
Abstract
Background
Pancreas disease (PD) is a contagious disease caused by salmonid alphavirus (SAV) with significant economic and welfare impacts on salmon farming. Previous work has shown that higher resistance against PD has underlying additive genetic components and can potentially be improved through selective breeding. To better understand the genetic basis of PD resistance in Atlantic salmon, we challenged 4506 smolts from 296 families of the SalmoBreed strain. Fish were challenged through intraperitoneal injection with the most virulent form of the virus found in Norway (i.e., SAV3). Mortalities were recorded, and more than 900 fish were further genotyped on a 55 K SNP array.
Results
The estimated heritability for PD resistance was 0.41 ± 0.017. The genetic markers on two chromosomes, ssa03 and ssa07, showed significant associations with higher disease resistance. Collectively, markers on these two QTL regions explained about 60% of the additive genetic variance. We also sequenced and compared the cardiac transcriptomics of moribund fish and animals that survived the challenge with a focus on candidate genes within the chromosomal segments harbouring QTL. Approximately 200 genes, within the QTL regions, were found to be differentially expressed. Of particular interest, we identified various components of immunoglobulin-heavy-chain locus B (IGH-B) on ssa03 and immunoglobulin-light-chain on ssa07 with markedly higher levels of transcription in the resistant animals. These genes are closely linked to the most strongly QTL associated SNPs, making them likely candidates for further investigation.
Conclusions
The findings presented here provide supporting evidence that breeding is an efficient tool for increasing PD resistance in Atlantic salmon populations. The estimated heritability is one of the largest reported for any disease resistance in this species, where the majority of the genetic variation is explained by two major QTL. The transcriptomic analysis has revealed the activation of essential components of the innate and the adaptive immune responses following infection with SAV3. Furthermore, the complementation of the genomic with the transcriptomic data has highlighted the possible critical role of the immunoglobulin loci in combating PD virus.
Similar content being viewed by others

Background
Pancreas disease (PD) is a severe contagious disease of farmed Atlantic salmon (Salmo salar) and rainbow trout (Oncorhynchus mykiss), affecting fish during the sea-water phase of the production. The aetiological agent, salmonid alphavirus (SAV), is a single-stranded RNA virus belonging to the Togaviridae family [1], and it has become a pathogen of high economic concern in the salmon farming countries such as Norway, Scotland and Ireland. So far, six different subtypes of this virus, SAV1 to SAV6, have been identified [2,3,4], where outbreaks caused by SAV3 have only been reported in the Norwegian sea-waters to date [5,6,7]. SAV3 was first detected and described, at its genomic details, based on the isolates collected from the west coast of Norway in 2003 and 2004 [8] where now constitute an endemic region for this disease. Since 2007, PD has become a notifiable disease in Norway, and strict national regulations have been in place for more efficient confinement of the spread of the virus. Since then, the number of PD cases throughout Norway, due to SAV3 infection, has remained relatively constant with about 100 outbreaks per year [9].
The economic losses due to PD outbreaks can be substantial. Based on an economic model, the estimated direct associated cost in 2007 with a PD outbreak for 500,000 smolts in Norway was about 14.4 million Norwegian Kroners (NOK; approximately €1.45 M) [10]. The same study also found that the saleable biomass due to this disease was reduced by 70% and the production costs increased with 6 NOK per kg. An updated analysis from the data, based on 2013 sale prices, suggested that the direct cost of PD outbreak for 1,000,000 smolts to be about 55.4 M NOK (approximately €5.53 M) [11]. These figures suggest that although different methods of prevention, such as vaccination, improving management and optimised production conditions, have caused the mortality during an outbreak to decline, still, the financial losses due to PD infections have increased.
SAV usually infects salmon at the smolt stage during the first year in the sea. Clinical manifestation of PD infection might include sudden loss of appetite and lethargy, reduction in growth, abnormal swimming behaviour and increased mortality [12]. Mortality due to PD infection can vary from negligible to very high, with an expected average mortality of around 7%, based on data collected from 2006 to 2008 [13]. Histopathological examination of infected animals often exhibits loss of exocrine pancreatic tissue, cardiac degeneration, inflammation and subsequent degeneration and inflammation of the skeletal muscle [14]. Following infection, mainly if the infection occurs during the later stages of production, a significant reduction in growth and deterioration in the feed conversion ratio can be expected [15]. Further, the fish that survive the outbreak can become more susceptible to other pathogens and secondary infections [16].
So far, a few studies have aimed to evaluate a host immune response to infection with PD [17]. It has been demonstrated that following an infection, the virus will stimulate the expression of innate immunity through pattern recognition receptors (PRR) such as toll-like receptors (TLRs) and RNA helicases. Subsequently, this stimulation triggers antiviral effectors and the type I interferons (IFN I) signalling pathway, where the most significant changes in the gene expression response of a host can be observed in the infected heart. The stimulation of these genes and genetic pathways, where in turn, further enhances the innate immune responses [18, 19]. Subsequent inflammation in the affected tissues, triggered by the products of immune-related genes like chemokines and cytokines is then expected [17]. Activation of adaptive immune responses and production of neutralizing antibodies takes place following PD infection [20,21,22].
A possible solution and a common practice to enhance resistance against PD is vaccination. Several vaccines including a multivalent vaccine by MSD Animal Health (AquaVac® PDt), a monovalent vaccine by PHARMAQ (ALPHA JECT micro® 1PD) and a DNA vaccine by Elanco (Clynav) are a few examples. Although the efficiency of vaccines has been questioned regarding their failure to eradicate the disease, in addition to their high costs, various side effects and negative impacts on slaughter quality, vaccination helps to reduce the mortality and alleviate some of the associated signatures of the infection, such as weight loss and feed conversion rate [23].
An early indication of a possible genetic basis for PD resistance was reported through epidemiological observations in PD response between different strains of Atlantic salmon [16, 24, 25]. Subsequent works have shown that higher tolerance against PD has indeed some underlying additive genetic components and this trait can potentially be improved through selective breeding. Norris et al. [26] estimated the heritability of 0.08 for PD resistance (0.21 on the liability scale), based on mortality data from a field outbreak due to SAV1 infection. In a controlled challenge trial, Gonen et al. [27] investigated the genetic basis of PD resistance in two different strains of Atlantic salmon (i.e., SalmoBreed and Mowi) and two different physiological stages, fry and post-smolt, against SAV3. Their estimates suggest a moderate to high heritability of 0.26–0.34 for resistance to PD. Further, using a 6 K Illumina iSelect SNP array (Centre for Integrative Genetics) as well as a low-density panel of genetic markers, Gonen et al. [27] have reported a suggestive quantitative trait locus (QTL) at the distal end of chromosome 3 (ssa03) in both populations. In addition to ssa03, their analysis also suggests putative QTL on ssa02, ssa04, ssa07, ssa14 and ssa23.
