Biol. 1991;218(1):129–40.
Kim KS, Pelton JG, Inwood WB, Andersen U, Kustu S, Wemmer DE. The rut pathway for pyrimidine degradation: novel chemistry and toxicity problems. J Bacteriol. 2010;192(16):4089–102.
Cole J. Nitrate reduction to ammonia by enteric bacteria: redundancy, or a strategy for survival during oxygen starvation? FEMS Microbiol Lett. 1996;136(1):1–11.
Shah NB, Duncan TM. Aerobic growth of Escherichia coli is reduced, and ATP synthesis is selectively inhibited when five C-terminal residues are deleted from the subunit of ATP synthase. J Biol Chem. 2015;290(34):21032–41.
Lemieux MJ, Huang Y, Wang DN. Glycerol-3-phosphate transporter of Escherichia coli: structure, function and regulation. Res Microbiol. 2004;155(8):623–9.
Friedrich T, Dekovic DK, Burschel S. Assembly of the Escherichia coli NADH:ubiquinone oxidoreductase (respiratory complex I). Biochim Biophys Acta. 2016;1857(3):214–23.
Bansal T, Englert D, Lee J, Hegde M, Wood TK, Jayaraman A. Differential effects of epinephrine, norepinephrine, and indole on Escherichia coli O157:H7 chemotaxis, colonization, and gene expression. Infect Immun. 2007;75(9):4597–607.
Gray AN, Egan AJ, Van't Veer IL, Verheul J, Colavin A, Koumoutsi A, et al. Coordination of peptidoglycan synthesis and outer membrane constriction during Escherichia coli cell division. Elife. 2015;4.
Niki H, Imamura R, Kitaoka M, Yamanaka K, Ogura T, Hiraga S. E.coli MukB protein involved in chromosome partition forms a homodimer with a rod-and-hinge structure having DNA binding and ATP/GTP binding activities. EMBO J. 1992;11(13):5101–9.
Kawamukai M, Matsuda H, Fujii W, Nishida T, Izumoto Y, Himeno M, et al. Cloning of the fic-1 gene involved in cell filamentation induced by cyclic AMP and construction of a delta fic Escherichia coli strain. J Bacteriol. 1988;170(9):3864–9.
Taylor DE. Bacterial tellurite resistance. Trends Microbiol. 1999;7(3):111–5.
Sharma VK, Akavaram S, Schaut RG, Bayles DO: Comparative genomics reveals structural and functional features specific to the genome of a foodborne Escherichia coli O157:H7. BMC Genomics 2019, 20(1):doi:https://doi.org/10.1186/s12864-019-5568-6.
Kato A, Ohnishi H, Yamamoto K, Furuta E, Tanabe H, Utsumi R. Transcription of emrKY is regulated by the EvgA-EvgS two-component system in Escherichia coli K-12. Biosci Biotechnol Biochem. 2000;64(6):1203–9.
Sharma VK, Zuerner RL. Role of hha and ler in transcriptional regulation of the esp operon of enterohemorrhagic Escherichia coli O157:H7. J Bacteriol. 2004;186(21):7290–301.
Branchu P, Matrat S, Vareille M, Garrivier A, Durand A, Crepin S, et al. NsrR, GadE, and GadX interplay in repressing expression of the Escherichia coli O157:H7 LEE pathogenicity island in response to nitric oxide. PLoS Pathog. 2014;10(1):e1003874.
Kendall MM, Gruber CC, Rasko DA, Hughes DT, Sperandio V. Hfq virulence regulation in enterohemorrhagic Escherichia coli O157:H7 strain 86-24. J Bacteriol. 2011;193(24):6843–51.
Dong T, Schellhorn HE. Global effect of RpoS on gene expression in pathogenic Escherichia coli O157:H7 strain EDL933. BMC Genomics. 2009;10:349. https://doi.org/10.1186/1471-2164-10-349.
Laaberki MH, Janabi N, Oswald E, Repoila F. Concert of regulators to switch on LEE expression in enterohemorrhagic Escherichia coli O157:H7: interplay between Ler, GrlA, HNS and RpoS. Int J Med Microbiol. 2006;296(4–5):197–210. https://doi.org/10.1016/j.ijmm.2006.02.017.
Porter ME, Mitchell P, Roe AJ, Free A, Smith DG, Gally DL. Direct and indirect transcriptional activation of virulence genes by an AraC-like protein, PerA from enteropathogenic Escherichia coli. Mol Microbiol. 2004;54(4):1117–33.
