
Abstract
Background
Heterosis breeding is the most useful method for yield increase around the globe. Heterosis is an intriguing process to develop superior offspring to either parent in the desired character. The biomass vigor produced during seedling emergence stage has a direct influence on yield heterosis in plants. Unfortunately, the genetic basis of early biomass vigor in cotton is poorly understood.
Results
Three stable performing F1 hybrids varying in yield heterosis named as high, medium and low hybrids with their inbred parents were used in this study. Phenotypically, these hybrids established noticeable biomass heterosis during the early stage of seedling growth in the field. Transcriptome analysis of root and leaf revealed that hybrids showed many differentially expressed genes (DEGs) relative to their parents, while the comparison of inbred parents showed limited number of DEGs indicating similarity in their genetic constitution. Further analysis indicated expression patterns of most DEGs were overdominant in both tissues of hybrids. According to GO results, functions of overdominance genes in leaf were enriched for chloroplast, membrane, and protein binding, whereas functions of overdominance genes in root were enriched for plasma membrane, extracellular region, and responses to stress. We found several genes of circadian rhythm pathway related to LATE ELONGATED HYPOCOTYL (LHY) showed downregulated overdominant expressions in both tissues of hybrids. In addition to circadian rhythm, several leaf genes related to Aux/IAA regulation, and many root genes involved in peroxidase activity also showed overdominant expressions in hybrids. Twelve genes involved in circadian rhythm plant were selected to perform qRT-PCR analysis to confirm the accuracy of RNA-seq results.
Conclusions
Through genome-wide comparative transcriptome analysis, we strongly predict that overdominance at gene expression level plays a pivotal role in early biomass vigor of hybrids. The combinational contribution of circadian rhythm and other metabolic process may control vigorous growth in hybrids. Our result provides an important foundation for dissecting molecular mechanisms of biomass vigor in hybrid cotton.
Similar content being viewed by others

Background
Cotton is the prime fiber crop, comprises of more than 50 species, and evolved around 10–20 million years ago (MYA) [1]. The upland cotton (Gossypium hirsutum L.) has been cultivated in tropical and temperate regions of the world, contributing 95% of natural textile fiber, and also a substantial source of edible oil. The global population is increasing at an alarming rate, putting agriculture sector under extreme pressure to ensure food security. Another major constraint for sustainable crop production is hasty climate change. To mitigate these challenges, plant breeders are working hard to increase yield and resistant against the pest, disease, biotic and abiotic stresses in crops. In this regard, a major breakthrough was achieved in rice, maize, sunflower, vegetables, and fruits through heterosis breeding. Hybrid rice produced through the utilization of heterosis gave 10–20% more yield than conventional varieties in China [2]. Hybrids of cotton are developed and cultivated mainly in China as well as in India. The development of hybrid cotton at commercial level was not started until the 1980s in China. Since then, hybrids have been harbored with Bt toxin gene as a result area of cultivation was increased after the mid-2000s with the advantage of more yield and resistance [3]. Manual crossing (emasculation and pollination) is one of the major hurdles causing the sluggish pace of hybrid cotton seed development. However, the cytoplasmic male sterile system has been proven an economic tool for commercial hybrid seed production in upland cotton in recent times [4, 5].
Heterosis breeding is not a modern day tool to increase crop yield. In the last century, people had learned crossed fertilized plant produced more output than the self-fertilized. However, the term heterosis was coined by Gorge. H Shull [6]. Heterosis/hybrid vigor is a biological phenomenon to produce superior offspring’s in characteristics of growth, biomass, stature, fertility, disease resistance, and stress management than either parent [7,8,9]. In contrast, inbreeding depression reveals that continuous self-fertilization declined the crop yield [10]. Allopolyploid cotton contains more than two sets of chromosomes. In spite of this, many researchers reported cotton has significant heterosis in yield contributing and fiber quality traits [11,12,13,14]. Interspecific crosses between upland and Pima cotton produce better fiber traits but disease incidence was a major problem. In contrast, intraspecific hybrids gave stability in production, improved fiber traits, and ensure seed purity [15]. In most cases, positive heterosis is required but for plant height, maturity, and gossypol content negative heterosis is desired in cotton.
The genetic basis of crop heterosis have been intensively researched almost for a century with different approaches. Many researchers tried to explain with the so-called hypothesis of dominance [16,17,18], overdominance [19,20,21], epistasis [22, 23], and genetic distance [24, 25]. Later on, many studies in agronomic crops provide strong evidence that the genetic basis of heterosis is perturbed. It varies with different species, stages, and traits [26,27,28]. Generally, crop hybrids show heterosis at two key stages of development e.g. vegetative and reproductive. The biomass produced in the vegetative stage controls the fate of final output, as it provides the energy basis for architecture development and adaptation to adverse conditions. Moreover, early growth advantages increased nutrients acquisition and competitiveness in plants. Leaf area, nodes, and vegetative branches are established during early stage of growth in cotton, and these eventually provide energy basis for the development of fruit branches, flower buds, and bolls. A previous study in cotton reported that during reproductive stage any disturbance in net assimilates can disturb the source and sink direction and finally affect the fiber quality traits [29]. To be concise, any pitfall in vegetative growth directly affects the final output in agronomic crops. Many studies were performed to investigate the molecular basis of vegetative or biomass heterosis in hybrids of rice [30, 31], maize [32, 33], wheat [34], and Brassica [35]. But, less is conducted in cotton. Now, the whole genome sequence of upland cotton is available online to the researchers [36, 37]. Furthermore, high-throughput sequencing technologies have enabled researchers to investigate the molecular mechanism of crop heterosis as a routine task [38, 39].
We designed this study to understand the genetic basis of early biomass vigor in intraspecific hybrid cotton. Most of past studies focused on a single hybrid and both parents to analyze gene expression differences. However, we sequenced root and leaf tissues of three contrasting hybrids together with their inbred parents for better understanding the genetic aspects. Through genome-wide comparative transcriptome analysis, we aimed to identify DEGs, gene expression patterns, and overview the biological pathways that mediate early biomass vigor in cotton. Our result provides a foundation to understand the preliminary biological basis of biomass heterosis. Furthermore, these data resources will be important to find candidate genes of biomass vigor in hybrid cotton in the future.
Results
Biomass heterosis exist in contrasting hybrids at the seedling stage
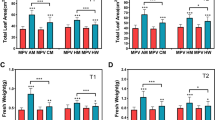
Three contrasting hybrids having high (H), medium (M), and low (L) parent heterosis in yield traits together with their four inbred parents (denoted as A, B, C, and D) were used to investigate the early biomass vigor in the field. We observed high and medium hybrids did not show statistically significant difference in fresh biomass (g/plant) and dry biomass (g/plant) relative to their mid-parent values (MPV) at 10 days after emergence of seedling (DAE) and 20 DAE (Additional file 25: Figure S1 and Additional file 26: Figure S2). However, high hybrid showed highly significant difference in fresh biomass compared with its MPV at 30 DAE (Fig. 1). According to the results, low hybrid showed significant difference in fresh biomass compared with its MPV at 10 DAE (Additional file 25: Figure S1). Similar results were obtained at 20 DAE (Additional file 26: Figure S2) and 30 DAE (Fig. 1). High hybrid showed more biomass accumulation and low hybrid showed decreased biomass accumulation compared to their parents. However, medium hybrid did not show any difference in biomass accumulation compared with both parents. These results indicate biomass heterosis is established in hybrids as compared to their parents during early seedling growth.