The goal of this study was to investigate the genetic architecture of PD resistance in Atlantic salmon, based on mortality data, in much greater detail than previous reports. Within a controlled infection environment and by testing approximately 4500 pedigreed fish challenged against SAV3, we first aimed to estimate the heritability of resistance against this disease. We then genotyped a subset of animals using a 55 K SNP array to identify and narrow down the genomic regions containing QTL. Finally, to identify putative candidate genes of importance within these QTL regions and also to better understand the host’s response to SAV3 infection, we compared the transcriptomic data from the animals that survived the challenge versus those that were in a moribund state early during the trial.
Results and discussion
Challenge and mortality
A total of 4506 PIT-tagged animals with pedigree information, representing 296 full-sib families, contributed to the data following the challenge with SAV3. As expected from a SAV3 intraperitoneal injection (i.p.) model of infection [17], mortality initiated at 7 days post-infection (dpi) (Fig. 1a), and the trial was terminated after 29 dpi when the disease reached its mature phase, approximately 1 week after the mortality was plateaued. The mortality peaked at 13 dpi and started to stabilize from day 21 (Fig. 1b). At this stage, the cumulative mortality had reached approximately 47%. By the end of the experiment, a total of 2151 (approximately 48%) animals died due to PD infection.

a The cumulative per cent mortality curve and b histogram of mortality profile of SAV3 infection following injection. Each bar shows the number of dead fish per day from the start (0 dpi) to the termination of the challenge (29 dpi)
Heritability estimation
We observed considerable variation in the rates of survival between the challenged families, ranging from 3 to 100% (Fig. 2). The pedigree-based and the SNP-based heritability were estimated to be 0.41 ± 0.017 and 0.32 ± 0.081, respectively. The heritability estimates in this study are considerably higher than the estimate reported previously, based on a field data outbreak following a SAV1 infection [26]. The estimate from the field outbreak, however, is probably an underestimation of the true heritability of the trait, considering the challenges associated with a thorough collection and early diagnosis of the infected animals, from a cage in a natural setting. Further, to accurately monitor, collect and record all mortalities that are due to a specific pathogen from the field is not a trivial task. Mortalities in the field can be due to other causes or diseases such as heart and skeletal muscle inflammation (HSMI), with manifested symptoms similar to those of PD [17]. We also have little knowledge of how various environmental factors in natural settings shape the immune response and how such a response can influence the outcome of a disease. Little is known on how sequential exposure to various biotic or abiotic elements in an environment can benefit or harm the fish and how gene by environment interactions or even the interactions between different microbes can influence the trajectory of disease progression. One might also expect substantial differences in estimates of additive genetic variance and heritability for resistance against different subtypes of the virus, i.e., SAV1 versus SAV3.

Per cent survival, per family, by the end of the challenge, at 29 dpi. A total of 296 full-sib families, shown on the x-axis and sorted based on survival rate, were used in this trial
In contrast, in a controlled experiment, all mortalities, with high certainty, can be attributed to the specific pathogen of interest. Furthermore, one can monitor mortalities regularly, and various environmental parameters can be controlled and adjusted. In this regard, our experiment and estimates are more comparable and similar to those reported by Gonen et al. [27]. In their study, they challenged two different strains of Atlantic salmon, the Mowi strain, where fish were at the fry stage and the SalmoBreed strain, as post-smolts. Their estimated heritability on the binary scale ranged from 0.26–0.34. Therefore, the finding in our study supports the reported estimates by Gonen et al. [27], in that resistance to SAV3 has a moderate to high additive genetic components, and selective breeding can be an efficient and attractive tool for reducing and managing the outbreaks due to this virus.
Genome-wide association study
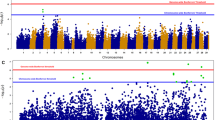
Approximately 49 K genetic markers and 903 fish, comprising of 430 dead and 473 survivors, passed all the quality control measures. The proportion of the dead and survivors among the genotyped animals was the same as that for the entire challenged population (i.e., approximately 48% mortality and 52% survivals). The genotyped fish were from 65 families, with a minimum of 10 and an average of 14 animals per family. The inflation factor was estimated to be 1.07, an indication of no confounding effect due to population stratification. A total of 17 genetic markers, nine located on chromosome ssa03 and eight on chromosome ssa07 exceeded the genome-wide significance level of p < 1.024e− 06 (Fig. 3a). The QTL on ssa03 spans a region of approximately 27 Mbp, from 63.8 to 90.8 Mbp (Fig. 3b). On the SNP chip array, there are about 655 informative genetic markers within this chromosomal segment of ssa03, which collectively explained about 31% of the additive genetic variance in our dataset (\( {h}_{RGH}^2 \) = 0.100 ± 0.05). The two most significant SNPs on this genomic block are at both ends of the segment, each explaining approximately 3.4% of the phenotypic variance, suggesting the possibility of two different QTL on ssa03 (Fig. 3b; Table 1). The first SNP (ssa03: 63,829,838), is a synonymous variant, located in a member of dedicator of cytokinesis gene family (LOC106601059; Dock7). The members of this gene family are evolutionary conserved and are involved in intracellular signalling networks [28]. The second significant SNP (ssa03: 90,830,374) is an intergenic variant, flanked by a cytokine encoding gene, Metrnl (meteorin-like protein) [29]. The expression of Metrnl is regulated through products of different immune-related genes such as IL-4, IL-12 and IFN-γ, and is associated with inflammation response [29]. This gene showed an approximately 5-fold elevation in expression among animals that survived the challenge (Supplementary Table 1) compared to those that died early in the trial.

a Manhattan plot of association analysis to PD resistance in Atlantic salmon. The figure shows the -log10 (p-value) of the test statistics for each SNP plotted against the physical positions of the markers on the chromosomes. The black and the orange lines indicate the genome-wide and the chromosome-wide significance threshold cut-off levels, respectively. b Zoomed-in Manhattan plot of ssa03 showing the distribution and association of genetic markers on this chromosome
The finding of a QTL on ssa03 corroborates well with the reported QTL on the same chromosome in the two populations investigated by Gonen et al. [27]. In that study, an association on ssa03 was identified in both Mowi and the SalmoBreed strains, even though the fish were genotyped using either a sparse SNP panel (on the Mowi strain – challenged as fry) or a 4 K SNP array (SalmoBreed population – challenged as post-smolts). Further, the position of the QTL in both studies is consistent, with the most significantly associated markers located at the end of the chromosome. These independent findings, which are based on different populations, different year-classes, different challenge models and different developmental stages of the fish, validate the presence of a significant QTL on this chromosome. Our data further helps to refine and narrow down the location of the causative mutation to about 27 Mbp region of ssa03.
