
Abstract
Background
Enteric Escherichia coli survives the highly acidic environment of the stomach through multiple acid resistance (AR) mechanisms. The most effective system, AR2, decarboxylates externally-derived glutamate to remove cytoplasmic protons and excrete GABA. The first described system, AR1, does not require an external amino acid. Its mechanism has not been determined. The regulation of the multiple AR systems and their coordination with broader cellular metabolism has not been fully explored.
Results
We utilized a combination of ChIP-Seq and gene expression analysis to experimentally map the regulatory interactions of four TFs: nac, ntrC, ompR, and csiR. Our data identified all previously in vivo confirmed direct interactions and revealed several others previously inferred from gene expression data. Our data demonstrate that nac and csiR directly modulate AR, and leads to a regulatory network model in which all four TFs participate in coordinating acid resistance, glutamate metabolism, and nitrogen metabolism. This model predicts a novel mechanism for AR1 by which the decarboxylation enzymes of AR2 are used with internally derived glutamate. This hypothesis makes several testable predictions that we confirmed experimentally.
Conclusions
Our data suggest that the regulatory network underlying AR is complex and deeply interconnected with the regulation of GABA and glutamate metabolism, nitrogen metabolism. These connections underlie and experimentally validated model of AR1 in which the decarboxylation enzymes of AR2 are used with internally derived glutamate.
Similar content being viewed by others

Background
Escherichia coli can act as both a commensal and potential pathogen. Pathogenic strains of E. coli cause a range of diseases including urinary tract infections, pneumonia, meningitis, and enteric infections. Survival of enteric E. coli strains requires contending with the highly acidic environment of the human digestive tract. The stomach, with a pH as low as 1.5, provides protection against microbial infection. E. coli is known to be unusually tolerant to acid, rivaling the tolerance of Helicobacter pylori [1–3]. This tolerance may contribute to the unusually low dose required for an E. coli infection, requiring only ~102 cell dosage for infection [4–10]. Thus, although not a virulence factor specific to pathogenesis, innate acid adaptation systems are nonetheless essential for the survival of both pathogenic and non-pathogenic enteric E. coli [3, 11–13].
Multiple acid resistance (AR) mechanisms have been described for E. coli [1–3, 7, 11, 14]. Four of the five primary systems utilize a pyridoxal-5’-phosphate (PLP)-dependent amino acid decarboxylase with an externally derived amino acid to consume a proton and generate a by-product and CO2. A corresponding anti-porter exchanges the amino acid and by-product across the membrane. The glutamate-dependent system named as AR2 or GDAR, is the most robust system, allowing up to 80% survival after 2 h in extremely low pH and producing GABA as by-product [11]. Other amino acid dependent systems are the arginine-dependent system (AR3 or ADAR) the lysine-dependent system (AR4 or LDAR), and the more recently discovered ornithine-dependent system (ODAR) [15]. The first described AR system, AR1, is an oxidative AR system repressed by glucose that is σS-dependent and does not require an externally-derived amino acid [2, 4, 11]. Despite the fact that AR1 was the first discovered AR system, its mechanism has still not been determined.
The main transcriptional regulatory elements of amino acid-dependent AR have been characterized. GadE is the primary regulator of AR2 and serves as a transcriptional activator for genes encoding two glutamate decarboxylase isoforms (gadA and gadB) and the glutamate/GABA antiporter (gadC) [16–18]. Transcriptional activation of gadA/B requires heterodimerization of GadE with RcsB [19]. Regulation of gadE, in turn, is complex and involves the activities of multiple circuits whose effects are integrated by binding to the large intergenic region upstream of gadE [2, 16, 20, 21]. AdiY is the primary regulator of AR3 that, together with CysB, coordinately regulates the corresponding arginine decarboxylase gene (adiA) [22]. CadC is the primary regulator of AR4, regulating both the lysine decarboxylase (cadA) and antiporter (cadB) genes [23]. The regulatory network for ODAR is not well-defined. With the exception of the binding of GadE-RcsB to the cadBA promoter, it is not known whether or how the regulation different system AR systems and adaptations are coordinated. Elements of AR2 can be induced by non-acid stimuli including treatment with acetate and entry into stationary phase [11]. In addition, acid stress leads to adaptations beyond the amino acid-dependent AR machinery including expression of the electron transport chain, the envelope stress response and alterations in membrane permeability to protons, a formate hydrogen lyase system that reduces protons to hydrogen gas, and reversals in the cell potential that may drive a chloride/proton antiporter, and numerous metabolic processes [1, 2, 24–26]. The regulatory mechanisms underlying these expression changes have not been established, and the coordination of these and other acid responses with broader cellular metabolism has not been fully explored.
Despite extensive mapping of genes and their regulatory elements in E. coli, only a small fraction of its TFs have been studied on a genomic scale. Chromatin-immunoprecipitation followed by sequencing (ChIP-Seq) enables genome-wide mapping of TF binding sites and has been applied extensively to eukaryotes [27]. In every organism in which ChIP-Seq has been applied, many more binding sites for even well studied TFs have been reproducibly discovered [28]. Surprisingly, these approaches have been used sparingly in E. coli. ChIP-microarray (or ChIP-chip) data has been described for only 19 of 297 E. coli TFs while higher resolution ChIP-Seq and ChIP-exo data have been described for only a handful [29–39]. In bacteria, ChIP-Seq identifies binding sites with high reproducibility and spatial resolution frequently sufficient to identify multiple binding sites within a single promoter but cannot establish if these sites have functions [40–42]. The integration of ChIP-Seq and gene expression data following TF perturbations allows us to identify binding sites that have putative regulatory effects, distinguishing between direct and indirect regulatory effects. This approach has been used to map the transcriptional regulatory network for Mycobacterium tuberculosis (MTB) [42] and networks in S. Typhimurium [43, 44] and E. coli [31].
As part of an on-going effort to comprehensively map the transcriptional regulatory network of E. coli, we have performed ChIP-Seq on a large number of E. coli TFs. We report here the results for 4 TFs with interactions relevant to AR: CsiR, Nac, NtrC, and OmpR. We identified all previously reported in vivo direct interactions for these TFs and confirmed several others previously inferred from gene expression data. Our data further demonstrated that nac and csiR directly modulate AR, and lead to a regulatory network model in which all four TFs participate in coordinating acid resistance, glutamate metabolism, and nitrogen metabolism. This model predicts a novel mechanism for AR1 by which the decarboxylation enzymes of AR2 are used with internally derived glutamate. This hypothesis makes several testable predictions that we confirmed experimentally.
Methods
Bacterial strains and culture conditions
Single gene knock-out strains (Δnac, ΔcsiR, ΔgadC, ΔgadE, ΔgadA, ΔgadB) were obtained from the Keio collection and verified via PCR. The ΔgadAΔgadB KO and the ΔgabDTP/ΔcsiR KO was created by using the one step gene-inactivation technique by Datsenko and Wanner [45] using ΔgadA and ΔcsiR as a background strains respectively. Cells are grown and maintained in LB media with kanamycin and chloramphenicol. For wild-type, E. coli strain K-12 MG1655 was used.