Phenotypic heterosis observed in F1 hybrids at 30 days after emergence of seedling (DAE). a Fresh biomass observed in hybrids relative to their mid-parent value (MPV). Here ** is used for significant difference at p < 0.01 and * at p < 0.05. b Dry biomass observed in hybrids compared with their MPV
Transcriptome sequencing, mapping and global expressions in root and leaf
Three contrasting F1 hybrids and their four inbred parents were used to perform RNA sequencing in this study. Root and leaf tissues of the same plant with three biological replicates were used to build 42 cDNA libraries for RNA Illumina sequencing. The brief detail of sequencing, mapping and gene expressions of the individual library is presented in Additional file 1. The overall sequencing means of valid reads was 98.8% with a value of 90% exon region mapping. The value of Q20% was 99.6% in our sequencing results. In the root of A, B, C, D, H, M, and L genotypes, mean of valid reads was approximately 51, 54, 50, 52, 43, 46, and 45 million, respectively (Table 1). The mean of valid reads in the leaf of A, B, C, D, H, M, and L genotypes was ~ 50, 49, 42, 44, 45, 43, and 45 million, respectively. On an average, more than 89% valid reads were mapped to G. hirsutum TM-1 reference genome in this sequencing. However, the mapped percentage in root samples was lower than leaf. The mean of multi-mapped and splice reads was more than 28 and 31%, respectively. In this study, the root tissue of each genotype showed higher number of total expressed genes compared to leaf. For example, approximately 77 thousand genes were expressed in root of A, while it was ~ 75 thousand in leaf (Additional file 1). We also found total numbers of expressed genes in both tissues of F1 hybrids were higher than the inbred parents (Additional file 1).
Differentially expressed genes in root and leaf transcriptome
Here, we analyzed the dynamic changes of root and leaf transcriptome in all F1 hybrids and their inbred parents. The expression levels significantly different at p ≤ 0.05 and with log2 (fold change) > 1 or log2 (fold change) < − 1 designated as differentially expressed genes in each hybrid parent triad. The total number of DEGs (Up + down) among each hybrid parent triad is represented in Fig. 2a. The comparative analysis in the root of hybrid (H) compared with both parents revealed H with maternal parent (A) and paternal parent (B) respectively showed 2917 and 2828 total number of DEGs (Fig. 2a: Additional file 2). The comparison of both parents ARvsBR showed 1154 total number of DEGs. In the leaf transcriptome of H, the comparison of ALvsHL showed 3807 total number of DEGs, whereas the comparison of BLvsHL showed a total of 2797 DEGs (Fig. 2a: Additional file 3). Furthermore, the comparison of both parents ALvsBL showed 1013 total number of DEGs. Distribution of DEGs in H-hybrid revealed major portion of genes was unique and less was common in each tissue (Fig. 2b and c). For instance, 2631 genes in roots and 2993 in leaf were unique in the combination of H with A. Unique DEGs in comparison of B and H was 2648 in root and 1974 in leaf. A large portion of differential gene expression is probably major determinant of better performance in high hybrid.

Total DEGs and their distribution in root and leaf of each hybrid parent triad. a shows total number of up and down regulated DEGs. Here, R: root, L: leaf, A: maternal parent, and B, C, D represents three paternal parents of high (H), medium (M), and low (L) hybrids, respectively. b in root c in leaf represents distribution of unique and common DEGs in high hybrid parent triad. Similarly, d in root e in leaf shows distribution of unique and common DEGs in medium hybrid parent triad. f in root g in leaf represents unique and common DEGs distribution in low hybrid parent triad
The root transcriptome of medium hybrid (M) revealed comparison of M with maternal parent (A) and paternal parent (C), respectively showed 2577 and 2144 total number of DEGs (Fig. 2a: Additional file 4). It was observed the comparison of ALvsML had 3910 total number of DEGs in the leaf of M hybrid, whereas the comparison of CLvsML showed 2170 total number of DEGs (Fig. 2a: Additional file 5). Moreover, the combination of both parents had 1067 DEGs in root and 1586 in leaf. Although results of unique and common DEGs distribution in M hybrid identified more unique but less common genes. However, unique DEGs were more in the comparison of M with A than with C (Fig. 2d and e). The comparative analysis revealed total number of DEGs between low hybrid (L) and its maternal parent (A) was 3580 in the root (Fig. 2a: Additional file 6). The combination of L and its paternal parent (D) had 3215 total number of DEGs. Furthermore, the comparison of ALvsLL and DLvsLL, respectively showed 5827 and 2623 total number of DEGs (Fig. 2a: Additional file 7). Distribution of DEGs exposed that the majority of genes were unique, whereas less was overlap (Fig. 2f and g). For example, A versus L had 2647 unique DEGs in root and 4795 in leaf. Unique DEGs in DvsL were 2413 in root and 1714 in leaf. More interestingly, both parents of low hybrid had higher genetic differences among each other than those of medium and high hybrids. Overall, the result of comparative analysis of DEGs between hybrids and parents revealed that hybrids had different genomic constituent relative to their parents. However, comparison of both parents indicated that they have few genetic differences among them.
F1 hybrids exhibited overdominant gene expressions
Allopolyploids have been found to exhibit expression level dominance [35, 40]. So, to address the magnitude and directionality of expressions in interspecific F1 upland cotton hybrids, DEGs of root and leaf transcriptome were divided into 12 possible groups as described by Rapp et al., [41]. Genes in groups 1–2 showed additive expression. The expression of genes in groups 3–4 was termed as male expression level dominance (M-ELD), wherein the expression of genes in groups 5–6 was named as female expression level dominance (F-ELD). The expression of genes in groups 7–9 called downregulated overdominance, wherein the expression of genes in groups 10–12 was named as upregulated overdominance (Fig. 3a). The result of expression patterns analysis in high hybrid (H) in both tissues detected limited number of genes was additive expressions. Male and female parent like expression groups also had few genes. However, it was found overdominant upregulated (group12) and downregulated (group 8) groups had the highest number of genes in both tissues (Fig. 3b: Additional file 8). The analysis results of medium hybrid (M) also dissected overdominant up and downregulated groups had the highest number of genes in both tissues (Fig. 3c: Additional file 9). The analysis result of low hybrid (L) was also similar to H and M hybrids (Fig. 3d: Additional file 10). However, groups of parent-like expression also had many genes in L hybrid as compared to H and M hybrids. The result of expression patterns analysis directed that overdominance at the gene expression level contributes to early biomass vigor in hybrid cotton.