On chromosome ssa07, the QTL lies in a genomic region of approximately 9.12 Mbp, from 38.6 to 47.8 Mbp. There are approximately 340 informative SNPs within this segment of the chromosome, where based on the regional heritability estimates, they accounted for approximately 33% of the additive genetic variation (\( {h}_{RGH}^2 \) = 0.106 ± 0.05). The most strongly associated SNP on this genomic block is an intergenic SNP (ssa07: 45,525,972), explaining about 5.8% of the variation (Table 1). This SNP is in a short distance from the 3′ untranslated region of an uncharacterized gene with a putative protein structure similar to the products of extensins. In plants, extensin proteins, through the strengthening of the cell walls, play an essential role in the defence mechanism against pathogens, preventing tissue damages and are involved in wound responses [30,31,32]. In our transcriptome data, the products of this gene showed a 1.75-fold increase in expression among the survived animals (data not shown). On the downstream of this SNP, there are also a few genes of interest, including fibroblast growth factor receptor oncogene partner 2 (FGFR1OP2), a gene with potential roles in the wound-healing pathway [33, 34]. By the end of the experiment, the transcription level of this gene increased by 2-fold (data not shown). Gonen et al. [27] reported a suggestive QTL on ssa07, detected only on the Mowi strain. However, due to the low resolution of their mapping SNP panel, it is not possible to localize the position of QTL and make a comparative assessment with the QTL on ssa07 identified in this work. Further, Gonen et al. [27] did not find any association on this chromosome in the SalmoBreed strain, possibly due to QTL not segregating in the 2009 year-class of this population.
While no marker on ssa06 passed the genome-wide significant threshold, two markers on the p arm of this chromosome (ssa06: 3,319,000 and ssa06: 3,846,000) passed the chromosome-wide significance level. The proportion of phenotypic variation explained by each of these markers is about 2%. This finding is interesting, as the p arm of ssa06 shares homeology (i.e., shared ancestry due to the whole genome duplication event at the base of all extant salmonids) with the q arm of ssa03 [35], where we have mapped the QTL on chromosome 3. Therefore, one might speculate the possibility of a duplicated QTL on the homeologous chromosomal regions of these chromosomes. At the same time, we cannot dismiss the possibility of this peak being a by-product of miss-assembly in the reference genome, where some duplicated contigs or scaffolds from ssa03 have mistakenly collapsed with sequences on ssa06. This idea is further supported by relatively lower average LD (r2) within the QTL segment of ssa06 (0.46) compared to that of ssa03 (0.54).
Analysis of transcriptomic data
We obtained, on average, 15 million paired-end reads transcriptome sequence data, per animal, from the apex of the heart. Following trimming and filtering the low-quality reads, sequences with multiple hits against the genome and removal of rRNA and Illumina adapter contaminations, approximately 10 million uniquely aligned paired-end reads per fish retained for assessment of the gene expression profiles. Both cluster and principal component analysis (PCA) of the gene expression data, separated and grouped animals based on the survival status of the fish (Fig. 4a and b), confirming significant changes in the transcriptional regulation of a vast number of genes during infection and between the moribund and survived animals. In total, we identified 7431 differentially expressed transcripts between the two groups. Approximately, half of these transcripts showed a higher level of expression in the animals that survived the challenge (3480 transcripts) while the other half (3951 transcripts) had higher expression in the moribund animals (Supplementary Table 1). The functional enrichment analysis of these gene sets suggests a broad range of genes with different functional properties to be enriched within each group (Supplementary Table 2).

a Heatmap of the transcriptome sequence data between the moribund animals (M) at 11 dpi and the animals that survived (S) the challenge. The x-axis represents animals that were sampled during the two stages. The y-axis shows the expression profiles of differentially expressed genes between the two groups (Supplementary Table 1). b Principle component clustering of the same transcriptome data
The transcriptome of the most severely sick fish had signatures of high levels of expression of genes associated with innate immune response and IFN-related antiviral genes. On the other hand, fish, which survived till the termination of the challenge, exhibited higher expression of genes specific for adaptive immunity (Supplementary Table 1). Similar differences exist in Atlantic salmon with the early and late mortality following infection with the infectious salmon anaemia virus – ISAV [36]. As previously reported, the expression of genes involved in the innate antiviral responses shows a strong correlation with loads of SAV [17], and therefore, the observed transcriptional differences between the moribund and the survived fish, in this study, were expected.
The PRR machinery is essential for triggering various components of the immune response. In an Atlantic salmon, infected with a virus, RNA helicase Rig-I is usually among the genes with the most active response. In our study, this gene had significantly higher expression in moribund fish. Across the animals that survived the infection, we found significantly higher levels of expression in a Toll-like receptor (TLR13) and the NOD-like receptor gene (NLR family member X1), both of which being part of the PRR. In response to a viral infection, TLR13 can activate a MyD88-dependent pathway, an essential element for signalling within the immune cells, resulting in the activation of nuclear factor κB (NF-κB) and type I IFN via interferon regulatory factor 7 (IRF7) [37]. IRF4 and IRF8, which found to have higher expression in the resistant animals, control termination of pre-B-cell receptor signalling, and therefore, promote differentiation to small pre-B-cells undergoing light-chain gene rearrangements [38]. Besides, a panel of genes involved in the regulation of IFN-γ, a member of the type II class of IFN with a critical role in T-cell mediated adaptive immunity, had elevated their expressions in the animals that survived the challenge (AXL, CD2 (LOC106575562), CD226, CD276 (LOC106587232), CD3E, HMGB1, IRF8 (LOC106562284), TNFR5 (LOC106575195), ZFPM1; Supplementary Table 1).
Chemokines are important in triggering inflammation in virus-infected tissues. Many genes involved in the chemokine-mediated signalling showed significantly higher levels of expression in the moribund animals. Some examples are chemokine ligand 17 (CCL17; LOC106613702), chemokine ligand 19 (CCL19; LOC106563358), chemokine receptor-like 2 (CCRL2; LOC106600445), chemokine ligand 1 (CXCL1; LOC106580387), chemokine ligand 11 (CXCL11; LOC106564720), chemokine ligand 4 (CCL4; LOC106600447), chemokine ligand 25 (CCL25; LOC106613704) and chemokine ligand 20 (CCL20; LOC106570861). These genes play essential roles in the recruitment of leukocytes to sites of infection and inflammation and guiding the lymphocytes throughout the body [39]. It is important to note that excessive recruitment of chemokine gene products can result in damages to the tissue. For a number of these genes, we identified two or more gene copies, located on different chromosomes, with significant expression profile differences between the moribund and the survived animals. For example, three copies of CCL19, on ssa01, ssa11 and ssa14, all had higher levels of expression at 11 dpi. At least two of these chromosomes, ssa01 and ssa11, are known to share ancestral homeologous regions [35]. Similarly, duplicated copies of CCL25 on the homeologous segments of ssa10 and ssa16 and CXCL11 on the homeologous regions of ssa02 and ssa12 exhibited elevated levels of expression in animals with early mortality.
The most significantly down-regulated gene, not only among the immune-related genes but within the entire dataset, is the complement component C1q-like protein 2, with almost no expression among the animals that survived the challenge (log2-fold change of 9.877). The role of this gene in salmonids, which have also shown to be down-regulated in several other viral infections, is not very well understood. However, it is unlikely that its lack of expression is a sign of suppression of a classical complement pathway, but rather an indication of a connecting bridge between the innate and the acquired immunity [40, 41]. Such a hypothesis is further supported considering that several other complement components, such as C1S (LOC106605669), C1QC, CLU (LOC106581046), C6 and CD55, had increased their levels of expression by the end of the challenge.