ChIP-Seq
TFs were ligated into pT7-FLAG-4 vector (Sigma-Aldrich) for Flag-tagging and inducible expression. Plasmids were cloned into E. coli MG1655 strains and checked for kanamycin selection. Fidelity of the clones were validated through sequencing. Western blot verified production of inducible Flag-tagged TF using 1 mM IPTG. ChIP assays were performed by induction of strains in LB media starting at OD600 0.2 with 1 mM IPTG for 2 h. Cells were fixed with formaldehyde and glycine and sheared through sonication before immunoprecipitation with anti-FLAG monoclonal antibody. Further pull-down was done using agarose protein G beads. Reverse cross-linking of samples was performed by incubation with Proteinase K. DNA purification was carried out using DNA purification kit (Qiagen). Library preparation was done using standard Illumina TruSeq ChIP Sample Preparation protocols. ChIP replicate experiments presented here were performed by students as part of final projects for course BE605 in Biomedical Engineering at Boston University. Multiplexed sequencing was performed on an Illumina GAIIx Sequencer that generated single 50 bp reads. Total reads generated for the sequencing runs ranged from 3.5 – 22 million reads with an average of 10.62 million reads. ChIP-Seq control samples were wild-type strains with and without empty vectors subjected to the same immunoprecipitation protocol.
ChIP-qPCR
40 ml E. coli cells expressing C-terminally FLAG-tagged Nac were grown in Gutnick Medium [46] at 30 °C and supplemented with 2 mM NH4Cl. Cultures were harvested 60 min after growth ceased (nitrogen depleted), at an OD600 between 0.6 and 0.7. ChIP was performed as previously described [35]. To serve as an “input” control, 20 μl chromatin were also de-crosslinked by boiling for 10 min and cleaned up using a PCR purification kit (Qiagen). ChIP and input samples were analyzed using an ABI 7500 Fast real time PCR machine. Enrichment of ChIP samples was calculated relative to a control region within the transcriptionally silent bglB gene and normalized to input DNA. Occupancy units represent background-subtracted fold-enrichment.
RNA-Seq
RNA-Seq was performed following induction of Nac and CsiR using the same TF inducible E. coli strains used in ChIP-Seq as described above. Control experiments under identical conditions were also performed on WT E. coli. 50 mL of TF-inducible strains were induced with 1 mM IPTG for 2 h starting at OD600 0.2 in LB media. Total RNA extraction was performed using TRIzol® reagent (LifeTechnologies). Samples were subjected to 1-h DNAse digestion and purified using RNeasy spin columns (Qiagen). Samples were processed using Ribo-Zero rRNA removal kits and library preparation was done using NEB Next ultra-directional RNA library prep kit for Illumina. Multiplexed sequencing was performed on an Illumina GAIIx Sequencer that generated single 40 bp reads. Total coverage for the sequencing runs ranged from 8–14 million reads with an average of 10 million reads.
OmpR RT-PCR
50 mL of the ompR-inducible strains were grown in LB media starting at OD600 0.2 with 1 mM IPTG for 2 h. For ΔcsiR and Δnac strains, strains were subject to AR2 acid challenge conditions described below. Total RNA extraction was performed using TRIzol® reagent (LifeTechnologies). Samples were subjected to 1-h DNAse digestion and purified using RNeasy spin columns (Qiagen). Samples were analyzed using BioRad CFX96 Real-Time System C1000 Thermal Cycler. Gene expression was calculated using the ΔΔCt method with rpoD as a reference gene.
Data analysis
The analysis of ChIP-Seq data to identify binding sites was performed as previously described [28, 42, 47]. Reads were aligned to E. coli genome (Genbank entry U00096.2). Binding sites were compared to reported binding sites from EcoCyc [48] after manually curating reported regulatory interactions for those with experimental evidence for binding (Additional file 1: Table S1). Binding sites were assigned to potential gene targets based on proximity to potential promoters and taking into account operon structure from EcoCyc. Genes with start codons within 500 bp of a binding site were considered as potential targets. In the case of divergent promoters the gene closest to the binding site was considered to be the target unless gene expression data or known promoter structure indicated an alternative target or potential regulation of both divergently transcribed genes.
Determination of binding sequence motifs was performed using MEME SUITE tool (version 4.10.2) [49]. A 4th-order markov model based on the whole genome sequence served as background bfile to create more accurate motifs.
For the analysis of RNA-Seq data, Bowtie2 [50] was used to align raw reads to the E. coli genome (Genbank entry U00096.2) and samtools [51] was used to obtain BAM files. R scripts (Bioconductor GenomicRanges [52] package and custom-written scripts) were used to calculate raw read counts per gene and RPKMs. Differential expression was calculated as the ratio of RPKMs after TF induction to RPMKs in control experiments with WT E. coli.
Acid challenge assays
Acid challenge (AR) assays were all adapted from the protocol described by Castanie-Cornet et al. [11].
Testing AR1
Cultures are grown overnight in LB media buffered at pH 5.5 with 100 mM mopholinethanesulfonic acid (MES) at 37 °C. A negative control sample was also cultured overnight in EG media at pH 7.0 in 37 °C. New 1:1000 diluted test cultures are made in E-minimal media with 0.5% glucose (EG media) adjusted to pH 2.5 and pH 7.0 respectively while the negative control was diluted by 1:1000 into EG-media at pH 2.5. All diluted cultures were incubated for 2 h in 37 °C and were then plated in LB plates. The number of colony-forming units (CFUs) after overnight plate incubation at 37 °C were counted to determine survival. Kanamycin (50 μg/mL) was added to media for the knockout strains.
Testing AR2/GDAR
Cultures are grown overnight in LB media with 0.5% glucose at pH 7.0 in 37 °C. New 1:1000 diluted test cultures are made in E-minimal media with 0.5% glucose (EG media) at pH 2.5 supplemented with 1 mM L-glutamate. A negative control culture was also prepared without L-glutamate supplement. Test cultures were incubated for 2 h in 37 °C and were then plated in LB plates. The number of colony-forming units (CFUs) after overnight plate incubation at 37 °C were counted to determine survival. Kanamycin (50 μg/mL) was added to media for the knockout strains.
Induction of gadE for AR rescue
GadE was cloned into a pZE11 expression vector under the control of the pLtetO promoter [53]. This construct was transformed into WT, ΔcsiR, Δnac and ΔgadE strains. GadE induction was carried out by addition of anhydrotetracycline (aTc) during the incubation at 37 °C step. Acid challenge was performed according to procedure above.
For nac and ntrC physiological induction
E. coli MG1655 WT strains were grown on N−C− minimal media supplemented with 0.4% glucose to mid-exponential phase (OD600 = 0.5). 5 mM glutamine was added as control [54].
For csiR natural physiological induction
E. coli MG1655 WT strains were grown on LB media to lag and mid-exponential phases (OD600 0.1 and 0.5 respectively). The sample in lag phase served as control [55].
For ompR physiological induction
E. coli MG1655 WT strains were grown on LB media mid-exponential phase (OD600 = 0.5) with 20% sucrose. A sample without sucrose was used as control [56].
For TF artificial induction
Inducible TF strains were incubated in LB media at 37 °C with 1 mM IPTG for 2 h.
RNA extraction and qRT-PCR
For the samples in the above section, total RNA extraction was performed using RNeasy Protect Bacteria kit (Qiagen). Samples were subjected to 1-h TURBO DNAse digestion and purified using AMPure RNAclean XP beads. qRT-PCR was performed using BioRad CFX96 Real-Time System C1000 Thermal Cycler using gene-specific primers. Gene expression was calculated using the ΔΔCt method with rpoD serving as a reference gene.
Results
Validation of Binding Site Mapping
Our regulatory network mapping strategy utilized transcription factors tagged with FLAG and under inducible control (Methods) [28, 42, 57–60]. Importantly, control ChIP-seq experiments in strains lacking FLAG-tagged proteins revealed minimal non-specific binding in E. coli. The use of an inducible promoter system ensures expression of targeted TFs, which allowed us to study the binding of all TFs in the same standard reproducible condition. While the induction of TFs raises potential concerns about overexpression artifacts, we confirmed the accuracy of this approach in E. coli for the TFs studied in this report in several ways. First, we identified all previously experimentally validated in vivo direct interactions from EcoCyc [48] with high spatial accuracy (Additional file 1: Table S1 and Table S2). Second, motifs inferred from our binding data are consistent with those previously described (Additional file 1: Figure S1). Third, our data for NtrC are consistent with previously published data for NtrC induced from its native promoter [30] (Additional file 1: Figure S3). Finally, our results for binding site accuracy in E. coli are consistent with the results of TF mapping in Mycobacterium tuberculosis and related Mycobacteria [42, 57–59].