Gonyar LA, Kendall MM. Ethanolamine and choline promote expression of putative and characterized fimbriae in enterohemorrhagic Escherichia coli O157:H7. Infect Immun. 2014;82(1):193–201.
Jordan DM, Cornick N, Torres AG, Dean-Nystrom EA, Kaper JB, Moon HW. Long polar fimbriae contribute to colonization by Escherichia coli O157:H7 in vivo. Infect Immun. 2004;72(10):6168–71.
Torres AG, Milflores-Flores L, Garcia-Gallegos JG, Patel SD, Best A, La Ragione RM, et al. Environmental regulation and colonization attributes of the long polar fimbriae (LPF) of Escherichia coli O157:H7. Int J Med Microbiol. 2007;297(3):177–85.
de Souza CS, Torres AG, Caravelli A, Silva A, Polatto JM, Piazza RM. Characterization of the universal stress protein F from atypical enteropathogenic Escherichia coli and its prevalence in Enterobacteriaceae. Protein Sci. 2016;25(12):2142–51.
Nachin L, Nannmark U, Nystrom T. Differential roles of the universal stress proteins of Escherichia coli in oxidative stress resistance, adhesion, and motility. J Bacteriol. 2005;187(18):6265–72.
Da Re S, Ghigo JM. A CsgD-independent pathway for cellulose production and biofilm formation in Escherichia coli. J Bacteriol. 2006;188(8):3073–87.
Wang Z, Wang J, Ren G, Li Y, Wang X. Influence of Core oligosaccharide of lipopolysaccharide to outer membrane behavior of Escherichia coli. Mar Drugs. 2015;13(6):3325–39.
Gualdi L, Tagliabue L, Landini P. Biofilm formation-gene expression relay system in Escherichia coli: modulation of sigmaS-dependent gene expression by the CsgD regulatory protein via sigmaS protein stabilization. J Bacteriol. 2007;189(22):8034–43.
Ogasawara H, Yamamoto K, Ishihama A. Role of the biofilm master regulator CsgD in cross-regulation between biofilm formation and flagellar synthesis. J Bacteriol. 2011;193(10):2587–97.
Dueholm MS, Albertsen M, Otzen D, Nielsen PH. Curli functional amyloid systems are phylogenetically widespread and display large diversity in operon and protein structure. PLoS One. 2012;7(12):e51274.
Brombacher E, Baratto A, Dorel C, Landini P. Gene expression regulation by the Curli activator CsgD protein: modulation of cellulose biosynthesis and control of negative determinants for microbial adhesion. J Bacteriol. 2006;188(6):2027–37.
Sharma VK, Bearson BL. Hha controls Escherichia coli O157:H7 biofilm formation by differential regulation of global transcriptional regulators FlhDC and CsgD. Appl Environ Microbiol. 2013;79(7):2384–96.
Sharma VK, Bayles DO, Alt DP, Looft T, Brunelle BW, Stasko JA. Disruption of rcsB by a duplicated sequence in a curli-producing Escherichia coli O157:H7 results in differential gene expression in relation to biofilm formation, stress responses and metabolism. BMC Microbiol. 2017;17(1):56.
Uhlich GA, Chen CY, Cottrell BJ, Hofmann CS, Dudley EG, Strobaugh TP, et al. Phage insertion in mlrA and variations in rpoS limit curli expression and biofilm formation in Escherichia coli serotype O157: H7. Microbiology (Reading). 2013;159(Pt 8):1586–96.
Andreozzi E. Gunther NWt, Reichenberger ER, Rotundo L, Cottrell BJ, Nunez a, Uhlich GA: Pch genes control biofilm and cell adhesion in a clinical serotype O157:H7 isolate. Front Microbiol. 2018;9:2829.
Cecchini G, Schroder I, Gunsalus RP, Maklashina E. Succinate dehydrogenase and fumarate reductase from Escherichia coli. Biochim Biophys Acta. 2002;1553(1–2):140–57.
Pletnev P, Osterman I, Sergiev P, Bogdanov A, Dontsova O. Survival guide: Escherichia coli in the stationary phase. Acta Nat. 2015;7(4):22–33.