The 12 possible gene expression patterns in F1 hybrids compared to their parents in root and leaf. a Expression patterns of 12 groups, M: male parent, F1: hybrid, and F: female parent. b Total number of genes in each group in root and leaf of high (H) hybrid. c Total number of genes in each group in root and leaf of medium (M) hybrid. d Total number of genes in each group in root and leaf of low (L) hybrid
Functional annotations of overdominant genes
To understand the functions of genes with overdominant expressions in biomass heterosis, GO and KEGG enrichment analysis was implemented in root and leaf of hybrids. GO enrichment analysis (p-value < 0.01) of overdominant genes in root of high hybrid (H) relative to its parents revealed most of the upregulated genes were involved in functions related to plasma membrane, regulation of transcription/DNA-template, and extracellular region. Conversely, downregulated genes were enriched in plasmodesma, integral component of plasma membrane, and vacuole (Additional file 27: Figure S3a, Additional file 11). KEGG pathway enrichment analysis (p-value < 0.05) of overdominant genes showed most of the upregulated genes were enriched in porphyrin & chlorophyll metabolism, phenylpropanoid biosynthesis, and oxidative phosphorylation pathways (Fig. 4a: Additional file 12). But, the majority of downregulated genes were enriched in starch & sucrose metabolism, phenylpropanoid biosynthesis, and circadian rhythm plant (Fig. 4b: Additional file 12). In the leaf of H hybrid, most of the overdominant upregulated genes were involved in functions of plasma membrane, protein serine/threonine kinase activity, and plasmodesma, while downregulated genes were involved in cellular functions of chloroplast, membrane, and extracellular region (Additional file 27: Figure S3a: Additional file 13). Enriched pathways of overdominant genes found upregulated genes were enriched in plant-pathogen interaction, plant hormone signal transduction, and circadian rhythm plant (Fig. 4a: Additional file 14). In contrast, significant pathways for downregulated genes were plant hormone signal transduction, carbon metabolism, and circadian rhythm plant (Fig. 4b: Additional file 14).

Enriched pathways of overdominant DEGs in root and leaf of F1 hybrids. H: High, M: Medium, and L: Low a Pathways of upregulated overdominant DEGs. b Pathways of downregulated overdominant DEGs
The results of GO enrichment analysis of overdominant genes in the root of medium hybrid (M) compared with its parents uncovered that upregulated genes were enriched in plasma membrane, extracellular region and oxidation-reduction process. In contrast, downregulated genes were involved in following functions e.g. integral component of the plasma membrane and response to salt stress (Additional file 27: Figure S3b: Additional file 15). Pathway enrichment analysis of overdominant genes found upregulated genes were enriched in phenylpropanoid biosynthesis, amino & nucleotide sugar metabolism, and porphyrin & chlorophyll metabolism (Fig. 4a: Additional file 16). For downregulated genes, enriched pathways were circadian rhythm plant, glycosaminoglycan degradation, and ascorbate and aldarate metabolism (Fig. 4b: Additional file 16). GO enrichment analysis of overdominant genes in leaf of M hybrid revealed most of upregulated genes performed functions related to chloroplast, chloroplast stroma, and plastid. However, downregulated genes involved in the following functions such as chloroplast, protein binding, and ATP binding (Additional file 27: Figure S3b: Additional file 17). Pathway enrichment analysis of overdominant genes revealed most of upregulated genes were found in carbon metabolism, phagosome, and circadian rhythm plant (Fig. 4a: Additional file 18). On the other hand, downregulated genes were enriched in planted hormone signal transduction, endocytosis, and carbon metabolism (Fig. 4b: Additional file 18).
GO enrichment analysis of overdominant genes in the root of low hybrid (L) compared with its parents found following enriched terms e.g. plasma membrane, extracellular region, and oxidation-reduction process. The functions of downregulated genes were enriched for chloroplast, regulation of transcription/DNA-template, and transcription factor activity/sequence-specific DNA binding (Additional file 27: Figure S3c: Additional file 19). Enriched pathways of overdominant upregulated genes were phenylpropanoid biosynthesis, porphyrin & chlorophyll metabolism, and pentose & glucuronate interconversions (Fig. 4a: Additional file 20). On the other hand, most of downregulated genes were enriched in circadian rhythm plant, carbon metabolism, and starch & sucrose metabolism (Fig. 4b). In the leaf of L hybrid, GO enrichment analysis of overdominant genes showed functions of upregulated genes were enriched in chloroplast, protein folding, and embryo development ending in seed. In contrast, majority of the downregulated genes were involved in cellular function of chloroplast, membrane, and chloroplast stroma (Additional file 27: Figure S3c: Additional file 21). The pathway enrichment analysis of overdominant genes uncovered majority of upregulated genes were involved in protein processing in the endoplasmic reticulum, spliceosome, and circadian rhythm plant (Fig. 4a: Additional file 22). For downregulated genes, enriched pathways were carbon metabolism, plant hormone signal transduction, and circadian rhythm plant (Fig. 4b: Additional file 22).
The comparison of overdominance genes between contrasting hybrids
The results of KEGG revealed that most of the overdominant genes of high, medium and low hybrids relative to their parents were enriched in similar pathways. We performed comparison analysis between high, medium and low hybrids to see unique and common overdominant expressed genes in the above mentioned pathways. The results showed that 44 genes in leaf and 29 genes in root were common in comparison of all hybrids (Additional file 28: Figure S4: Additional file 23). On the other hand, many genes were also overlapped between comparisons of two hybrids. The reason behind these results might be the common female parent in hybrids. The expression level of genes that showed overdominant expression in all hybrids relative to their inbred parents is presented in Fig. 5. We found most of root genes were belonging to circadian rhythm, phenylpropanoid biosynthesis, and porphyrin & chlorophyll metabolism, while leaf genes were enriched in circadian rhythm, plant hormone signal transduction, and carbon metabolism. Majority of these genes were involved in functions related to DNA binding, oxidation-reduction process, heme binding, and response to oxidative stress (Additional file 29: Figure S5). We presumed phenomenal changes in these pathways associated genes may be transformed biomass vigor in hybrids of cotton.

Expression heatmap of all common overdominance genes of F1 hybrids. H: High, M: Medium, L: Low, A, B, C, and D represent four inbred parents
The circadian rhythm, metabolic processes, and biomass vigor
Through compression analysis between hybrids, we found circadian rhythm plant pathway contained many overdominant genes in root and leaf of all hybrids. Genes in this pathway had tissue-specific expression except of three (Gh_A12G1061, Gh_A12G1062, and Gh_D12G1185). In this pathway, six genes (Gh_A11G0926, Gh_A12G1061, Gh_A12G1062, Gh_D12G1185, Gh_D12G1184, and Gh_D11G1068) related to MYB domain transcription factor, encoding LHY protein showed downregulation in root of hybrids (Additional file 30: Figure S6). In contrast, five genes (Gh_A12G1061, Gh_A12G1062, Gh_D12G1185, Gh_A09G1504, and Gh_D09G1515) encoding LHY protein showed downregulated expression in leaf of hybrids. The results indicated four genes (Gh_A08G0451, Gh_D01G0200, Gh_D07G0867, and Gh_D11G1518) associated with transcript factor CO-like showed upregulation in leaf of hybrids (Additional file 30: Figure S6). Genes (Gh_A05G0944 and Gh_D05G1029) named as CIA2 (CHLOROPLAST IMPORT APPARATUS 2) associated with PSEUDO-RESPONSE REGULATOR9 (PRR9) also showed similar results in leaf of hybrids (Additional file 29: Figure S5). In addition to circadian rhythm, several genes (Gh_A03G0944, Gh_A09G1415, Gh_A11G1859, Gh_A12G1915, and Gh_D02G1327) in phenylpropanoid biosynthesis related to peroxidase superfamily protein, and many genes from porphyrin &chlorophyll metabolism showed upregulated expressions in root of hybrids. Furthermore, many genes of plant hormone signal transduction pathways linked to AUX/IAA transcriptional regulator family protein and genes from carbon metabolism pathway also found with downregulated expressions in leaf of hybrids. To be concise, all these results suggest genes in circadian rhythm together with other metabolic process performed overdominance that might change root and leaf growth, and ultimately lead to altered biomass vigor in hybrid cotton.