In contrast, the gene with the highest fold-change increase, by the end of the experiment, was a gene with an eotaxin-like product. Eotaxins have an essential function in stimulating and chemotaxis of eosinophil, a specialized pro-inflammatory white blood cell, [42, 43], but are also crucial in the recruitment of helper T-cells [44], mast cells [45], and directing the killer lymphocytes and monocytes to the sites of inflammation [46]. We further found strong evidence for the activation of specific adaptive immunity genes among survivors. As a few examples, several genes associated with cellular immune response and T-cell activation (CD2 (LOC106575562), CD3E and CD28 (LOC106575093) have significantly elevated expression in the fish that survived the challenge. Similarly, CD74 (LOC106604844), which plays a crucial role in MHC class II antigen processing through stabilizing peptide-free class II alpha/beta heterodimer, was also found to have been expressed about 2-fold higher in these animals. Tumour necrosis factor ligand superfamily member 11 (TNFSF11), a gene involved in stimulating naïve T-cell proliferation that regulates the interactions between T-cell and dendritic cells, was also expressed 2.5-fold higher in survived animals. Since the peak of acquired immune responses in SAV infected salmon usually occurs after three to 4 weeks post-infection, the transcriptional differences between the moribund and survivors were also likely affected by the sampling time. However, the ability to survive until the development of adaptive immunity against SAV may be a critical factor that determines resistance against PD.
Candidate immune-related genes in the QTL regions
The QTL region on ssa03 contains more than 900 annotated genes of which we found 135 with different expression patterns between the two groups. On ssa07, the genomic segment containing QTL has about 156 expressed genes, of which 62 had significantly modified their expression profiles. A heatmap of genes with putative immune-related functions and different expression profiles within these QTL segments are presented in Supplementary Figure 1a and b. These genes are candidates that can potentially affect an animal’s resistance to PD infection. The most notable of these genes, however, are multiple transcripts from the immunoglobulin heavy chain (IGH) locus on ssa03 and the immunoglobulin light chain locus on ssa07. The transcripts exhibited 4 to 20 times higher levels of expression in the animals that survived the test.
The immunoglobulin molecules in Atlantic salmon consist of an assemblage of two heavy and two light chains, made up of constant and variable domains. The duplicated IGH loci in Atlantic salmon reside on the homeolog of two chromosomes, ssa03 (IGH-B) and ssa06 (IGH-A) [47]. The initial size estimation of IGH-B locus suggested a genomic region of 700 Kb [47], but in the current assembly of the Atlantic salmon genome, the clusters of IGH segments span a much larger region, covering from 77.5 to 79.3 Mb. In addition to the constant segments (CH) of all three types of IG (i.e., D, M and T), the IGH-B locus contains 28 DH (diversity) genes, eight JH (junction) genes and numerous VH (variable) genes, of which 36 genes are functional. In our data, about 92% of the IGH transcripts exclusively map to IGH-B, and almost all transcripts encode an immunoglobulin type M (IgM). On ssa07, a significant increase in the expression of two immunoglobulin constant (IGKC) and three variable domains (IGKV) were detected (Supplementary Figure 1b). In B-cells, antibodies are expressed and assembled through somatic recombination of variable segments. Recombination is followed by enzymatic insertion and deletion of nucleotides in the variable region, generating a broad diversity of antibodies, which can recognise a diverse array of antigens. The findings presented in the current study, and the fact that the two QTL are in close genomic proximities to the immunoglobulin loci, suggest them as likely candidates for conferring higher resistance against PD virus in this species.
Although there are various lines of evidence regarding the crucial role of antibodies in defence against viruses, the early activation of antiviral immunity and inflammatory responses does not necessarily prevent the propagation of the pathogen and the subsequent development of the disease. Clearance of pathogens and disease symptoms usually take place shortly after the initiation of the adaptive immunity [17, 36]. In the case of PD, B-cells can combat SAV in different ways. Marked increase of the IG transcripts in the infected hearts is an indication of the recruitment of B-cells, which can either produce antibodies locally or act as phagocytic cells [48]. The membrane-bound form of salmon IgM lacks the third exon of the constant region. Considering that the expression levels of transcripts from all the exons of the constant regions were similar in our dataset, one might expect that the secreted form of IgM was predominant. Immunization of Atlantic salmon with a vaccine can cause a marked increase of polyspecific antibodies, which precede the appearance of antigen-specific antibodies [49]. Mutations in IGH locus may affect the time-course of antibody responses, their antigen-binding properties or the size and diversity of antibodies repertoire. Each of these factors can be important for conferring higher resistance against SAV. As it stands, however, our knowledge of the genetic variations in the immunoglobulin loci is limited, and the coverage of genetic markers spanning these regions in genomic studies conducted so far in Atlantic salmon has not been optimal. Further, considering the duplicated nature of the salmon genome and the repetitive structure of these loci, there is a need for additional sequencing and genome assemblage, fine mapping, and a basic understanding and comparative analysis of these genes across different populations.
Conclusions
The findings reported in this study provide knowledge, necessary for helping us better understand the genetic basis of resistance against PD. First, our data confirm that PD resistance has a high heritable, additive genetic component. The estimated heritability of 0.41 on the observed binary scale, is among the highest estimates for any disease resistance in Atlantic salmon. One might also expect this finding to be irrespective of the mode of infection, as recently we have shown that resistance against SAV3 is not influenced by the means at which the virus enters the host, whether it is through injection or via cohabitation [50]. Second, it seems that the majority of the genetic variation for PD survival can be explained by two significant QTL, one located on ssa03 and the other on ssa07. These QTL collectively explained more than 60% of the additive genetic variation, in our population, respectively. Therefore, it would be possible to both implement and complement a breeding program for improving PD resistance using marker-assisted selection based on information within these QTL regions. Third, we have narrowed down the location of QTL on each of these chromosomes and complemented genomic analysis with transcriptomic data. Within the QTL containing segments, we have identified several candidate genes with potential impacts on an animal’s response to PD infection. In particular, a locus with multiple components of immunoglobulin heavy chain genes on ssa03 and a few immunoglobulin chain loci on ssa07 are of specific interest. Notably, resistance to PD seemed to be associated with increased transcription of antibodies producing secretory IgM. The finding of the potential role of antibodies in both genetic and acquired resistance against PD provides an attractive platform for close collaboration and synergy in vaccine development and breeding programs.