Analysis of Regulatory Interactions
Our ChIP-Seq data identify a large number of previously undetected binding sites (Additional file 1: Table S1, Additional files 2, 3, 4 and 5) including binding over a range of coverage enrichment, potentially reflecting differences in binding affinity [42]. In addition, although binding within intergenic regions is enriched over what would be expected by chance, a large number of binding sites in genes were also identified. This has been commonly reported for other ChIP-Seq studies in bacteria [28, 47]. To assess the potential transcriptional functions of these newly identified binding sites, we analyzed transcriptomic data following the perturbation of each TF (Additional files 2 and 3). For Nac and CsiR, we performed RNA-Seq after TF induction using the same strains used for ChIP-Seq and present the genes most likely affected based on our binding and expression data (see Tables 1 and 2, Methods). We also analyzed previously published microarray data for an E. coli strain in which a mutation in the NtrC-activating kinase, NtrB, upregulates NtrC [54]. This publication compared microarray data for NtrC upregulation to an ntrC deletion strain. Since induction of NtrC also induces nac, these data reveal genes directly or indirectly induced by both TFs. We did not assess the impact of OmpR on RNA levels genome-wide, but rather we performed gene-specific RT-PCR. We also performed RT-PCR following TF perturbations to validate additional specific interactions, as described below. Using the combination of ChIP-Seq and transcriptomic data, we identified potential direct regulatory interactions as described in the Methods. We first describe our results in detail for each TF, and then describe a global regulatory network arising from this analysis that links acid resistance with central metabolism.
CsiR
CsiR is reported to repress csiD via a σs promoter upstream of the csiD-ygaF-gabD-gabT-gabP operon, though mutation of csiR does not directly impact regulation of gabDTP, potentially due to two internal promoters near gabD [55]. GabDTP are involved in the inter-conversion of GABA and alpha-ketoglutarate (α-KG). It has been postulated that these genes may play a role in linking acid resistance to the TCA cycle (Additional file 1: Figure S5) through the metabolism of GABA derived from AR2, although this has not been confirmed [55]. Moreover, no direct binding of CsiR to any operon has been reported [55, 61]. Our ChIP-Seq data confirms the expected binding site for CsiR in the csiD promoter (Fig. 1). Consistent with previous results, CsiR induction represses csiD while no significant expression changes were observed for ygaF or gabDTP. Our data also reveal a surprising number of novel binding sites of potential relevance to AR (see Table 2). We observe binding to gadX and ydeO, and RNA-Seq following CsiR induction indicates strong activation of both. CsiR also binds to the divergent promoter between gadW and gadY. We also observe weak repression of gadW and no evident effect on gadY, suggesting that this binding site operates on the gadW promoter (Additional file 2 ). GadX, YdeO, GadW and GadY are four regulators that form a complex circuit capable of activating the core AR2 genes gadE, gadA, gadB, and gadC (Fig. 2) [62, 63]. We see strong activation of all of these AR2 genes after induction of CsiR (see Table 2). We also observe that CsiR binds to the AR4 regulator cadC. No significant effect of csiR induction on cadC was evident in our data, though cadBA was moderately repressed. We further observe that induction of CsiR results in repression of the AR3 genes adiY and adiA, though this appears to reflect indirect regulation as no CsiR binding was seen.

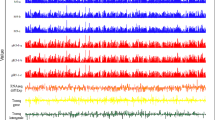
Mapping E. coli transcriptional regulatory interactions using ChIP-Seq. Examples of identified binding sites for csiR, nac, ntrC, and ompR. Each panel plots the total read coverage (blue), forward read coverage (green), and reverse read coverage (red). The maximum coverage for each plot is given by the number on the y-axis in units of coverage normalized to mean coverage. Multiple biological replicate experiments are shown for 3 TFs as noted on leftmost y-axes. ChIP-Seq coverage plots are shown for 8 separate genomic regions. The start location of each region is provided at the bottom left x-axes. The tick marks on the bottom x-axes are spaced 500 bp apart. Different regions are plotted at different scales for clarity. Previously described binding sites from EcoCyc are shown as black ticks below the coverage plot in each panel

ChIP-Seq mapping and transcriptomics reveal regulatory links between AR systems and cellular metabolic pathways. Map of selected direct binding sites potentially associated with AR. Novel TF binding is displayed as colored dashed lines. Novel regulatory links confirmed with gene expression data are shown as solid colored lines. Black lines signify previously reported known binding and regulation. Circle terminators indicate unconfirmed or indeterminate regulatory effect
NtrC and Nac
NtrC and Nac are the two principal regulators of nitrogen metabolism [64–66]. Nitrogen availability is sensed by monitoring levels of intracellular glutamine, which are linked to glutamate levels through nitrogen assimilation pathways (Additional file 1: Figure S5). Under low nitrogen conditions, the regulator NtrC is activated by phosphorylation by NtrB and modulates numerous σ-54 dependent genes. Consequently, Nac is induced and in turn modulates a set of σ-70 genes [54]. This coordinated activity of NtrC and Nac in low nitrogen affects components in the two major ammonia assimilation pathways: the glutamate synthase (GOGAT) pathway consisting of glnA and gltBD, and the glutamate dehydrogenase (GDH) pathway consisting of gdhA. NtrC activation leads to the glnA induction, while Nac represses gltBD [54]. Our data confirm all the previously known binding sites for Nac and identify numerous additional potential regulatory interactions (Additional file 1: Table S1, Additional file 3). As with CsiR, these include a surprising number associated with AR2 (Fig. 2). Our data also shows the reported repression of gltBD by Nac while also possibly repressing gdhA (Additional file 3, Fig. 2).
For Nac, we observe strong binding within the gadE gene, and both our RNA-seq data and published expression data for the perturbation of NtrC and Nac [54] indicate that Nac induction activates gadE expression (Table 2). Further confirmation of this binding site using ChIP-qPCR on natively tagged Nac in Gutnick media (see Methods) showed a 4.4-fold enrichment increase of the occupancy at the site within gadE. We also identify binding and apparent regulation by Nac for several genes in the circuitry upstream of GadE (Fig. 2). These data are consistent with previous reports indicating gadBC and gadA induction by acid in the absence of an σ-s and potentially dependent on σ-70 [67, 68]. We identify two Nac binding sites associated with the csiD-ygaF-gabD-gabT-gabP operon. In addition to the previously reported regulatory site upstream of gabD [54], we also identify a site upstream of csiD. We further identify two binding sites associated with the sucABCD-sdhCDAB operon whose genes catalyze the TCA reactions between α-KG, succinate and fumarate (Additional file 2).
Our data also recapitulate the known regulatory interactions of NtrC, as noted above (Additional file 1: Table S1 and Figure S2–S4). Although different methods and conditions were utilized, a comparison of our data with ChIP-Seq of NtrC by Brown et al. [30] reveals substantial agreement between the two datasets and with previously biochemically identified binding sites (Additional file 1: Figure S2–S4). Our data refine binding sites reported in the previous manuscript and extend these results with additional detected sites (Additional file 4). In particular, we identify weak binding and apparent repression by NtrC of the speF/potE operon, the first potential direct regulatory link for ODAR identified. No direct binding of NtrC to elements of AR2 was detected.