Bertin Y, Deval C, de la Foye A, Masson L, Gannon V, Harel J, et al. The gluconeogenesis pathway is involved in maintenance of enterohaemorrhagic Escherichia coli O157:H7 in bovine intestinal content. PLoS One. 2014;9(6):e98367.
Bezanson G, Mader D, Fillmore S, Bach S, Delaquis P. Reaction of surrogate Escherichia coli serotype O157:H7 and non-O157 strains to nutrient starvation: variation in phenotype and transcription of stress response genes and behavior on lettuce plants in the field. J Food Prot. 2019;82(11):1988–2000.
Lyte M, Bailey MT. Neuroendocrine-bacterial interactions in a neurotoxin-induced model of trauma. J Surg Res. 1997;70(2):195–201.
Kim D, Langmead B, Salzberg S. HISAT: a fast spliced aligner with low memory requirements. Nat Methods. 2015;12:357–60.
Anders S, Pyl PT, Huber W: HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31(2):166–169.
Love MI, Huber W, Anders H. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15(550).
Szklarczyk D, Gable A, Lyon D, Junge A, Wyder S, Huerta-Cepas J, Simonovic M, Doncheva N, Morris J, Bork P et al: STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucelic Acids Research 2019, 47(Database):607-613.
Datsenko KA, Wanner BL. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci U S A. 2000;97(12):6640–5.
Castanie-Cornet MP, Penfound TA, Smith D, Elliott JF, Foster JW. Control of acid resistance in Escherichia coli. J Bacteriol. 1999;181(11):3525–35.
Vogel HJ, Bonner DM. Acetylornithinase of Escherichia coli: partial purification and some properties. J Biol Chem. 1956;218(1):97–106.
Aziz RK, Bartels D, Best AA, DeJongh M, Disz T, Edwards RA, et al. The RAST server: rapid annotations using subsystems technology. BMC Genomics. 2008;9:75.
Perna NT, Plunkett G 3rd, Burland V, Mau B, Glasner JD, Rose DJ, et al. Genome sequence of enterohaemorrhagic Escherichia coli O157:H7. Nature. 2001;409(6819):529–33.
Acknowledgements
We thank Lindsay Andersen for technical support in the completion of this study. This work was supported by USDA-ARS CRIS project 5030-32000-112-00D. Mention of trade names or commercial products in this article is solely for the purpose of providing specific information and does not imply recommendation or endorsement by the U.S. Department of Agriculture. USDA is an equal opportunity provider and employer.
Funding
This work was supported by USDA, ARS CRIS funds. Mention of trade names or commercial products in this article is solely for the purpose of providing specific information and does not imply recommendation or endorsement by the U.S. Department of Agriculture.
Author information
Authors and Affiliations
Contributions
VS designed the study, performed experiments, and prepared the draft of the manuscript; SA performed comparative genomic analysis, performed experiments, and contributed to manuscript writing; DB analyzed data and contributed to writing of the manuscript. All authors read and approved the final draft of the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Table S1.
List of 5509 genes with reads mapped to the reference genome in response to growth of E. coli O157:H7 strain NADC 6564 in response to norepinephrine.
Additional file 2: Table S2.
List of genes upregulated in response to growth of E. coli O157:H7 strain NADC 6564 in response to norepinephrine.
Additional file 3: Table S3.
List of genes downregulated in response to growth of E. coli O157:H7 strain NADC 6564 in the presence of norepinephrine.
Additional file 4: Fig. S1.
Comparison of the growth rate and viable bacterial cell counts of E. coli O157:H7 strain NADC 6564 grown in the absence or presence of norepinephrine. (A) Bacterial growth was measured by taking A600 readings over a 24 h period for strain NADC 6564 grown in DMEM lacking (green curve) or containing norepinephrine (red curve). Each growth curve was generated by plotting means (± SD) of A600 readings of three independent cultures whereby triplicate of each culture were analyzed for growth and (B) Viable cell counts were determined by plating 10-fold serial dilutions of strain NADC 6564 grown in the absence (green bar) or presence (red bar) of norepinephrine as described in materials and methods. The error bars represent standard deviation of the mean of three independent assays. *** p = 0.0005.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Sharma, V.K., Akavaram, S. & Bayles, D.O. Genomewide transcriptional response of Escherichia coli O157:H7 to norepinephrine. BMC Genomics 23, 107 (2022). https://doi.org/10.1186/s12864-021-08167-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-021-08167-z