qRT-PCR analysis
Twelve overdominant genes from circadian rhythm plant were selected to validate RNA-seq data by real-time qRT- PCR analysis. All selected genes had overdominance expression in high, medium, and low hybrids relative to their parents in RNA-seq results. We selected five genes (Gh_A12G1061, Gh_A12G1062, Gh_D12G1184, Gh_D11G1068, and Gh_D13G1939) from root and one gene (Gh_A09G1504) from leaf with downregulated expressions in hybrids, while six genes (Gh_A08G0451, Gh_D01G0200, Gh_D11G1518, Gh_D05G1029 Gh_D07G0867, Gh_A05G0944, and Gh_D12G1525) were selected from leaf with upregulated expressions in hybrids. It was found that the expression trends as calculated by qRT-PCR were consistent with RNA-seq data, thus confirming the accuracy of the RNA-seq results in this study (Fig. 6).

qRT-PCR of 12 DEGs involved in the circadian rhythm pathway and showed overdominance performance in all hybrids relative to their parents. In each figure, H: High, M: Medium, L: Low and A, B, C, and D represents four inbred parents. First five genes represent expression in root, while later genes represent expression in leaf
Discussion
Overview of early biomass vigor and transcriptome sequence
Heterosis refers to the superiority of offspring in the desired character or traits than either parent. Hybrid breeding effectively utilized the magic of heterosis to produce better quality and quantity of crops worldwide. Both intraspecific and interspecific crosses of cotton showed heterosis in yield contributing traits. However, success of significant heterosis in intraspecific crosses of upland cotton is not an easy task for breeders. Despite different complexities, our team has successfully developed nine upland cotton hybrids. To be honest, researchers are working hard to resolve the complex genetic basis of crop heterosis. Still, a lot of efforts are required particularly in the complex genome of tetraploid cotton. The biomass produced during the early stages of growth plays significant role in yield increments in plants. Keeping this in view, we performed this comprehensive study to investigate the genetic basis of early biomass heterosis in hybrid cotton by comparative transcriptome analysis of contrasting hybrids and their inbred parents.
In this study, three intraspecific upland cotton hybrids varying in yield heterosis named as high (H), medium (M) and low (L) hybrids and their inbred parents were used to investigate early biomass heterosis. According to the results of the field evaluation, these hybrids showed phenotypic heterosis in biomass accumulation than both parents during the early stage of seedlings growth. More biomass accumulation in high hybrid possibly related to more efficiency in nutrient uptake and photosynthetic process. Moreover, early growth advantages seem to be important to achieve heterosis in other traits. A previous research found intraspecific hybrid plants produced extreme phenotypes than both parents in the diversity of traits such as morphology, physiology, and biochemical attributes of tissues or organs [42]. A study in rice measuring the seedling traits observed that hybrids showed mid and better parent heterosis in all seedling traits expect height [30]. Wheat hybrids showed strong hybrid vigor compared to both inbred lines during the seedling stage [34]. Increased cotyledon size was recorded even a few days after sowing in Arabidopsis hybrids [43].
As a genetic perspective, the root and leaf transcriptome sequencing was performed to generate large scale cDNA sequence data and profile transcriptome changes in contrasting hybrids and their inbred parents. The trancriptomic data uncovered that total number of expressed genes were somehow higher in hybrids than all parents especially in root. More expressed genes in hybrids may have a relationship with an altered role in the diversity of key functions including anchorage the plant, absorption of water and nutrients, photosynthetic process, and many other metabolic processes. Root arises first from radical, so more expressed genes may be having a correlation with more functional changes during the early stage. The root vigor during early stages of seedling growth has been documented in hybrids of maize and wheat [44, 45]. In this study, comparative analysis among hybrids and parents showed many DEGs in hybrids compared with either parent in both root and leaf tissues. However, comparisons of parents had only limited number of DEGs, thus indicating similarity in their genomic constituent. These results predict crossing of genetically similar inbred lines leads to changes in the genetic organization of hybrids. At the early stages of growth, these changes caused differential expression of genes as a consequence biomass heterosis has witnessed in hybrids. The results of many recent studies reported differences in genomic expressions between hybrids and parental lines have a direct relationship with the level of heterosis [46, 47]. Therefore, superior performance is supposed to be the result of global differences of gene expressions between homozygous inbred lines and heterozygous hybrids.
Overdominant gene expressions mediate early biomass vigor in cotton
To assess the gene expression patterns in hybrids compared with parents, the expressions of DEGs in F1 hybrids are statistically divided into 12 groups. We observed expression of few genes was additive in both tissues of hybrids. The male or female parent like expression groups also had few numbers of genes. In contrast to our results, it was stated in cotton that synthetic and natural allopolyploids showed expression level dominance and follow the expression patterns of one of two diploids parents [41, 48]. However, Yoo et al., [40] suggested that these changes became more balanced in natural allopolyploid cotton on evolutionary timescale by higher rates of novel gene expression patterns as well as homoeolog silencing. They also found domesticated cotton had more novel expressed genes than its wild mate. Therefore, provides striking evidence that strong artificial selection leads to new gene expression patterns in crops. In our study, the expression patterns of most DEGs were overdominant in both tissues of contrasting hybrids. These results demonstrated that there are higher expressions of heterozygous alleles in the hybrids that altered the biomass vigor in hybrids relative to their parents. Previously, Zhao et al., [49] researched the mechanism of cotton heterosis during four different growing and developmental stages. They observed quantitative and qualitative gene expressions between hybrids and parents and suggested overdominant gene expressions had a positive influence on heterosis. Incomplete dominance of deleterious alleles contributes extensively to trait variation and heterosis in maize [50]. In rice, a study to dissect the genetic basis of 38 agronomic traits concluded numerous superior alleles with positive dominance effect caused heterotic phenotypes [51]. However, a recent study reported allelic specific expression or imbalance expression of two parental alleles in hybrids caused heterosis in rice [52].
Circadian clock and metabolic genes play critical role in early biomass vigor of cotton
To a particular interest, the functions of most of the overdominant genes in hybrids were enriched to chloroplast, protein binding, plasma membrane, extracellular region, and responses to stress. The result of KEGG analysis exposed most of the overdominant genes of contrasting hybrids relative to their parents were enriched in similar pathways. These pathways were circadian rhythm, phenylpropanoid biosynthesis, porphyrin & chlorophyll metabolism, plant hormone signal transduction, and carbon metabolism. Interestingly, many genes involved in circadian rhythm plant had overdominant expressions in hybrids in both root and leaf. qRT-PCR results also verify this (Fig. 6). Circadian clock controls biological processes of plants through transcriptional and post transcriptional regulation of many output processes [53, 54]. For example, genes expression changes in circadian rhythm have an influence on photosynthesis, carbohydrate, and accumulation of chlorophyll, starch and sugar. Through repression facilitated by evening loop protein complex, this clock also regulates hypocotyl growth and flowering in plants [55]. Many genes in our study related to MYB domain transcription factor, LHY encoding protein showed downregulated expressions in both tissues of hybrids. In contrast, two genes named CIA2 had upregulated expressions in leaf. These two genes are associated with morning loop protein complex known as PRR9. These results predict that changes in circadian clock-associated genes might lead to the more rapid growth of root and leaf in hybrids compared with parents.