OmpR
OmpR is a response regulator known to regulate several genes involved in osmotic stress adaptation [69, 70]. Recently, it was also shown that an OmpR mutant is unable to survive even mild acid stress [71]. OmpR is thought to be regulated by IHF, Crp, and ppGpp. We identify a novel Nac binding site in the divergent promoter between ompR and greB. Our gene expression analysis suggests this site may repress both genes (Additional file 5). Our ChIP-Seq mapping of OmpR detected all sites with experimental evidence for binding in vivo, verifies several sites for which there was no previous evidence of binding, and identifies 46 previously undetected sites (Additional file 1: Table S1, Additional file 5). EcoCyc also includes binding of OmpR to the promoter of bolA based on in vitro binding data [72]. However, this binding site was not detected in a more recent in vivo study [73], and we do also do not identify this site in our in vivo data.
Of note, the OmpR binding sites we identified include binding upstream of the csiD-ygaF-gabD-gabT-gabP GABA metabolism operon, and upstream of yagU, a gene coding for an inner membrane protein required for AR [24]. RT-PCR following induction of ompR resulted in a 2-fold increased expression of yagU compared to WT. A recent publication describing the ChIP-chip mapping of OmpR in both E. coli and Salmonella typhimurium reported binding of OmpR upstream of CadBA [73]. Our data do not support this conclusion (Additional file 1: Table S2).
A Regulatory Network Linking Acid Resistance to Broader Cellular Metabolism
Collectively, our data suggest interactions between the regulation of different AR systems, GABA and glutamate metabolism, nitrogen metabolism, and the TCA cycle (Fig. 2, Additional file 1: Figure S5). This regulatory cross talk is mirrored in the known metabolic connectivity between these pathways (Fig. 2). These data suggest that Nac and CsiR may modulate AR, and their links to gadE and the network upstream of gadE suggest a role in AR2 specifically.
To test this possibility, we examined the phenotype of ΔcsiR and Δnac mutants in acid challenge under different AR conditions using well-described experimental protocols for inducing each system, along with corresponding positive and negative controls (Fig. 3a) [2, 7, 11, 14]. Single gene knockout strains were acquired from the Keio collection and sequence-verified [45]. Neither ΔcsiR nor Δnac displayed altered growth in standard non-acid conditions (Fig. 3b). However, when acid challenged in pH 2.5 after induction of AR2, both ΔcsiR and Δnac displayed significantly decreased colony recovery (Fig. 3b) and survival (Additional file 1: Figure S6). We further tested both strains in AR1-inducing conditions. Surprisingly, deletion of either csiR or nac fully abolished growth and survival under AR1 (Fig. 3b, Additional file 1: Figure S6).

Validation of a Proposed Mechanism for AR1. We hypothesized that AR1 may be mediated by the AR2 machinery using an internal source of glutamate. Our regulatory network implicates both nac and csiR in this process. We tested this hypothesis by examining the phenotype of several deletion mutants in acid stress assays using published protocols for inducing AR1 or AR2, along with positive and negative controls (Castanie-Cornet et al. [11]; Lin et al [4]). Acid stress assays consisted of overnight culture, acid challenge at pH 2.5 for 2 h, followed by plating, overnight incubation, and colony counting (Methods). a Example plates for one experiment for selected mutants comparing AR1 conditions to AR2 conditions. b Summary of colony counts averages for all mutants across all experiments for AR1, AR2, and for two non-acidic control growth conditions (for which strains were plated directly after overnight incubation without acid challenge) for 3 replicates (n = 3). Colony counts provided to allow comparison to control WT data. Resulting counts were tested at a significance level of α = 0.05 (* p-value < 0.05). Plots of % survival for AR1 and AR2 are provide in Additional file 1: Figure S6 c RT-PCR of gadE in WT, ΔcsiR, and Δnac from colonies recovered after acid challenge following AR2 induction (n = 3 for all). d AR Rescue of KO strains via induction of gadE showing the summary of colony counts averages for WT, ΔcsiR, Δnac and ΔgadE with gadE induced in AR1 and AR2 conditions for 3 replicate experiments (n =3). Numbers on the x-axis above strain names indicate amount of aTc added during AR challenge in ng/μL. Resulting counts were tested at a significance level of α = 0.05 (* p-value < 0.05)
A Proposed Mechanism for AR1
The connectivity of the regulatory network, the experimentally confirmed impact of ΔcsiR and Δnac on both AR2 and AR1, and the metabolic connections between AR and central metabolism imply the possibility that AR under different conditions is modulated by both the intracellular and extracellular availability of key intermediates. This led us to a specific hypothesis concerning the mechanism for AR1. AR1 differs from other systems in that it does not require a specific external amino acid supplement. Given the connections between Nac, CsiR, AR2, and the internal metabolism of glutamate suggested by our data, we hypothesized that AR1 utilizes the decarboxylation mechanism of AR2 with an internal glutamate source. Our proposed mechanism for AR1 makes several specific and testable predictions. In particular, if AR1 uses the decarboxylation mechanism of AR2, it should require the decarboxylases GadA and/or GadB and the protein that induces these, GadE, but not require the glutamate transporter GadC.
To test these specific predictions we acquired and sequence-verified ΔgadE, ΔgadA, ΔgadB, and ΔgadC gene deletion strains from the Keio collection. We further generated a ΔgadAΔgadB strain in which both AR2 decarboxylase genes were deleted since deletion of either gadA or gadB does not fully eliminate AR2 [11]. None of the deletions impacted growth in standard non-acid conditions (Fig. 3b). Moreover, all three AR2-associated genes are required for AR2, as expected (Fig. 3b). We see essentially no colony formation after extreme acid stress under AR2, although mutants have no impact in growth in neutral pH. AR2 also requires glutamate as expected. AR2 resistance is still present in ΔgadA or ΔgadB.
Consistent with our model for AR1, GadE and GadA/GadB are required for resistance in AR1-inducing conditions despite the absence of external glutamate during the overnight pre-incubation and the 2 h of acid stress (Fig. 3b). In addition, as predicted, GadC is not required for AR1 [11]. Although deletion of GadC decreased survival during AR1 relative to WT, all ΔgadC experiments for AR1 resulted in colonies while none for AR2 did. Moreover, deletion of either gadA or gadB individually was sufficient to render AR1 ineffective. None of the studied mutants impacted the neutral positive control, nor the AR1 specific negative control in which cells were pre-incubated with glucose. These data indicate that AR1 requires both the glutamate decarboxylation genes of AR2, and the primary regulator of these genes, but not the AR2 transporter for extracellular glutamate.
GadE Expression Can Explain Δnac and ΔcsiR Phenotypes and Limited AR1 Efficacy
Given the model that AR1 utilizes an internal source of glutamate with the AR2 decarboxylase machinery, we hypothesized that both CsiR and Nac could modulate AR1 as a consequence of their regulation of AR2 via gadE. As described above, both nac and csiR induction increases gadE expression, with the former effect acting through direct binding of Nac to gadE while the latter presumably through binding and regulation of the circuit upstream of gadE. This implies that ΔcsiR and/or Δnac may impact AR by decreasing expression of GadE. We confirmed this experimentally in two ways.
First, we used RT-qPCR to measure gadE RNA in each of the deletion mutants relative to rpoD in colonies recovered after AR2 induction and acid stress. As shown in Fig. 3c, gadE expression was significantly decreased in both ΔcsiR and Δnac, as predicted, though not entirely abolished. As AR2 was also reduced but not abolished in these strains, the residual level of gadE expression appears sufficient to confer a degree of acid resistance via AR2. In contrast, the residual level of gadE expression in ΔcsiR and Δnac appears insufficient to support AR1.