Methods
SalmoBreed population and challenge test
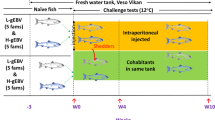
In November 2015, 4506 PIT-tagged (passive integrated transponder) smolts were transported from NOFIMA MARINE AS in Sunndalsøra, Norway (https://nofima.no/en/) to the challenge facility in VESO Vikan (https://www.veso.no/about-us; Namsos, Norway). The group consisted of all families from the entire 2015 year-class of the SalmoBreed nucleus, approximately 15 individuals per family, to assure reliable estimates of genetic parameters [51]. At the parr stage, all fish were PIT-tagged, weighted and reared in the same tank till smoltification when they were transferred to the challenge facility. Following arrival at the VESO Vikan, fish were kept in a single tank and acclimatized at 12 °C brackish water and 24:00 h light regime for 8 days. After acclimatization, using a common-garden approach, all fish were i.p. injected with 0.1 mL of a cell culture containing SAV3 at a concentration of 104.8 TCID50 per mL and the challenge continued in the same tank. The SAV3 isolate, R-1-2007 (GenBank: LT630447.1), used in this study, is one of the most pathogenic forms of the virus found in Norway [52]. Random mortalities were collected throughout the trial and examined for assessing the cause of death. The experiment aimed to target 50% mortality, in order to maximize the phenotypic variation expected from a case-control study (i.e., var. (trait) = p × (1 - p), where p is the frequency of surviving animals). At the end of the experiment, approximately 4 weeks after the infection when the mortalities stabilized, the remaining fish were euthanized with an overdose of anaesthesia (100 mg/L, Finquel®Vet, ScanAqua AS, Årnes, Norway).
Pedigree-based genetic estimations
The pedigree-based variance components, i.e., the additive genetic (\( {\sigma}_a^2 \)), common-environmental family (\( {\sigma}_c^2 \)) and residual variances (\( {\sigma}_e^2 \)), were estimated using a threshold model implemented in the THRGIBBS1F90 module from the BLUPF90 package [53]. The Gibbs sampling scheme was run for 120,000 iterations with the first 20,000 discarded as burn-in and then set to save every 100th sample. The model used was as follows:
where y is a vector of PD survival status, recorded as a binary trait, X, Z1 and Z2 are design matrices assigning the phenotype to the fixed effect, tag weight (b), and the random variables, the common-environmental full-sib family prior to tagging (c) and the animal (u) effects. The residuals are defined in the e vector. The random effects are assumed to have the following distributions, \( \mathrm{c}\sim N\left(0,\mathbf{I}{\sigma}_c^2\right) \), \( u\sim N\left(0,\mathbf{A}{\sigma}_u^2\right) \) and \( e\sim N\left(0,\mathbf{I}{\sigma}_e^2\right) \), where I is an identity matrix, A is the additive genetic numerator relationship matrix, \( {\sigma}_{\mathrm{c}}^2 \), \( {\sigma}_{\mathrm{u}}^2 \) and \( {\sigma}_e^2 \) are the variances of full-sib family effect, additive genetic and residual respectively. The pedigree-based heritability was estimated as:
Genotyping and genotype quality assessment
Adipose fin-clip from 930 of the challenged animals, representing 65 families, were collected throughout the trial. With an average of 15 fish per family, the choice of family selection was set to maximize the genetic diversity, based on the pedigree information, while utilizing the within-family genetic variations [51, 54,55,56]. DNA extraction and genotyping were performed by IdentiGEN (https://identigen.com/; Dublin, Ireland). We used a 55 K custom made Affymetrix Axiom array and the Affymetrix Axiom analysis suite software to genotype the fish and call the SNPs. Additional genotype quality measures and filtering were applied using the SNP & Variation Suite v8.8.3 (SVS; Golden Helix Inc., Bozeman, MT, USA www.goldenhelix.com). Samples and SNPs with call rates < 98%, Hardy-Weinberg p-value (Fisher’s exact test) < 10–10 or minor allele frequency < 0.05% were excluded, leaving 903 animals and about 49 K markers for the subsequent genomic analysis.
Estimation of SNP-based heritability and the QTL effect size
The restricted maximum likelihood method, implemented in the GCTA software [57], was used to estimate variance components and calculate the SNP-based heritability. We applied a model with variables similar to the one used for estimating the pedigree-based genetic parameters except that i. the gender effect, predicted according to the Y-specific linked markers [58], was also included into the model and ii. the A matrix was replaced with the genomic relationship matrix (i.e., the G matrix), computed as described previously [57, 59]. We then used the software package DISSECT to estimate the proportion of genetic variation that is explained by the genomic loci harbouring the QTL (\( {h}_{RGH}^2 \)) [40, 41] by first dividing SNPs between the genetic markers located within the regions of interest (Rk) and those covering the rest of the genome (Gk). The genomic relationship matrices (\( {\mathbf{G}}^{R_k} \) and \( {\mathbf{G}}^{G_k} \)) and variances (\( {\sigma}_{g^{R_k}}^2\ \mathrm{and}\ {\sigma}_{g^{G_k}}^2 \)) were then computed and fitted using the genetic markers within each group [41].
Genome-wide association study
SVS was used to conduct the genome-wide association study (GWAS) using a mixed linear model. The methodological details of the association analysis have been described previously [59]. In brief, we first pruned the quality-controlled markers (window size 40, window increment 5 and r2 threshold 0.5) to construct an n × n genetic distance relationship matrix based on the identity-by-state (IBS). The relationship information was then used to perform a single-locus mixed-model association analysis, using the full set of quality-controlled markers, by applying Efficient Mixed-Model Association eXpedited (EMMAX) [60, 61]. The model used for the analysis can generally be described as:
where y is the vector of survival scores, X, Z1, Z2 and Z3 are the incidence matrices assigning phenotype to the vectors of fixed effects (b), i.e., the animal weight at tagging and gender, the common-environmental family effect (c), additive genetic effect (u; \( \mathrm{Var}(u)={\sigma}_u^2\mathbf{K};\mathbf{K}=\mathrm{IBS}\Big), \) allele substitution effect of the SNP (a), assigned to the matrix for marker genotypes and the residual (e;\( \mathrm{Var}(e)={\sigma}_e^2\mathbf{I};\mathbf{I}= \) identity matrix). The genome-wide and chromosome-wide significance thresholds were set as 0.05/NG and 0.05/NC, respectively, where NG is the total number of quality-controlled genetic markers in the study, and NC is the number of SNPs per [62, 63]. Although EMMAX only performs standard linear regression, even on binary data, its suitability for assessing case-control studies have been thoroughly investigated and validated [60]. Nonetheless, we further compared its results with analysis based on a logistic mixed model, using the GMMAT R package [64], where we applied a logit link function for binary traits and found the results to be almost identical.
We estimated the linkage disequilibrium (LD) within the QTL regions, for SNP pairs separated by up-to 100 kb, using the r2 function in plink v1.9 [65]. The inflation factor (lambda) was computed as the ratio of the median of the observed Chi-squared test statistics to the expected value, in the absence of stratification (0.456) [66]. The proportion of the phenotypic variation explained by each SNP was calculated as:
where mrssh0 is the Mahalanobis total sum of squares, proportional to the variance of the data, and mrssj is the Mahalanobis residual sum of squares for marker j [61, 67, 68].