It is a well-known fact that in the morning-phased loop, LHY and CCA1 (CIRCADIAN CLOCK ASSOCIATED 1) activate the expression of PSEUDO-RESPONSE REGULATOR 7 (PRR7) and PRR9 genes to maintain clock length and amplitude in plants [56]. Ni et al., [57] reported that during the day, epigenetic repression of CCA1 and LHY induced the expression of TIMING OF CAB EXPRESSION1 (TOC1), GIGANTEA (GI), and downstream genes that contain evening elements. These caused changes in circadian-mediated physiological and metabolic pathways. As a result, growth and biomass vigor were produced in allotetraploids and F1 hybrids of Arabidopsis. Overexpression of ZmCCA1b in maize disturbed circadian rhythms resulting in decreased chlorophyll content, plant height, and node elongation of stems thus, biomass heterosis was altered [58]. In our study, four genes associated with transcript factor CO-like showed upregulated expressions in the leaf of hybrids. These genes are believed to be involved in flowering time in response to daylight. Light responsive circadian regulators switch vegetative growth to flowering and maybe this resulting in different level of biomass vigor in hybrids. Such type of regulator in rice named as Ghd7 affects plant height, heading date, and final yield [59]. Furthermore, CO-like genes control flowering in response to day length in Arabidopsis [60, 61]. According to our results, clock associated genes showed differential expression in a tissue-specific way as these were not common between root and leaf. Previous experimentation in Arabidopsis reported roots and shoots circadian clock is clearly different from each other, therefore plant clock is organ specific [62].
In addition to circadian rhythm, other pathways, such as plant hormone signal transduction, carbon metabolism, phenylpropanoid biosynthesis, and porphyrin & chlorophyll metabolism may also contribute to biomass vigor in cotton. As, altered expression of circadian rhythm genes also disturbs the other biological pathways such as phytohormone signaling, stress response, and energy metabolism [63]. We found many genes of AUX/IAA transcriptional regulator family protein with downregulated expressions in leaf of hybrids. Auxin hormone plays a diversity of functions in plants. It regulates leaf initiation, leaf shape, and shoots apical meristems. Genes involved in IAA biosynthesis showed decreased expression in B. napus hybrids and play role in hybrid vigor [27]. According to a study in Arabidopsis, the gene activity of IAA was frequently increased in hybrids that promote leaf cell numbers [64]. Furthermore, many genes of peroxidase superfamily protein showed upregulated expression in root of hybrids in our results. We assume changes in these genes may be contributing to root growth in hybrids. Peroxidases are thought to be involved in a variety of physiological functions in plants e.g. lignification, suberization, cross-linking of cell wall proteins, and wound healing. A previous study has shown peroxidase participates in auxin metabolism [65]. Cell wall related peroxidase effect the root elongation in Arabidopsis [66]. Taken together all, our result predict genes involved in circadian rhythm and other metabolic processes showed overdominance in hybrids resulting in altered biomass vigor in cotton.
Conclusions
It is notable that F1 hybrids of upland cotton showed biomass vigor during the early stages of vegetative growth. The genome-wide comparative transcriptome study of hybrids and inbred lines determined many DEGs compared to either parent in root and leaf of hybrids. Further analysis indicated expression pattern of most DEGs was overdominant in both tissues of hybrids. These results strongly suggest at expression level overdominance mediate early biomass vigor in hybrids of cotton. Phenomenal changes in genes of circadian rhythm and other metabolic processes may be enabled vigorous growth of hybrids relative to their parents. However, how overdominant expressed genes involved in these pathways lead to the different level of biomass heterosis in hybrids needed further investigation. Our comprehensive study gave new insights to understand the preliminary molecular mechanism of biomass heterosis. Our results and large scale data will be potential source for further molecular research on biomass vigor in hybrid cotton.
Methods
Plant materials and estimation of early biomass heterosis
In 2015, our research group crossed five maternal and six paternal inbred lines to produce 30 intrapecific upland cotton hybrids with North Carolina mating design II (NCII). After two-year field experimentation at three locations, the level of heterosis for boll number per plant, boll weight, lint percentage, lint yield, and seed cotton yield were investigated in hybrids and parents. Results can be seen in our previous study [67]. According to results of the field test, when one maternal inbred line named SJ48 (denoted as A) crossed with three paternal inbred lines viz. Z98–15 (B), 851–2 (C), and DT-8 (D), these three crosses constantly produced high, medium, and low heterosis in all yield contributing traits, respectively. We selected these three stable performing F1 hybrids (H, M, and L) and their inbred parents for biomass study. In 2018, all seven genotypes were planted at the east cotton breeding farm of Cotton Research Institute of the Chinese Academy of Agricultural Science, Anyang, China. Randomized complete block design with three replications was used to arrange plant materials. Seeds were sown in late April. To overview phenotypic biomass heterosis, we harvested five plants and consider each plant as a replicate from each genotype on 10 days after emergence of seedling (DAE), 20 DAE, and 30 DAE. Fresh and dry biomass were calculated for each genotype on these intervals. Fresh biomass of the whole plant was measured immediately after harvesting. The dry biomass of the whole plant was calculated after overnight oven drying at 100 °C.
RNA extraction, Illumina sequencing, and data analysis
For transcriptomic analysis, total RNA was extracted from each genotype. For RNA extraction, seedlings were raised in standard Hoagland solution in the lab. When true leaves were fully expanded then root and leaf tissue of the same plant was harvested in three biological replications. All harvested samples were instantly placed in liquid nitrogen and stored at − 80 °C before use. Total RNA was isolated using the Sigma Spectrum Plant Total RNA kit (Sigma-Aldrich, USA) according to the manufacturer’s protocol. The total RNA quantity and purity were analyzed on Bioanalyzer 2100 and RNA 6000 Nano LabChip Kit (Agilent, CA, USA) with RIN number > 7.0. Then the cleaved RNA fragments were reverse-transcribed to create the 42 individual final cDNA libraries (21 for root and 21 for leaf tissue for each of the seven genotypes) in accordance with the protocol for the mRNA-Seq sample preparation kit (Illumina, San Diego, USA), the average insert size for the paired-end libraries was 300 bp (±50 bp). The paired-end sequencing on an Illumina Hiseq 4000 was performed at the (LC-bio, China) following the vendor’s recommended protocol. Before assembly, reads that contained adaptor contamination, undetermined bases, and low-quality bases were removed. HISAT2 [68] was used to map reads to the genome of (http://mascotton.njau.edu.cn/info/1054/1118.htm). The mapped reads of each sample were assembled using StringTie [69]. The final transcriptome was generated using Perl scripts. StringTie [69] and Ballgown [70] were used to calculate the expression levels of all transcripts. StringTie [69] was used to perform expression level for mRNAs by calculating FPKM. The differentially expressed mRNAs were selected with log2 (fold change) > 1 or log2 (fold change) < − 1 and with statistical significance (p-value < 0.05) by R package Ballgown [70]. The expression level dominance analysis was performed by using EdgeR software as described by Rapp et al., [41]. GO enrichment and KEGG pathway enrichment analysis was performed on the DEGs using the Goatools software (https://github.com/tanghaibao/Goatools) and KOBAS software (http://kobas.cbi.pku.edu.cn) [71], with a corrected p-value ≤0.05 as the threshold and rich factor.
qRT-PCR analysis
For, quantitative real-time RT-PCR, we used the same total RNA to generate first standard cDNA that was used in RNA sequencing. PrimerScript RT Reagent Kit (Perfect Real Time, TaKaRa, Japan) was used to generate cDNA for each replication. Primers for qPCR were designed using the Oligo7 software, synthesized commercially (Biotechnology, Beijing, China), and are shown in Additional file 24. Protocol of qRT-PCR was same as described in our previous study [4]. Three biological replicates were used for each sample and the actin gene was used for normalization. Gene relative expression levels were calculated using the 2−ΔΔCt method.