Second, to confirm that decreased gadE expression levels are sufficient to explain the AR phenotypes of ΔcsiR and Δnac, we rescued AR in these strains by inducing gadE ectopically. We cloned gadE into an inducible vector under the control of the tetO operator. This vector was then introduced into ΔcsiR, Δnac, ΔgadE and WT strains (see Methods). As shown in Fig. 3d, induction of gadE in ΔcsiR and Δnac during acid challenge was capable of restoring WT levels of AR2 survival and providing substantial AR1 survival. Induction of gadE also restored WT levels of AR2 survival in the ΔgadE background, indicating that the functionality of induced gadE was not detectably altered.
Surprisingly, induction of gadE in ΔcsiR, Δnac, and ΔgadE during AR1 conditions resulted in significantly more colony recovery than observed in WT strains (see Fig. 3d, blue bars). This suggests increasing GadE expression could increase the efficacy of AR1. This is supported by significantly increased colony recovery during AR1 when gadE was induced in a WT background. In contrast, AR2 survival was not substantially increased. Together these data confirm the role of gadE in both AR2 and AR1, indicate that decreased gadE expression is sufficient to explain the impact of ΔcsiR and Δnac on AR2 and AR1, and suggest that gadE expression may be a limiting factor in AR1 but not AR2.
Discussion
The primary transcriptional regulatory elements of amino acid-dependent AR have been characterized, but little is known about whether or how different system AR systems and adaptations are coordinated, or how AR is coordinated with broader cellular metabolism. We have utilized a combination of ChIP-Seq and transcriptomics to map the potential regulatory interactions of four transcriptions factors that appear to coordinate acid resistance, glutamate metabolism, and nitrogen metabolism: CsiR, Nac, NtrC, and OmpR. Taken together, our data suggest that the regulatory network underlying AR is complex and interconnected with the regulation central metabolism (Fig. 2, Additional file 1: Figure S5).
Our findings led to an experimentally confirmed mechanism for AR1. AR1 differs from other systems in that it does not require a specific external amino acid supplement. The network model inferred from our data implies that AR under different conditions is modulated by both the intracellular and extracellular availability of key intermediates. Given the connections between nac, csiR, AR2, and the internal metabolism of glutamate suggested by our data, we hypothesized that AR1 utilizes the decarboxylation mechanism of AR2 with an internal glutamate source. Although it has been proposed that internal GABA and glutamate metabolism might cooperate with the GadA and GadB decarboxylases in AR [55], this has not been confirmed, nor has it been linked to a mechanism for AR1. Our proposed mechanism for AR1 made several specific and testable predictions. In particular, if AR1 uses the decarboxylation mechanism of AR2, it should require the decarboxylases GadA and/or GadB and the protein that induces these, GadE, but not require the glutamate transporter GadC. We confirmed these predictions experimentally (Fig. 3).
Our experimental results also confirm the functional importance of the regulatory links we identified between nac, csiR and AR. Deletion of either nac or csiR substantially diminished the efficacy of AR2 during acid challenge, and abolished AR1. Our regulatory network model predicted that deletion of nac and csiR would decrease the expression of gadE during acid challenge, and we confirmed this via RT-PCR (Fig. 3c). We further confirmed that this decrease in gadE expression was sufficient to explain the AR phenotypes observed. Induction of gadE in ΔcsiR and Δnac resulted in robust survival in both AR2 and AR1 conditions (Fig. 3d).
Induction of gadE in ΔcsiR and Δnac restored AR2 survival to WT levels (Fig. 3c). In AR1 conditions by contrast, gadE induction in these backgrounds resulted in significantly more survival relative to WT. We also observed substantially greater AR1 survival when gadE was induced in a WT strain, while AR2 survival was not substantially increased. Thus, increasing gadE expression is sufficient to increase AR1 efficacy, but not AR2. This suggests that gadE expression may be limiting in AR1, but is not limiting in AR2. However, differences in gadE expression are not sufficient to fully explain the difference between AR2 and AR1 efficacy. At corresponding levels of gadE expression, we see consistently greater survival in AR2 relative to AR1. This was observed in both ΔcsiR and Δnac, where the residual level of gadE expression was sufficient to confer measurable protection from AR2, but none from AR1. Similarly, gadE induction in all background strains tested resulted in greater survival in AR2 relative to AR1. We hypothesize that differences in the levels of intracellular glutamate available to gadA/B may explain part of these results.
Our data provide new perspective on previously published results. Although previous studies have demonstrated that deletion of either σ54 or ntrC leads to increased expression of GDAR and increased acid resistance [74, 75], the lack of direct interactions between NtrC and known regulators of AR2 suggests this effect is indirect. The role of Nac in AR2 suggests that part of this effect may be through the known σ70–dependent regulation of nac by NtrC. However, the activation of GDAR by ΔntrC cannot be easily explained by this link alone. Previous studies have also demonstrated that NtrC, RcsB, and GadX regulate the locus of enterocyte effacement (LEE) pathogenicity island in enterohemorrhagic E. coli, indicating that the coordination of nitrogen metabolism and AR can play both non-specific (through acid resistance) and specific (through LEE) roles in E. coli pathogenesis [74–77].
The link between AR1 and AR2, and the potential role for Nac and CsiR in mediating this link, raise many questions that remain to be investigated. First, questions remain about the mechanism of regulation of gadE by Nac. Our ChIP-Seq data reproducibly identifies binding of Nac to the GadE coding region (Fig. 1). Extensive genic binding has been previously reported for bacteria [28] and many experimentally confirmed examples of transcriptionally functional binding of TFs within coding regions in bacteria have been reported [59, 78–82]. Our RNA-Seq data (Tables 1 and 2) and previously published data [54] confirm that increased expression of Nac increases gadE mRNA levels. However, whether this change in mRNA levels is mediated through transcription initiation, transcription elongation, or mRNA stability remains to be determined. Second, if intracellular glutamate is the basis for AR1, as our hypothesis and data suggest, the source(s) of this glutamate remain to be determined. One speculative source is the conversion of α-KG from the TCA cycle to glutamate via GabD or GabT, possibly consistent with the regulation of the suc/sad operon by Nac [55]. Required maintenance of TCA cycle intermediates during growth on glucose may then contribute to the glucose repression of AR1. Third, what is the fate of GABA during AR1 if glutamate is being converted into GABA by GadA or GadB? We speculate that the decreased effectiveness of AR1 in the ΔgadC strain suggests the need to export GABA. Finally, the specific timing and roles of the newly identified regulatory links during acid, the roles of σ-70 and σ-s, and the role of non-transcriptional regulatory mechanisms also remain to be determined.
Conclusions
We have presented a comprehensive genome-wide mapping of four TFs in E. coli using a combination of ChIP-Seq and transcriptomics: CsiR, Nac, NtrC, and OmpR. Our data identified all previously in vivo confirmed direct interactions and revealed several others previously inferred only from gene expression data. Our data also reveal novel regulatory interactions that appear to coordinate carbon and nitrogen metabolism with acid resistance. We have experimentally verified that CsiR and Nac, which are known carbon and nitrogen metabolism regulators respectively, modulate acid resistance through the transcriptional regulation of gadE, the master regulator of AR2. Our data also led to a model for the mechanism of the first described acid resistance mechanism, AR1. Our model predicts that AR1 utilizes the decarboxylation enzymes of AR2 but with internally derived glutamate. We have experimentally verified predictions arising from this model. Together our data provide new insight into the mechanisms of acid resistance in E. coli, and reveal an interconnected regulatory network that coordinates acid resistance with broader cellular metabolism.