Analysis of the transcriptome sequence data
We investigated the host transcriptomic response to SAV3 infection by collecting the apex of the heart from six challenged fish at a moribund state during the early stage of the challenge (11 dpi) and six individuals that survived till the termination of the experiment (29 dpi). Heart rather than pancreas was chosen as the target tissue, as it is usually more damaged due to infection and it is less affected by cross-tissue contamination during sampling [12, 50]. The collected tissue samples were immediately immersed in RNALater (Ambion) and stored in − 20 °C. The Qiagen RNeasy Plus mini kit was used for extracting total RNA. Library preparation of and sequencing were performed by the Norwegian High-Throughput Sequencing Centre in Oslo, using the standard protocols outlined by Illumina (www.illumina.com). Sequencing was done on an Illumina HiSeq 4000 platform as paired-end (PE) 150 bp reads.
The initial sequence quality assessment was done by fastQC (www.bioinformatics.babraham.ac.uk/projects/fastqc/). The sequencing adapters and low-quality nucleotides were removed by Trimmomatic (v.0.36) [69]. The remaining sequences were then aligned to the salmon reference genome assembly ICSASG_v2 (https://www.ncbi.nlm.nih.gov/assembly/GCF_000233375.1/) using TopHat (v.2.0.13) [70]. Only reads with a single hit against the genome were used for gene expression assessment. The transcriptome profile information was assessed against the RefSeq annotation, GCF_000233375.1 (https://www.ncbi.nlm.nih.gov/assembly/GCF_000233375.1). The expression data were scaled using the median of the geometric means of fragment counts across all samples [71]. Estimation of the magnitude of gene expression and evaluation of differential levels of transcription was performed by Cuffdiff. For a gene to be assigned as differentially expressed, we set a minimum of 1.5-fold change in expression and an adjusted p-value of less than 0.05. More information regarding details of sequence handling, assessment and analysis have been outlined previously [72, 73].
Availability of data and materials
All the sequence data have been submitted to the short sequence reads archive under the SRA accession number PRJNA590166. The GWAS summary statistics data for all the key findings of the study is available in Table 1. The raw genotype data is not publicly available as it is the property of a commercial enterprise. Requests to access the raw genotype material should be directed to the corresponding author.
Abbreviations
- dpi:
-
Days post-infection
- GWAS:
-
Genome-wide association study
- FPKM:
-
Fragments per kilobase of transcript per million mapped reads
- HSMI:
-
Heart and skeletal muscle inflammation
- IBS:
-
Identity-by-state
- i.p.:
-
Intraperitoneal injection
- ISAV:
-
Infectious salmon anaemia virus
- LD:
-
Linkage disequilibrium
- Mbp:
-
Mega base-pair
- PCA:
-
Principal component analysis
- PD:
-
Pancreas disease
- PRR:
-
Pattern recognition receptor
- QTL:
-
Quantitative trait locus
- SAV:
-
Salmonid alphavirus
- SNP:
-
Single nucleotide polymorphism
References
Weston JH, Welsh MD, McLoughlin MF, Todd D. Salmon pancreas disease virus, an alphavirus infecting farmed Atlantic salmon, Salmo salar L. Virology. 1999;256:188–95.
Hjeltnes B, Bang Jensen B, Bornø G, Haukaas A. In: Walde CS, editor. Fiskehelserapporten 2018: Veterinærinstituttet, Norway; 2019.
Fringuelli E, Rowley HM, Wilson JC, Hunter R, Rodger H, Graham DA. Phylogenetic analyses and molecular epidemiology of European Salmonid alphaviruses (SAV) based on partial E2 and nsP3 gene nucleotide sequences. J Fish Dis. 2008;31:811–23.
Graham DA, Fringuelli E, Rowley HM, Cockerill D, Cox DI, Turnbull T, et al. Geographical distribution of salmonid alphavirus subtypes in marine farmed Atlantic salmon, Salmo salar L., in Scotland and Ireland. J Fish Dis. 2012;35:755–65.
Weston JH, Graham DA, Branson E, Rowley HM, Walker IW, Jewhurst VA, et al. Nucleotide sequence variation in Salmonid alphaviruses from outbreaks of salmon pancreas disease and sleeping disease. Dis Aquat Org. 2005;66:105–11.
Jansen MD, Bang Jensen B, McLoughlin MF, Rodger HD, Taksdal T, Sindre H, et al. The epidemiology of pancreas disease in salmonid aquaculture: a summary of the current state of knowledge. J Fish Dis. 2017;40:141–55.
Karlsen M, Gjerset B, Hansen T, Rambaut A. Multiple introductions of Salmonid alphavirus from a wild reservoir have caused independent and self-sustainable epizootics in aquaculture. J Gen Virol. 2014;95:52–9.
Hodneland K, Bratland A, Christie KE, Endresen C, Nylund A. New subtype of Salmonid alphavirus (SAV), Togaviridae, from Atlantic salmon Salmo salar and rainbow trout Oncorhynchus mykiss in Norway. Dis Aquat Org. 2005;66:113–20.
Jensen BB, Gu J, Sindre H. Fish health report 2018. 2019. https://www.vetinst.no/rapporter-og-publikasjoner/rapporter/2019/fish-health-report-2018.
Aunsmo A, Valle PS, Sandberg M, Midtlyng PJ, Bruheim T. Stochastic modelling of direct costs of pancreas disease (PD) in Norwegian farmed Atlantic salmon (Salmo salar L.). Prev Vet Med. 2010;93:233–41.
Pettersen JM, Rich KM, Jensen BB, Aunsmo A. The economic benefits of disease triggered early harvest: a case study of pancreas disease in farmed Atlantic salmon from Norway. Prev Vet Med. 2015;121:314–24.
Taksdal T, Olsen AB, Bjerkås I, Hjortaas MJ, Dannevig BH, Graham DA, et al. Pancreas disease in farmed Atlantic salmon, Salmo salar L., and rainbow trout, Oncorhynchus mykiss (Walbaum), in Norway. J Fish Dis. 2007;30:545–58.
Jansen MD, Taksdal T, Wasmuth MA, Gjerset B, Brun E, Olsen AB, et al. Salmonid alphavirus (SAV) and pancreas disease (PD) in Atlantic salmon, Salmo salar L., in freshwater and seawater sites in Norway from 2006 to 2008. J Fish Dis. 2010;33:391–402.
McLoughlin MF, Graham DA. Alphavirus infections in salmonids - a review. J Fish Dis. 2007;30:545–58.
Røsæg MV, Garseth ÅH, Brynildsrud OB, Jansen MD. Pancreas disease caused by Salmonid alphavirus subtype 2 reduces growth and feed conversion in farmed Atlantic salmon. Prev Vet Med. 2019;169:104699.
Rodger H, Mitchell S. Epidemiological observations of pancreas disease of farmed Atlantic salmon, Salmo salar L., in Ireland. J Fish Dis. 2007;30:157–67.