Availability of data and materials
The datasets generated or analyzed in this study are freely available with accession number GSE144676 at https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE144676.
Abbreviations
- CCA1:
-
Circadian clock associated1
- CO-LIKE:
-
Constans-like
- DAE:
-
Days after emergence
- DEGs:
-
Differentially expressed genes
- F-ELD:
-
Female expression level dominance;
- H:
-
High
- L:
-
Low
- LHY:
-
Late elongated hypocotyl
- M:
-
Medium
- M-ELD:
-
Male expression level dominance
- MPV:
-
Mid parent value
- PRR9:
-
Pseudo-response regulator 9
References
Seelanan T, Schnabel A, Wendel JF. Congruence and consensus in the cotton tribe (Malvaceae). Syst Bot. 1997;1:259–90.
Cheng SH, Zhuang JY, Fan YY, Du JH, Cao LY. Progress in research and development on hybrid rice: a super-domesticate in China. Ann Bot. 2007;100(5):959–66.
Zhang J, Wu M, Yu J, Li X, Pei W. Breeding potential of introgression lines developed from interspecific crossing between upland cotton (Gossypium hirsutum) and Gossypium barbadense: heterosis, combining ability and genetic effects. PLoS One. 2016;11(1):e0143646.
Wu J, Zhang M, Zhang B, Zhang X, Guo L, Qi T, Wang H, Zhang J, Xing C. Genome-wide comparative transcriptome analysis of CMS-D2 and its maintainer and restorer lines in upland cotton. BMC Genomics. 2017;18(1):454.
Zhang B, Zhang X, Liu G, Guo L, Qi T, Zhang M, Li X, Wang H, Tang H, Qiao X. A combined small RNA and transcriptome sequencing analysis reveal regulatory roles of miRNAs during anther development of upland cotton carrying cytoplasmic male sterile Gossypium harknessii (D2) cytoplasm. BMC Plant Biol. 2018;18(1):242.
Shull GH. The composition of a field of maize. J Hered. 1908;1:296–301.
Birchler JA, Auger DL, Riddle NC. In search of the molecular basis of heterosis. Plant Cell. 2003;15(10):2236–9.
Fu D, Xiao M, Hayward A, Jiang G, Zhu L, Zhou Q, Li J, Zhang M. What is crop heterosis: new insights into an old topic. J Appl Genet. 2015;56(1):1–13.
Schnable PS, Springer NM. Progress toward understanding heterosis in crop plants. Annu Rev Plant Biol. 2013;64:71–88.
Lewers K, St Martin S, Hedges B, Palmer R. Testcross evaluation of soybean germplasm. Crop Sci. 1998;38(5):1143–9.
Wu Y, Yin J, Guo W, Zhu X, Zhang T. Heterosis performance of yield and fibre quality in F1 and F2 hybrids in upland cotton. Plant Breed. 2004;123(3):285–9.
Rauf S, Khan TM, Nazir S. Combining ability and heterosis in Gossypium hirsutum L. Int J Agri Biol. 2005;7(1):109–13.
Ali I, Shakeel A, Saeed A, Nazeer W, Zia Z, Ahmad S, Mahmood K, Malik W. Combining ability analysis and heterotic studies for within-boll yield components and fibre quality in cotton. J Anim Plant Sci. 2016;26(1):156–62.
Khan NU, Hassan G, Kumbhar MB, Kang S, Khan I, Parveen A. Heterosis and inbreeding depression and mean performance in segregation generations in upland cotton. Edit Adv Board E. 2005;17(4):531–46.
Munir S, Hussain SB, Manzoor H, Quereshi MK, Zubair M, Nouman W, Shehzad AN, Rasul S, Manzoor SA. Heterosis and correlation in interspecific and intraspecific hybrids of cotton. Genet Mol Res. 2016;15:2.
Xiao J, Li J, Yuan L, Tanksley SD. Dominance is the major genetic basis of heterosis in rice as revealed by QTL analysis using molecular markers. Genetics. 1995;140(2):745–54.
Stuber CW, Lincoln SE, Wolff D, Helentjaris T, Lander E. Identification of genetic factors contributing to heterosis in a hybrid from two elite maize inbred lines using molecular markers. Genetics. 1992;132(3):823–39.
Davenport CB. Degeneration, albinism and inbreeding. Science. 1908;28(718):454–5.
Li Z-K, Luo L, Mei H, Wang D, Shu Q, Tabien R, Zhong D, Ying C, Stansel J, Khush G. Overdominant epistatic loci are the primary genetic basis of inbreeding depression and heterosis in rice. I. Biomass and grain yield. Genetics. 2001;158(4):1737–53.
Tian M, Nie Q, Li Z, Zhang J, Liu Y, Long Y, Wang Z, Wang G, Liu R. Transcriptomic analysis reveals overdominance playing a critical role in nicotine heterosis in Nicotiana tabacum L. BMC Plant Biol. 2018;18(1):48.
East EM. Heterosis. Genetics. 1936;21(4):375.
Yu S, Li J, Xu C, Tan Y, Gao Y, Li X, Zhang Q, Maroof MS. Importance of epistasis as the genetic basis of heterosis in an elite rice hybrid. Proc Natl Acad Sci. 1997;94(17):9226–31.
Williams W. Heterosis and the genetics of complex characters. Nature. 1959;184(4685):527.
Zhang J, Abdelraheem A, Wu J. Heterosis, combining ability and genetic effect, and relationship with genetic distance based on a diallel of hybrids from five diverse Gossypium barbadense cotton genotypes. Euphytica. 2017;213(9):208.
Cheres M, Miller J, Crane J, Knapp S. Genetic distance as a predictor of heterosis and hybrid performance within and between heterotic groups in sunflower. Theor Appl Genet. 2000;100(6):889–94.
Li L, Lu K, Chen Z, Mu T, Hu Z, Li X. Dominance, overdominance and epistasis condition the heterosis in two heterotic rice hybrids. Genetics. 2008;180(3):1725–42.
Shang L, Wang Y, Cai S, Wang X, Li Y, Abduweli A, Hua J. Partial dominance, overdominance, epistasis and QTL by environment interactions contribute to heterosis in two upland cotton hybrids. G3. 2016;6(3):499–507.
Zhou G, Chen Y, Yao W, Zhang C, Xie W, Hua J, Xing Y, Xiao J, Zhang Q. Genetic composition of yield heterosis in an elite rice hybrid. Proc Natl Acad Sci. 2012;109(39):15847–52.