Abbreviations
- ADAR:
-
Arginine-dependent system
- AR:
-
Acid resistance
- aTc:
-
Anhydrotetracycline
- ChIP:
-
Chromatin-immunoprecipitation
- EG:
-
E-minimal with glucose
- GABA:
-
Gamma-aminobutyric acid
- GDAR:
-
Glutamate-dependent acid resistance system
- GDH:
-
Glutamate dehydrogenase pathway
- GOGAT:
-
Glutamate synthase pathway
- IPTG:
-
Isopropyl β-D-1-thiogalactopyranoside
- LB:
-
Luria-Bertani
- LDAR:
-
Lysine-dependent system
- LEE:
-
Locus of enterocyte effacement
- MTB:
-
Mycobacterium tuberculosis
- ODAR:
-
Ornithine-dependent acid resistance system
- PCR:
-
Polymerase chain reaction
- RT-PCR:
-
Reverse transcriptase PCR
- TCA:
-
The citric acid cycle
- TF:
-
Transcription factor
- WT:
-
Wild-type
References
Kanjee U, Houry WA. Mechanisms of acid resistance in Escherichia coli. Annu Rev Microbiol. 2013;67:65–81.
Foster JW. Escherichia coli acid resistance: tales of an amateur acidophile. Nat Rev Microbiol. 2004;2(11):898–907.
Richard H, Foster JW. Escherichia coli glutamate- and arginine-dependent acid resistance systems increase internal pH and reverse transmembrane potential. J Bacteriol. 2004;186(18):6032–41.
Lin J, Smith MP, Chapin KC, Baik HS, Bennett GN, Foster JW. Mechanisms of acid resistance in enterohemorrhagic Escherichia coli. Appl Environ Microbiol. 1996;62(9):3094–100.
Benjamin MM, Datta AR. Acid tolerance of enterohemorrhagic Escherichia coli. Appl Environ Microbiol. 1995;61(4):1669–72.
Conner DE, Kotrola JS. Growth and survival of Escherichia coli O157:H7 under acidic conditions. Appl Environ Microbiol. 1995;61(1):382–5.
Richard HT, Foster JW. Acid resistance in Escherichia coli. Adv Appl Microbiol. 2003;52:167–86.
Gorden J, Small PL. Acid resistance in enteric bacteria. Infect Immun. 1993;61(1):364–7.
Giannella RA, Broitman SA, Zamcheck N. Influence of gastric acidity on bacterial and parasitic enteric infections. A perspective. Ann Intern Med. 1973;78(2):271–6.
Price SB, Wright JC, DeGraves FJ, Castanie-Cornet MP, Foster JW. Acid resistance systems required for survival of Escherichia coli O157:H7 in the bovine gastrointestinal tract and in apple cider are different. Appl Environ Microbiol. 2004;70(8):4792–9.
Castanie-Cornet MP, Penfound TA, Smith D, Elliott JF, Foster JW. Control of acid resistance in Escherichia coli. J Bacteriol. 1999;181(11):3525–35.
Cotter PD, Gahan CG, Hill C. A glutamate decarboxylase system protects Listeria monocytogenes in gastric fluid. Mol Microbiol. 2001;40(2):465–75.
Sanders JW, Leenhouts K, Burghoorn J, Brands JR, Venema G, Kok J. A chloride-inducible acid resistance mechanism in Lactococcus lactis and its regulation. Mol Microbiol. 1998;27(2):299–310.
Lu P, Ma D, Chen Y, Guo Y, Chen GQ, Deng H, Shi Y. L-glutamine provides acid resistance for Escherichia coli through enzymatic release of ammonia. Cell Res. 2013;23(5):635–44.
Kashiwagi K, Suzuki T, Suzuki F, Furuchi T, Kobayashi H, Igarashi K. Coexistence of the genes for putrescine transport protein and ornithine decarboxylase at 16 min on Escherichia coli chromosome. J Biol Chem. 1991;266(31):20922–27.
Masuda N, Church GM. Regulatory network of acid resistance genes in Escherichia coli. Mol Microbiol. 2003;48(3):699–712.
Ma Z, Gong S, Richard H, Tucker DL, Conway T, Foster JW. GadE (YhiE) activates glutamate decarboxylase-dependent acid resistance in Escherichia coli K-12. Mol Microbiol. 2003;49(5):1309–20.
Hommais F, Krin E, Coppee JY, Lacroix C, Yeramian E, Danchin A, Bertin P. GadE (YhiE): a novel activator involved in the response to acid environment in Escherichia coli. Microbiology. 2004;150(Pt 1):61–72.
Castanie-Cornet MP, Cam K, Bastiat B, Cros A, Bordes P, Gutierrez C. Acid stress response in Escherichia coli: mechanism of regulation of gadA transcription by RcsB and GadE. Nucleic Acids Res. 2010;38(11):3546–54.
Sayed AK, Foster JW. A 750 bp sensory integration region directs global control of the Escherichia coli GadE acid resistance regulator. Mol Microbiol. 2009;71(6):1435–50.
Ma Z, Masuda N, Foster JW. Characterization of EvgAS-YdeO-GadE branched regulatory circuit governing glutamate-dependent acid resistance in Escherichia coli. J Bacteriol. 2004;186(21):7378–89.
Stim-Herndon KP, Flores TM, Bennett GN. Molecular characterization of adiY, a regulatory gene which affects expression of the biodegradative acid-induced arginine decarboxylase gene (adiA) of Escherichia coli. Microbiology. 1996;142(Pt 5):1311–20.
Kuper C, Jung K. CadC-mediated activation of the cadBA promoter in Escherichia coli. J Mol Microbiol Biotechnol. 2005;10(1):26–39.
Hayes ET, Wilks JC, Sanfilippo P, Yohannes E, Tate DP, Jones BD, Radmacher MD, BonDurant SS, Slonczewski JL. Oxygen limitation modulates pH regulation of catabolism and hydrogenases, multidrug transporters, and envelope composition in Escherichia coli K-12. BMC Microbiol. 2006;6:89.
Maurer LM, Yohannes E, Bondurant SS, Radmacher M, Slonczewski JL. pH regulates genes for flagellar motility, catabolism, and oxidative stress in Escherichia coli K-12. J Bacteriol. 2005;187(1):304–19.
Chang YY, Cronan Jr JE. Membrane cyclopropane fatty acid content is a major factor in acid resistance of Escherichia coli. Mol Microbiol. 1999;33(2):249–59.
Mikkelsen TS, Ku M, Jaffe DB, Issac B, Lieberman E, Giannoukos G, Alvarez P, Brockman W, Kim TK, Koche RP, et al. Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature. 2007;448(7153):553–60.
Galagan J, Lyubetskaya A, Gomes A. ChIP-Seq and the complexity of bacterial transcriptional regulation. Curr Top Microbiol Immunol. 2013;363:43–68.
Park DM, Kiley PJ. The influence of repressor DNA binding site architecture on transcriptional control. mBio. 2014;5(5):e01684–14.
Brown DR, Barton G, Pan Z, Buck M, Wigneshweraraj S. Nitrogen stress response and stringent response are coupled in Escherichia coli. Nat Commun. 2014;5:4115.
Myers KS, Yan H, Ong IM, Chung D, Liang K, Tran F, Keleş S, Landick R, Kiley PJ. Genome-scale analysis of escherichia coli FNR reveals complex features of transcription factor binding. PLoS Genet. 2013;9:e1003565.
Prieto AI, Kahramanoglou C, Ali RM, Fraser GM, Seshasayee AS, Luscombe NM. Genomic analysis of DNA binding and gene regulation by homologous nucleoid-associated proteins IHF and HU in Escherichia coli K12. Nucleic Acids Res. 2012;40(8):3524–37.