Johansen LH, Thim HL, Jørgensen SM, Afanasyev S, Strandskog G, Taksdal T, et al. Comparison of transcriptomic responses to pancreas disease (PD) and heart and skeletal muscle inflammation (HSMI) in heart of Atlantic salmon (Salmo salar L). Fish Shellfish Immunol. 2015;46:612–23.
Gahlawat SK, Ellis AE, Collet B. Expression of interferon and interferon - induced genes in Atlantic salmon Salmo salar cell lines SHK-1 and TO following infection with salmon alphavirus SAV. Fish Shellfish Immunol. 2009;26:672–5.
Xu C, Guo T-C, Mutoloki S, Haugland O, Marjara IS, Evensen O. Alpha interferon and not gamma interferon inhibits Salmonid alphavirus subtype 3 replication in vitro. J Virol. 2010;84:8903–12.
Grove S, Austbø L, Hodneland K, Frost P, Løvoll M, McLoughlin M, et al. Immune parameters correlating with reduced susceptibility to pancreas disease in experimentally challenged Atlantic salmon (Salmo salar). Fish Shellfish Immunol. 2013;34:789–98.
Graham DA, Jewhurst VA, Rowley HM, McLoughlin MF, Rodger H, Todd D. Longitudinal serological surveys of Atlantic salmon, Salmo salar L., using a rapid immunoperoxidase-based neutralization assay for salmonid alphavirus. J Fish Dis. 2005;28:373–9.
Graham DA, Jewhurst VA, Rowley HM, McLoughlin MF, Todd D. A rapid immunoperoxidase-based virus neutralization assay for salmonid alphavirus used for a serological survey in Northern Ireland. J Fish Dis. 2003;26:407–13.
Jensen BB, Kristoffersen AB, Myr C, Brun E. Cohort study of effect of vaccination on pancreas disease in Norwegian salmon aquaculture. Dis Aquat Org. 2012;102:23–31.
McLoughlin MF, Peeler E, Foyle KL, O’Ceallachain D, Geoghegan F. An epidemiological investigation of the re-emergence of pancreas disease in Irish farmed Atlantic salmon (Salmo salar L.) in 2002. 2003. https://oar.marine.ie/handle/10793/239.
McLoughlin MF, Graham DA, Norris A, Matthews D, Foyle L, Rowley HM, et al. Virological, serological and histopathological evaluation of fish strain susceptibility to experimental infection with salmonid alphavirus. Dis Aquat Org. 2006;72:125–33.
Norris A, Foyle L, Ratcliff J. Heritability of mortality in response to a natural pancreas disease (SPDV) challenge in Atlantic salmon, Salmo salar L., post-smolts on a west of Ireland sea site. J Fish Dis. 2008;31:913–20.
Gonen S, Baranski M, Thorland I, Norris A, Grove H, Arnesen P, et al. Mapping and validation of a major QTL affecting resistance to pancreas disease (Salmonid alphavirus) in Atlantic salmon (Salmo salar). Heredity (Edinb). 2015;115:405–14.
Cote J-F. Identification of an evolutionarily conserved superfamily of DOCK180-related proteins with guanine nucleotide exchange activity. J Cell Sci. 2002;115:4901–13.
Ushach I, Arrevillaga-Boni G, Heller GN, Pone E, Hernandez-Ruiz M, Catalan-Dibene J, et al. Meteorin-like/Meteorin-β is a novel immunoregulatory cytokine associated with inflammation. J Immunol. 2018;201:3669–76.
Wu Y, Fan W, Li X, Chen H, Takáč T, Šamajová O, et al. Expression and distribution of extensins and AGPs in susceptible and resistant banana cultivars in response to wounding and Fusarium oxysporum. Sci Rep. 2017;7:42400.
Balaji V, Smart CD. Over-expression of snakin-2 and extensin-like protein genes restricts pathogen invasiveness and enhances tolerance to Clavibacter michiganensis subsp. michiganensis in transgenic tomato (Solanum lycopersicum). Transgenic Res. 2012;21:23–37.
Fragkostefanakis S, Dandachi F, Kalaitzis P. Expression of arabinogalactan proteins during tomato fruit ripening and in response to mechanical wounding, hypoxia and anoxia. Plant Physiol Biochem. 2012;52:112–8.
Lin A, Hokugo A, Choi J, Nishimura I. Small cytoskeleton-associated molecule, fibroblast growth factor receptor 1 oncogene partner 2/wound inducible transcript-3.0 (FGFR1OP2/wit3.0), facilitates fibroblast-driven wound closure. Am J Pathol. 2010;176:108–21.
Lin A, Hokugo A, Nishimura I. Wound closure and wound management. Cell Adhes Migr. 2010;4:396–9.
Lien S, Koop BF, Sandve SR, Miller JR, Kent MP, Nome T, et al. The Atlantic salmon genome provides insights into rediploidization. Nature. 2016;533:200–5.
Jørgensen SM, Afanasyev S, Krasnov A. Gene expression analyses in Atlantic salmon challenged with infectious salmon anemia virus reveal differences between individuals with early, intermediate and late mortality. BMC Genomics. 2008;9:179.
Shi Z, Cai Z, Sanchez A, Zhang T, Wen S, Wang J, et al. A novel toll-like receptor that recognizes vesicular stomatitis virus. J Biol Chem. 2011;286:4517–24.
Busslinger M. Transcriptional control of early B cell development. Annu Rev Immunol. 2004;22:55–79.
Jin T, Xu X, Hereld D. Chemotaxis, chemokine receptors and human disease. Cytokine. 2008;44:1–8.
Nagamine Y, Pong-Wong R, Navarro P, Vitart V, Hayward C, Rudan I, et al. Localising loci underlying complex trait variation using regional genomic relationship mapping. PLoS One. 2012;7:e46501.
Canela-Xandri O, Law A, Gray A, Woolliams JA, Tenesa A. A new tool called DISSECT for analysing large genomic data sets using a Big Data approach. Nat Commun. 2015;6:10162.
Garcia-Zepeda EA, Rothenberg ME, Ownbey RT, Celestin J, Leder P, Luster AD. Human eotaxin is a specific chemoattractant for eosinophil cells and provides a new mechanism to explain tissue eosinophilia. Nat Med. 1996;2:449–56.
Menzies-Gow A, Ying S, Sabroe I, Stubbs VL, Soler D, Williams TJ, et al. Eotaxin (CCL11) and Eotaxin-2 (CCL24) induce recruitment of eosinophils, basophils, neutrophils, and macrophages as well as features of early- and late-phase allergic reactions following cutaneous injection in human atopic and nonatopic volunteers. J Immunol. 2014;169:2712–8.
Menzies-Gow A, Ying S. Eotaxin (CCL11) and eotaxin-2 (CCL24) induce recruitment of eosinophils, basophils, neutrophils, and macrophages as well as features of early-and late-phase. J Immunol. 2002;169:2712–8.
De Paulis A, Annunziato F, Di Gioia L, Romagnani S, Carfora M, Beltrame C, et al. Expression of the chemokine receptor CCR3 on human mast cells. Int Arch Allergy Immunol. 2001;124:146–50.