Xiangbin G, Youhua W, Zhiguo Z, Oosterhuis DM. Response of cotton fiber quality to the carbohydrates in the leaf subtending the cotton boll. J Plant Nutr Soil Sci. 2012;175(1):152–60.
Zhu D, Zhou G, Xu C, Zhang Q. Genetic components of heterosis for seedling traits in an elite rice hybrid analyzed using an immortalized F2 population. J Genet Genomics. 2016;43(2):87–97.
Zhai R, Feng Y, Wang H, Zhan X, Shen X, Wu W, Zhang Y, Chen D, Dai G, Yang Z. Transcriptome analysis of rice root heterosis by RNA-Seq. BMC Genomics. 2013;14(1):19.
Wang X, Cao H, Zhang D, Li B, He Y, Li J, Wang S. Relationship between differential gene expression and heterosis during ear development in maize (Zea mays L.). J Genet Genomics. 2007;34(2):160–70.
Romagnoli S, Maddaloni M, Livini C, Motto M. Relationship between gene expression and hybrid vigor in primary root tips of young maize (Zea mays L.) plantlets. Theor Appl Genet. 1990;80(6):769–75.
Liu YJ, Gao SQ, Tang YM, Gong J, Zhang X, Wang YB, Zhang LP, Sun RW, Zhang Q, Chen ZB. Transcriptome analysis of wheat seedling and spike tissues in the hybrid Jingmai 8 uncovered genes involved in heterosis. Planta. 2018;247(6):1307–21.
Shen Y, Sun S, Hua S, Shen E, Ye CY, Cai D, Timko MP, Zhu QH, Fan L. Analysis of transcriptional and epigenetic changes in hybrid vigor of allopolyploid Brassica napus uncovers key roles for small RNA s. Plant J. 2017;91(5):874–93.
Li F, Fan G, Lu C, Xiao G, Zou C, Kohel RJ, Ma Z, Shang H, Ma X, Wu J. Genome sequence of cultivated upland cotton (Gossypium hirsutum TM-1) provides insights into genome evolution. Nat Biotechnol. 2015;33(5):524.
Zhang T, Hu Y, Jiang W, Fang L, Guan X, Chen J, Zhang J, Saski CA, Scheffler BE, Stelly DM. Sequencing of allotetraploid cotton (Gossypium hirsutum L. acc. TM-1) provides a resource for fiber improvement. Nat Biotechnol. 2015;33(5):531.
Mutz K-O, Heilkenbrinker A, Lönne M, Walter J-G, Stahl F. Transcriptome analysis using next-generation sequencing. Curr Opin Biotechnol. 2013;24(1):22–30.
Rhoads A, Au KF. PacBio sequencing and its applications. Genomics Proteomics Bioinformatics. 2015;13(5):278–89.
Yoo M, Szadkowski E, Wendel J. Homoeolog expression bias and expression level dominance in allopolyploid cotton. Heredity. 2013;110(2):171.
Rapp RA, Udall JA, Wendel JF. Genomic expression dominance in allopolyploids. BMC Biol. 2009;7(1):18.
Rieseberg LH, Widmer A, Arntz AM, Burke JM. The genetic architecture necessary for transgressive segregation is common in both natural and domesticated populations. Philos Trans R Soc Lond Ser B Biol Sci. 2003;358(1434):1141–7.
Fujimoto R, Taylor JM, Shirasawa S, Peacock WJ, Dennis ES. Heterosis of Arabidopsis hybrids between C24 and col is associated with increased photosynthesis capacity. Proc Natl Acad Sci. 2012;109(18):7109–14.
Hoecker N, Keller B, Piepho H-P, Hochholdinger F. Manifestation of heterosis during early maize (Zea mays L.) root development. Theor Appl Genet. 2006;112(3):421–9.
Wang Z, Ni Z, Wu H, Nie X, Sun Q. Heterosis in root development and differential gene expression between hybrids and their parental inbreds in wheat (Triticum aestivum L.). Theor Appl Genet. 2006;113(7):1283–94.
Riddle NC, Jiang H, An L, Doerge R, Birchler JA. Gene expression analysis at the intersection of ploidy and hybridity in maize. Theor Appl Genet. 2010;120(2):341–53.
Li X, Wei Y, Nettleton D, Brummer EC. Comparative gene expression profiles between heterotic and non-heterotic hybrids of tetraploid Medicago sativa. BMC Plant Biol. 2009;9(1):107.
Flagel LE, Wendel JF. Evolutionary rate variation, genomic dominance and duplicate gene expression evolution during allotetraploid cotton speciation. New Phytol. 2010;186(1):184–93.
Zhao Y-L, Yu S-X, Xing C-Z, Fan S-L, Song M-Z, Ye W-W. Differential gene expression between hybrids and their parents during the four crucial stages of cotton growth and development. Agric Sci China. 2009;8(2):144–53.
Yang J, Mezmouk S, Baumgarten A, Buckler ES, Guill KE, McMullen MD, Mumm RH, Ross-Ibarra J. Incomplete dominance of deleterious alleles contributes substantially to trait variation and heterosis in maize. PLoS Genet. 2017;13(9):e1007019.
Huang X, Yang S, Gong J, Zhao Y, Feng Q, Gong H, Li W, Zhan Q, Cheng B, Xia J. Genomic analysis of hybrid rice varieties reveals numerous superior alleles that contribute to heterosis. Nat Commun. 2015;6:6258.
Shao L, Xing F, Xu C, Zhang Q, Che J, Wang X, Song J, Li X, Xiao J, Chen L-L. Patterns of genome-wide allele-specific expression in hybrid rice and the implications on the genetic basis of heterosis. Proc Natl Acad Sci. 2019;116(12):5653–8.
McClung CR. Plant circadian rhythms. Plant Cell. 2006;18(4):792–803.
Harmer SL. The circadian system in higher plants. Annu Rev Plant Biol. 2009;60:357–77.
Nusinow DA, Helfer A, Hamilton EE, King JJ, Imaizumi T, Schultz TF, Farré EM, Kay SA. The ELF4–ELF3–LUX complex links the circadian clock to diurnal control of hypocotyl growth. Nature. 2011;475(7356):398.
Farré EM, Harmer SL, Harmon FG, Yanovsky MJ, Kay SA. Overlapping and distinct roles of PRR7 and PRR9 in the Arabidopsis circadian clock. Curr Biol. 2005;15(1):47–54.
Ni Z, Kim E-D, Ha M, Lackey E, Liu J, Zhang Y, Sun Q, Chen ZJ. Altered circadian rhythms regulate growth vigour in hybrids and allopolyploids. Nature. 2009;457(7227):327.
Ko DK, Rohozinski D, Song Q, Taylor SH, Juenger TE, Harmon FG, Chen ZJ. Temporal shift of circadian-mediated gene expression and carbon fixation contributes to biomass heterosis in maize hybrids. PLoS Genet. 2016;12(7):e1006197.
Xue W, Xing Y, Weng X, Zhao Y, Tang W, Wang L, Zhou H, Yu S, Xu C, Li X. Natural variation in Ghd7 is an important regulator of heading date and yield potential in rice. Nat Genet. 2008;40(6):761.
Suárez-López P, Wheatley K, Robson F, Onouchi H, Valverde F, Coupland G. CONSTANS mediates between the circadian clock and the control of flowering in Arabidopsis. Nature. 2001;410(6832):1116.