Kahramanoglou C, Seshasayee AS, Prieto AI, Ibberson D, Schmidt S, Zimmermann J, Benes V, Fraser GM, Luscombe NM. Direct and indirect effects of H-NS and Fis on global gene expression control in Escherichia coli. Nucleic Acids Res. 2011;39(6):2073–91.
Fitzgerald DM, Bonocora RP, Wade JT. Comprehensive mapping of the Escherichia coli flagellar regulatory network. 2014.
Stringer AM, Currenti S, Bonocora RP, Baranowski C, Petrone BL, Palumbo MJ, Reilly AA, Zhang Z, Erill I, Wade JT. Genome-scale analyses of Escherichia coli and Salmonella enterica AraC reveal noncanonical targets and an expanded core regulon. J Bacteriol. 2014;196(3):660–71.
Seo SW, Kim D, Szubin R, Palsson BO. Genome-wide Reconstruction of OxyR and SoxRS Transcriptional Regulatory Networks under Oxidative Stress in Escherichia coli K-12 MG1655. Cell Rep. 2015;12(8):1289–99.
Seo SW, Kim D, O’Brien EJ, Szubin R, Palsson BO. Decoding genome-wide GadEWX-transcriptional regulatory networks reveals multifaceted cellular responses to acid stress in Escherichia coli. Nat Commun. 2015;6:7970.
Cho S, Cho YB, Kang TJ, Kim SC, Palsson B, Cho BK. The architecture of ArgR-DNA complexes at the genome-scale in Escherichia coli. Nucleic Acids Res. 2015;43(6):3079–88.
Beauchene NA, Myers KS, Chung D, Park DM, Weisnicht AM, Keles S, Kiley PJ. Impact of Anaerobiosis on Expression of the Iron-Responsive Fur and RyhB Regulons. mBio. 2015;6(6):e01947–15.
Gomes AL, Abeel T, Peterson M, Azizi E, Lyubetskaya A, Carvalho L, Galagan J. Decoding ChIP-Seq peaks with a double-binding signal refines binding peaks to single-nucleotide and predicts cooperative interaction. Genome Res. 2014;24(10):1686–97.
Lun DS, Sherrid A, Weiner B, Sherman DR, Galagan JE. A blind deconvolution approach to high-resolution mapping of transcription factor binding sites from ChIP-seq data. Genome Biol. 2009;10(12):R142.
Galagan JE, Minch K, Peterson M, Lyubetskaya A, Azizi E, Sweet L, Gomes A, Rustad T, Dolganov G, Glotova I, et al. The Mycobacterium tuberculosis regulatory network and hypoxia. Nature. 2013;499(7457):178–83.
Petrone BL, Stringer AM, Wade JT. Identification of HilD-regulated genes in Salmonella enterica serovar Typhimurium. J Bacteriol. 2014;196:1094–101.
Stringer AM, Currenti S, Bonocora RP, Baranowski C, Petrone BL, Palumbo MJ, Reilly AE, Zhang Z, Erill I, Wade JT. Genome-Scale Analyses of Escherichia coli and Salmonella enterica AraC Reveal Non-Canonical Targets and an Expanded Core Regulon. J Bacteriol. 2013;196(3):660–71.
Datsenko KA, Wanner BL. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci U S A. 2000;97(12):6640–5.
Gutnick D, Calvo JM, Klopotowski T, Ames BN. Compounds which serve as the sole source of carbon or nitrogen for Salmonella typhimurium LT-2. J Bacteriol. 1969;100(1):215–9.
Jaini S, Lyubetskaya A, Gomes A, Peterson M, Park ST, Raman S, Schoolnik G, Galagan J: Transcription Factor Binding Site Mapping Using ChIP-Seq. In: Molecular Genetics of Mycobacteria, Second Edition. Washington: American Society of Microbiology; 2014.
Keseler IM, Mackie A, Peralta-Gil M, Santos-Zavaleta A, Gama-Castro S, Bonavides-Martinez C, Fulcher C, Huerta AM, Kothari A, Krummenacker M, et al. EcoCyc: fusing model organism databases with systems biology. Nucleic Acids Res. 2013;41(D1):D605–612.
Bailey TL, Boden M, Buske FA, Frith M, Grant CE, Clementi L, Ren JY, Li WW, Noble WS. MEME SUITE: tools for motif discovery and searching. Nucleic Acids Res. 2009;37:W202–8.
Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9(4):357–9.
Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, Genome Project Data Processing S. The Sequence Alignment/Map format and SAMtools. Bioinformatics (Oxford, England). 2009;25(16):2078–9.
Lawrence M, Huber W, Pages H, Aboyoun P, Carlson M, Gentleman R, Morgan MT, Carey VJ. Software for computing and annotating genomic ranges. PLoS Comput Biol. 2013;9(8):e1003118.
Lutz R, Bujard H. Independent and tight regulation of transcriptional units in Escherichia coli via the LacR/O, the TetR/O and AraC/I1-I2 regulatory elements. Nucleic Acids Res. 1997;25(6):1203–10.
Zimmer DP, Soupene E, Lee HL, Wendisch VF, Khodursky AB, Peter BJ, Bender RA, Kustu S. Nitrogen regulatory protein C-controlled genes of Escherichia coli: scavenging as a defense against nitrogen limitation. Proc Natl Acad Sci U S A. 2000;97(26):14674–9.
Metzner M, Germer J, Hengge R. Multiple stress signal integration in the regulation of the complex sigma S-dependent csiD-ygaF-gabDTP operon in Escherichia coli. Mol Microbiol. 2004;51(3):799–811.
Cai SJ, Inouye M. EnvZ-OmpR interaction and osmoregulation in Escherichia coli. J Biol Chem. 2002;277(27):24155–61.
Ranganathan S, Bai G, Lyubetskaya A, Knapp GS, Peterson MW, Gazdik M, Gomes AL, Galagan JE, McDonough KA. Characterization of a cAMP responsive transcription factor, Cmr (Rv1675c), in TB complex mycobacteria reveals overlap with the DosR (DevR) dormancy regulon. Nucleic Acids Res. 2015;44(1):134–51.
Minch KJ, Rustad TR, Peterson EJ, Winkler J, Reiss DJ, Ma S, Hickey M, Brabant W, Morrison B, Turkarslan S, et al. The DNA-binding network of Mycobacterium tuberculosis. Nat Commun. 2015;6:5829.
Knapp GS, Lyubetskaya A, Peterson MW, Gomes AL, Ma Z, Galagan JE, McDonough KA. Role of intragenic binding of cAMP responsive protein (CRP) in regulation of the succinate dehydrogenase genes Rv0249c-Rv0247c in TB complex mycobacteria. Nucleic Acids Res. 2015;43(11):5377–93.
Jaini S, Lyubetskaya A, Gomes A, Peterson M, Park ST, Raman S, Schoolnik G, Galagan JE: Transcription Factor Binding Site Mapping Using ChIP-Seq. In: Molecular Genetics of Mycobacteria, 2nd Edition. Edited by Hatfull G, Jacobs WR, Jr. Washington: ASM Press; 2014.
Marschall C, Labrousse V, Kreimer M, Weichart D, Kolb A, Hengge-Aronis R. Molecular analysis of the regulation of csiD, a carbon starvation-inducible gene in Escherichia coli that is exclusively dependent on sigma s and requires activation by cAMP-CRP. J Mol Biol. 1998;276(2):339–53.
Sayed AK, Odom C, Foster JW. The Escherichia coli AraC-family regulators GadX and GadW activate gadE, the central activator of glutamate-dependent acid resistance. Microbiol-Sgm. 2007;153:2584–92.
Ma Z, Richard H, Tucker DL, Conway T, Foster JW. Collaborative regulation of Escherichia coli glutamate-dependent acid resistance by two AraC-like regulators, GadX and GadW (YhiW). J Bacteriol. 2002;184(24):7001–12.