Nakayama T, Watanabe Y, Oiso N, Higuchi T, Shigeta A, Mizuguchi N, et al. Eotaxin-3/CC chemokine ligand 26 is a functional ligand for CX3CR1. J Immunol. 2010;185:6472–9.
Yasuike M, de Boer J, von Schalburg KR, Cooper GA, McKinnel L, Messmer A, et al. Evolution of duplicated IgH loci in Atlantic salmon, Salmo salar. BMC Genomics. 2010;11:486.
Li J, Barreda DR, Zhang YA, Boshra H, Gelman AE, LaPatra S, et al. B lymphocytes from early vertebrates have potent phagocytic and microbicidal abilities. Nat Immunol. 2006;7:1116–24.
Lund H, Bakke AF, Sommerset I, Afanasyev S, Schriwer G, Thorisdottir A, et al. A time-course study of gene expression and antibody repertoire at early time post vaccination of Atlantic salmon. Mol Immunol. 2019;106:99–107.
Robinson NA, Krasnov A, Burgerhout E, Johnsen H, Moghadam HK, Hillestad B, et al. Response of the salmon heart transcriptome to pancreas disease: differences between high- and low-ranking families for resistance. Sci Rep. 2020;10:868.
Visscher PM, Goddard ME. A general unified framework to assess the sampling variance of heritability estimates using pedigree or marker-based relationships. Genetics. 2015;199:223–32.
Taksdal T, Bang Jensen B, Böckerman I, Mcloughlin MF, Hjortaas MJ, Ramstad A, et al. Mortality and weight loss of Atlantic salmon, Salmon salar L., experimentally infected with salmonid alphavirus subtype 2 and subtype 3 isolates from Norway. J Fish Dis. 2015;38:1047–61.
Misztal I, Tsuruta S, Lourenco D, Aguilar I, Legarra A, Vitezica Z. Manual for BLUPF90 family of programs. USA: Univ Georg Athens; 2018.
Hill WG. On estimation of genetic variance within families using genome-wide identity-by-descent sharing. Genet Sel Evol. 2013;46:62.
Ødegård J, Meuwissen THE. Estimation of heritability from limited family data using genome-wide identity-by-descent sharing. Genet Sel Evol. 2012;44:16–8.
Dagnachew B, Meuwissen T. Accuracy of within-family multi-trait genomic selection models in a sib-based aquaculture breeding scheme. Aquaculture. 2019;505:27–33.
Yang J, Lee SH, Goddard ME, Visscher PM. GCTA: a tool for genome-wide complex trait analysis. Am J Hum Genet. 2011;88:76–82.
Houston RD, Taggart JB, Cézard T, Bekaert M, Lowe NR, Downing A, et al. Development and validation of a high density SNP genotyping array for Atlantic salmon (Salmo salar). BMC Genomics. 2014;15:90.
Hillestad B, Moghadam HK. Genome-wide association study of Piscine Myocarditis virus (PMCV) resistance in Atlantic Salmon (Salmo salar). J Hered. 2019;110:720–6.
Kang HM, Sul JH, Service SK, Zaitlen NA, Kong SY, Freimer NB, et al. Variance component model to account for sample structure in genome-wide association studies. Nat Genet. 2010;42:348–54.
Segura V, Vilhjálmsson BJ, Platt A, Korte A, Seren Ü, Long Q, et al. An efficient multi-locus mixed-model approach for genome-wide association studies in structured populations. Nat Genet. 2012;44:825–30.
Duggal P, Gillanders EM, Holmes TN, Bailey-Wilson JE. Establishing an adjusted p-value threshold to control the family-wide type 1 error in genome wide association studies. BMC Genomics. 2008;9:516.
Sham PC, Purcell SM. Statistical power and significance testing in large-scale genetic studies. Nat Rev Genet. 2014;15:335–46.
Chen H, Wang C, Conomos MP, Stilp AM, Li Z, Sofer T, et al. Control for population structure and relatedness for binary traits in genetic association studies via logistic mixed models. Am J Hum Genet. 2016;98:653–66.
Chang CC, Chow CC, Tellier LC, Vattikuti S, Purcell SM, Lee JJ. Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience. 2015;4:7.
Utsunomiya YT, do Carmo AS, Carvalheiro R, HHR N, Matos MC, Zavarez LB, et al. Genome-wide association study for birth weight in Nellore cattle points to previously described orthologous genes affecting human and bovine height. BMC Genet. 2013;14:52.
Golden Helix I. SNP & Variation Suite ™ (Version 8.8.3) [Software]. Bozeman, MT: Golden Helix, Inc. Available from http://www.goldenhelix.com.
Vilhjalmsson BJ. Mixed model package for genome-wide association mapping. 2012. https://github.com/bvilhjal/mixmogam.
Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–20.
Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25:1105–11.
Anders S, Huber W. Differential expression analysis for sequence count data. Genome Biol. 2010;11:R106.
Moghadam HK, Johnsen H, Robinson N, Andersen Ø, Jørgensen EH, Johnsen HK, et al. Impacts of early life stress on the methylome and transcriptome of Atlantic salmon. Sci Rep. 2017;7:5023.
Boison SA, Gjerde B, Hillestad B, Makvandi-Nejad S, Moghadam HK. Genomic and transcriptomic analysis of amoebisc gill disease resistansce in Atlantic salmon (Salmo salar L.). Front Genet. 2019;10:68.
Acknowledgements
Not applicable.
Funding
The Research Council of Norway supported the transcriptome sequencing and analysis of this work under the Grant 226275 (Precision Breeding). The funding agency played no role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
B.H. and H.M. conceived and designed the experiment. B.H. supervised the challenge test and arranged for the collection of the samples for both transcriptome as well as SNP genotyping. H.M., S.M. and A.K. performed RNA extraction, quantification, bioinformatics and statistical analysis related to the transcriptomic data. H.M. and B.H. performed analysis of the genotype data. All authors contributed to the development of the manuscript and interpretation of the data and improvement of the paper. The author(s) read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The challenge trial was approved by the Food and Safety Authority in Norway, Department for National Assignments (FOTS ID: 8137–15/180369), which conforms to the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication 85–23, revised 1996).
Consent for publication
Not applicable.
Competing interests
No potential conflicts of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1: Supplementary Figure 1.
Heatmap of the immune-related genes, with different expression profiles between the moribund (M) and survived animals (S) within the QTL containing regions of a. ssa03 and b. ssa07.
Additional file 2: Supplementary Table 1.
Differentially expressed genes between the moribund animals at 11 dpi (sample_1) and animals that survived the challenge (sample_2). Supplementary Table 2. Functional enrichment analysis of differentially expressed genes between the moribund animals at 11 dpi and animals that survived the challenge.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Hillestad, B., Makvandi-Nejad, S., Krasnov, A. et al. Identification of genetic loci associated with higher resistance to pancreas disease (PD) in Atlantic salmon (Salmo salar L.). BMC Genomics 21, 388 (2020). https://doi.org/10.1186/s12864-020-06788-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-020-06788-4