Putterill J, Robson F, Lee K, Simon R, Coupland G. The CONSTANS gene of Arabidopsis promotes flowering and encodes a protein showing similarities to zinc finger transcription factors. Cell. 1995;80(6):847–57.
James AB, Monreal JA, Nimmo GA, Kelly CL, Herzyk P, Jenkins GI, Nimmo HG. The circadian clock in Arabidopsis roots is a simplified slave version of the clock in shoots. Science. 2008;322(5909):1832–5.
Wang J, Tian L, Lee H-S, Wei NE, Jiang H, Watson B, Madlung A, Osborn TC, Doerge R, Comai L. Genomewide nonadditive gene regulation in Arabidopsis allotetraploids. Genetics. 2006;172(1):507–17.
Groszmann M, Gonzalez-Bayon R, Lyons RL, Greaves IK, Kazan K, Peacock WJ, Dennis ES. Hormone-regulated defense and stress response networks contribute to heterosis in Arabidopsis F1 hybrids. Proc Natl Acad Sci. 2015;112(46):E6397–406.
Grambow H, Langenbeck-Schwich B. The relationship between oxidase activity, peroxidase activity, hydrogen peroxide, and phenolic compounds in the degradation of indole-3-acetic acid in vitro. Planta. 1983;157(2):132–7.
Passardi F, Tognolli M, De Meyer M, Penel C, Dunand C. Two cell wall associated peroxidases from Arabidopsis influence root elongation. Planta. 2006;223(5):965–74.
Li X, Shahzad K, Guo L, Qi T, Zhang X, Wang H, Tang H, Qiao X, Zhang J, Wu J. Using yield quantitative trait locus targeted SSR markers to study the relationship between genetic distance and yield heterosis in upland cotton (Gossypium hirsutum). Plant Breed. 2019;138(1):105–13.
Kim D, Langmead B, Salzberg SL. HISAT: a fast spliced aligner with low memory requirements. Nat Methods. 2015;12(4):357.
Pertea M, Pertea GM, Antonescu CM, Chang T-C, Mendell JT, Salzberg SL. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat Biotechnol. 2015;33(3):290.
Frazee AC, Pertea G, Jaffe AE, Langmead B, Salzberg SL, Leek JT. Ballgown bridges the gap between transcriptome assembly and expression analysis. Nat Biotechnol. 2015;33(3):243.
Xie C, Mao X, Huang J, Ding Y, Wu J, Dong S, Kong L, Gao G, Li C-Y, Wei L. KOBAS 2.0: a web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res. 2011;39(suppl_2):W316–22.
Acknowledgments
The authors appreciate the field staff for their anonymous work. Authors also thankful to the peer reviewers for their constructive suggestions which considerably improved this manuscript.
Funding
This work was supported by the National Key Research and Development Program of China (2016YFD0101400). The funding body had no role in the design, analysis, interpretation of data, and in writing the manuscript.
Author information
Authors and Affiliations
Contributions
CZX and JYW designed the experiments. LPG, TXQ, HT, XQQ, and HLW constructed hybrid plant population and field evaluation. KS, XXZ MZ, JJF, and BBZ performed data analysis. KS, JYW, and CZX contributed to the preparation of the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
All the cotton lines used and analyzed were public and available for non-commercial purpose. This article did not contain any studies with human participants or animals performed by any of the authors.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1.
Brief detail of sequencing, mapping and gene expressions of the individual library
Additional file 2.
Information on DEGs in root of high hybrid-parent triad
Additional file 3.
Information on DEGs in leaf of high hybrid-parent triad
Additional file 4.
Information on DEGs in root of medium hybrid-parent triad
Additional file 5.
Information on DEGs in leaf of medium hybrid-parent triad
Additional file 6.
Information on DEGs in root of low hybrid-parent triad
Additional file 7.
Information on DEGs in leaf of low hybrid-parent triad
Additional file 8.
Information on gene expression patterns in root and leaf of high hybrid relative to its parents
Additional file 9.
Information on gene expression patterns in root and leaf of medium hybrid relative to its parents
Additional file 10.
Information on gene expression patterns in root and leaf of low hybrid relative to its parents
Additional file 11.
Enriched GO terms of overdominant genes in root of high hybrid-parent triad
Additional file 12.
Enriched KEGG terms of overdominant genes in root of high hybrid-parent triad
Additional file 13.
Enriched Go terms of overdominant genes in leaf of high hybrid-parent triad
Additional file 14.
Enriched KEGG terms of overdominant genes in leaf of high hybrid-parent triad
Additional file 15.
Enriched Go terms of overdominant genes in root of medium hybrid-parent triads
Additional file 16.
Enriched KEGG terms of overdominant genes in root of medium hybrid-parent triad
Additional file 17.
Enriched Go terms of overdominant genes in leaf of medium hybrid-parent triad
Additional file 18.
Enriched KEGG terms of overdominant genes in leaf of medium hybrid-parent triad
Additional file 19.
Enriched Go terms of overdominant genes in root of low hybrid-parent triad
Additional file 20.
Enriched KEGG terms of overdominant genes in root of low hybrid-parent triad
Additional file 21.
Enriched Go terms of overdominant genes in leaf of low hybrid-parent triad
Additional file 22.
Enriched KEGG terms of overdominant genes in leaf of low hybrid-parent triad
Additional file 23.
Information of overdominant genes those were unique and common among comparisons of high, medium and low hybrids in root and leaf
Additional file 24.
List of Primers for quantitative RT-PCR
Additional file 25: Figure S1.
Phenotypic heterosis observed in all F1 hybrids at 10 days after emergence of seedling (DAE). a Fresh biomass in hybrids compared with their mid-parent value (MPV). b Dry biomass observed in hybrids compared with their MPV.
Additional file 26: Figure S2.
Phenotypic heterosis observed in all F1 hybrids at 20 days after emergence of seedling (DAE). Here * is used for significant difference at p < 0.0 a Fresh biomass in hybrids compared with their mid-parent value (MPV). b Dry biomass observed in hybrids compared with their MPV.
Additional file 27: Figure S3.
Most enriched GO terms of overdominant DEGs in root and leaf of F1 hybrids. a, b, and c shows GO terms with total number of genes for up and down overdominant DEGs of high (H), medium (M) and low (L) hybrids, respectively. Here, most enriched GO terms with p < 0.05 are only presented.
Additional file 28: Figure S4.
Venn diagram representing the comparison of overdominant genes between hybrids in root and leaf. L: Leaf, R: Root, H, M, and L represent high, medium, and low hybrids respectively. a Distribution of genes in root. b Distribution of genes in leaf.
Additional file 29: Figure S5.
Enriched GO terms for genes that showed overdominant expressions in all hybrids relative to their parents in root and leaf.
Additional file 30: Figure S6.
Functional annotations and mode of regulation for overdominant DEGs involved in circadian rhythm plant pathway. This figure is an interpretation of online available figure (https://www.kegg.jp/kegg-bin/show_pathway?ko04712+K12133).
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Cite this article
Shahzad, K., Zhang, X., Guo, L. et al. Comparative transcriptome analysis of inbred lines and contrasting hybrids reveals overdominance mediate early biomass vigor in hybrid cotton. BMC Genomics 21, 140 (2020). https://doi.org/10.1186/s12864-020-6561-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-020-6561-9