Bender RA, Snyder PM, Bueno R, Quinto M, Magasanik B. Nitrogen regulation system of Klebsiella aerogenes: the nac gene. J Bacteriol. 1983;156(1):444–6.
Muse WB, Bender RA. The nac (nitrogen assimilation control) gene from Escherichia coli. J Bacteriol. 1998;180(5):1166–73.
Bender RA. A NAC for regulating metabolism: the nitrogen assimilation control protein (NAC) from Klebsiella pneumoniae. J Bacteriol. 2010;192(19):4801–11.
Waterman SR, Small PL. Transcriptional expression of Escherichia coli glutamate-dependent acid resistance genes gadA and gadBC in an hns rpoS mutant. J Bacteriol. 2003;185(15):4644–7.
Castanie-Cornet MP, Foster JW. Escherichia coli acid resistance: cAMP receptor protein and a 20 bp cis-acting sequence control pH and stationary phase expression of the gadA and gadBC glutamate decarboxylase genes. Microbiology. 2001;147(Pt 3):709–15.
Yoshida T, Cai S, Inouye M. Interaction of EnvZ, a sensory histidine kinase, with phosphorylated OmpR, the cognate response regulator. Mol Microbiol. 2002;46(5):1283–94.
Yoshida T, Qin L, Egger LA, Inouye M. Transcription regulation of ompF and ompC by a single transcription factor, OmpR. J Biol Chem. 2006;281(25):17114–23.
Stincone A, Daudi N, Rahman AS, Antczak P, Henderson I, Cole J, Johnson MD, Lund P, Falciani F. A systems biology approach sheds new light on Escherichia coli acid resistance. Nucleic Acids Res. 2011;39(17):7512–28.
Yamamoto K, Hirao K, Oshima T, Aiba H, Utsumi R, Ishihama A. Functional characterization in vitro of all two-component signal transduction systems from Escherichia coli. J Biol Chem. 2005;280(2):1448–56.
Quinn HJ, Cameron AD, Dorman CJ. Bacterial regulon evolution: distinct responses and roles for the identical OmpR proteins of Salmonella Typhimurium and Escherichia coli in the acid stress response. PLoS Genet. 2014;10(3):e1004215.
Mitra A, Fay PA, Morgan JK, Vendura KW, Versaggi SL, Riordan JT. Sigma factor N, liaison to an ntrC and rpoS dependent regulatory pathway controlling acid resistance and the LEE in enterohemorrhagic Escherichia coli. PLoS One. 2012;7(9):e46288.
Riordan JT, Tietjen JA, Walsh CW, Gustafson JE, Whittam TS. Inactivation of alternative sigma factor 54 (RpoN) leads to increased acid resistance, and alters locus of enterocyte effacement (LEE) expression in Escherichia coli O157: H7. Microbiology. 2010;156(Pt 3):719–30.
Morgan JK, Vendura KW, Stevens Jr SM, Riordan JT. RcsB determines the locus of enterocyte effacement (LEE) expression and adherence phenotype of Escherichia coli O157: H7 spinach outbreak strain TW14359 and coordinates bicarbonate-dependent LEE activation with repression of motility. Microbiology. 2013;159(Pt 11):2342–53.
Shin S, Castanie-Cornet MP, Foster JW, Crawford JA, Brinkley C, Kaper JB. An activator of glutamate decarboxylase genes regulates the expression of enteropathogenic Escherichia coli virulence genes through control of the plasmid-encoded regulator, Per. Mol Microbiol. 2001;41(5):1133–50.
Koide T, Reiss DJ, Bare JC, Pang WL, Facciotti MT, Schmid AK, Pan M, Marzolf B, Van PT, Lo FY, et al. Prevalence of transcription promoters within archaeal operons and coding sequences. Mol Syst Biol. 2009;5:285.
Czaplewski LG, North AK, Smith MC, Baumberg S, Stockley PG. Purification and initial characterization of AhrC: the regulator of arginine metabolism genes in Bacillus subtilis. Mol Microbiol. 1992;6(2):267–75.
Mullin DA, Newton A. A sigma 54 promoter and downstream sequence elements ftr2 and ftr3 are required for regulated expression of divergent transcription units flaN and flbG in Caulobacter crescentus. J Bacteriol. 1993;175(7):2067–76.
Madan Babu M, Teichmann SA. Functional determinants of transcription factors in Escherichia coli: protein families and binding sites. Trends Genet. 2003;19(2):75–9.
Collado-Vides J, Magasanik B, Gralla JD. Control site location and transcriptional regulation in Escherichia coli. Microbiol Rev. 1991;55(3):371–94.
Acknowledgements
The authors gratefully acknowledge the support of Dan Harkness. We thank the Wadsworth Center Applied Genomic Technologies Core Facility for Sanger sequencing. We thank the Wadsworth Center Media Core Facility for liquid and solid media. We thank the Collins lab for providing the Keio strains.
Funding
This project was supported by grants from NIH (1R01GM117238) and NIH (R01) AI114648 to J.E.G. and J.T.W, and by a grant through the NIH Director’s New Innovator Award Program (1DP2OD007188) to J.T.W.
Availability of data and materials
Raw data files for all RNA-Seq and ChIP-Seq experiments are deposited in the NCBI Gene Expression Omnibus (GEO). Strains are available upon request.
Authors’ contributions
PA optimized the ChIP protocol, generated the E. coli clones and double KO strain, performed ChIP experiments, performed all acid challenge experiments, performed AR rescue experiments, wrote the paper and generated figures, performed analysis, and discussed results and implications; BH optimized the ChIP protocol, created E. coli clones and performed ChIP experiments; S. J. optimized the ChIP protocol, created E. coli clones, and performed ChIP experiments; A.L. analyzed ChIP-Seq sequence data; KH, JGC, IE, DH, LJ, MKS, AIS, JW, BGW, and WSW, performed ChIP experiments and performed acid challenge assays, and analyzed results; SLA, CB, SIB, DBB, DB, SC, DIC, JHC, KC, JC, DEG, DJG, JSG, CG, AKH, DVI, SRJ, JK, JL, HL, DL, QL, CPM, NM, SFM, CLM, JM, CSN, MP, HMP, AKR, DSR, NGR, JCS, HS, WRS, YS, MS, MMT, TT, NV, TEW, BHW, RY, AY, CY, YZ, and AJZ performed ChIP experiments and analyzed results; AMS performed ChIP-qPCR experiments; JWF helped plan experiments and discussed results and implications; JW discussed results and implications and edited the paper; SR optimized the ChIP protocol, oversaw ChIP experiments and discussed results and implications; NEB oversaw ChIP experiments, discussed results and implications, and edited the paper; W.W.W. oversaw the project, discussed results and implications, and edited the paper; and JEG conceived and oversaw project, wrote the paper and generated figures, performed analyses, and discussed results and implications. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable.
Author information
Authors and Affiliations
Corresponding author
Additional files
Additional file 1:
Supplementary text and materials of additional information presented in the paper. (DOCX 1478 kb)
Additional file 2:
Peaks called and bound and gene expression data for CsiR. (XLSX 410 kb)
Additional file 3:
Peaks called and bound and gene expression data for Nac. (XLSX 466 kb)
Additional file 4:
Peaks called for NtrC. (XLSX 11.1 kb)
Additional file 5:
Peaks called and gene expression data for ompR. (XLSX 28.0 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Aquino, P., Honda, B., Jaini, S. et al. Coordinated regulation of acid resistance in Escherichia coli . BMC Syst Biol 11, 1 (2017). https://doi.org/10.1186/s12918-016-0376-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12918-016-0376-y