
Abstract
Background
Chrysanthemum is a leading cut flower species. Most conventional cultivars flower during the fall, but the Chrysanthemum morifolium ‘Yuuka’ flowers during the summer, thereby filling a gap in the market. To date, investigations of flowering time determination have largely focused on fall-flowering types. Little is known about molecular basis of flowering time in the summer-flowering chrysanthemum. Here, the genome-wide transcriptome of ‘Yuuka’ was acquired using RNA-Seq technology, with a view to shedding light on the molecular basis of the shift to reproductive growth as induced by variation in the photoperiod.
Results
Two sequencing libraries were prepared from the apical meristem and leaves of plants exposed to short days, three from plants exposed to long days and one from plants sampled before any photoperiod treatment was imposed. From the ~316 million clean reads obtained, 115,300 Unigenes were assembled. In total 70,860 annotated sequences were identified by reference to various databases. A number of transcription factors and genes involved in flowering pathways were found to be differentially transcribed. Under short days, genes acting in the photoperiod and gibberellin pathways might accelerate flowering, while under long days, the trehalose-6-phosphate and sugar signaling pathways might be promoted, while the phytochrome B pathway might block flowering. The differential transcription of eight of the differentially transcribed genes was successfully validated using quantitative real time PCR.
Conclusions
A transcriptome analysis of the summer-flowering cultivar ‘Yuuka’ has been described, along with a global analysis of floral transition under various daylengths. The large number of differentially transcribed genes identified confirmed the complexity of the regulatory machinery underlying floral transition.
Similar content being viewed by others


Background
The transition to flowering is arguably the most critical switch in a plant’s life cycle [1]. It is triggered by a combination of environmental cues and endogenous signals [2]. The appropriate timing of the transition is crucial for reproductive success, and hence is a major production issue for ornamental plant cultivators [3]. In the model plant Arabidopsis thaliana, floral transition is controlled by an intricate regulatory network comprising six distinct pathways, namely the photoperiod, the autonomous, the vernalization, the gibberellin (GA), the ambient temperature and the age pathways [2, 4]. The outputs of the various pathways are integrated by the products of the floral integrator genes FLOWERING LOCUS T (FT) and SUPPRESSOR OF OVEREXPRESSION OF CONSTANS (SOC1), which promote flowering by inducing the expression of the floral meristem identity genes LEAFY (LFY) and APETALA1(AP1) [4–6]. The gene CO (encoding a BBX transcription factor) is a key component of the photoperiod pathway; it promotes flowering by up-regulating FT [7–9]. The autonomous and vernalization pathways activate flowering by down-regulating the floral repressor gene FLC, which encodes a MADS-box transcription factor acting to repress the floral integrators [1, 10]. An increase in GA synthesis promotes flowering, especially in plants experiencing short days (SD); early flowering results from the activation of SOC1 [4, 11]. Under an otherwise non-inductive photoperiod, the flowering of A. thaliana is also accelerated by a rise in the ambient temperature [12]. As the plant ages, SQUAMOSA PROMOTER BINDING LIKE (SPL) transcription factors promote flowering by inducing the expression of the floral integrators LFY, FRUITFULL (FUL) and SOC1 [4]. In addition to photoperiod and ambient temperature, flowering time can be affected by the content of trehalose-6-phosphate (T6P), acting through the sugar signaling pathway [13, 14].
Chrysanthemum (Chrysanthemum morifolium) is a popular ornamental plant worldwide [15, 16]. Most Chinese commercial cultivars will only flower if exposed to SD, so achieving year-round production requires flowering to be induced by controlling the daylength, which is a costly and energy-consuming measure [17, 18]. Even with precise control over photoperiod, premature flowering can be induced by higher than optimal ambient temperature, which is often experienced during the summer months [19]. The Japanese cultivar ‘Yuuka’ flowers naturally over the period July-September, filling a gap in the chrysanthemum cut flower market, its flowering is accelerated under SD comparing with the natural conditions [20, 21].
Some transcriptomic analysis relevant to flowering time regulation has been attempted in chrysanthemum and its close relatives. Some features of the chrysanthemum transcriptome related to photoperiod-responsive floral development have been explored using cDNA-AFLP technique [18]. In the cultivar ‘Fenditan’, a number of genes involved in the photoperiod pathway and flower organ determination have been identified using Illumina sequencing technology [22]. Meanwhile, an analysis of Chrysanthemum lavandulifolium (Fisch. ex Trautv) succeeded in identifying 211 flowering-related genes, along with a large number of genes which were differentially transcribed between immature plants and those on the cusp of flowering. Since the constitutive expression in C. morifolium of CsFTL3, a FT-like gene harbored by Chrysanthemum seticuspe, resulted in acceleration in flowering under non-inductive conditions, this gene has been considered to be an important regulator of the photoperiod flowering pathway [19]. The product of CsAFT, a gene encoding an anti-florigenic FT/TFL1 family protein, acts as a systemic floral inhibitor, determining flowering time under SD [23]. The suppression of CmBBX24 (encoding a BBX transcription factor) causes plants to flower earlier than expected [24]. Other than these few examples, the molecular basis of the response to photoperiod in summer-flowering chrysanthemums has not been deduced in any detail.
Here, a comparison between the RNA-Seq acquired transcriptomes of ‘Yuuka’ plants exposed to contrasting photoperiods was used to characterize the transcriptome associated with floral transition, with a view to clarifying the molecular basis of flowering time, which would be helpful to regulate flowering in chrysanthemum to achieve year-round production and the identified important regulatory genes will be useful for flowering-related transgenic breeding.
Results
Transcriptome sequencing and read assembly
To generate a broad survey of genes involved in floral transition induced by different photoperiod, six mRNA libraries were constructed respectively from six different samples of leaves and shoots apical meristem, and denoted CK, S1, S2, L1, L2 and L3 (Fig. 1). The total number of raw reads obtained from the six libraries (CK, S1, S2, L1, L2 and L3) was, respectively, 55,342,446, 58,118,768, 56,587,092, 54,554,772, 55,128,750 and 54,538,450. After filtering, approximately 316 × 106 clean reads (28.42 × 109 nt) were generated. The raw sequence data has been deposited in the NCBI Sequence Read Archive database under accession number SRP076366. The average Q20 and GC percentages were, respectively, 97.72 and 42.86 % (Table 1). After assembly, 115,300 non-redundant Unigenes (57,718 distinct clusters and 57,582 distinct singletons) were recognized; these were of average length 1030 nt and associated with an N50 (genome splicing quality) of 1588 nt (Additional file 1: Table S1).

The source of the mRNA prepared from ‘Yuuka’ plants grown under either SD or LD. CK: plants sampled prior to their exposure to a differential photoperiod; S1, S2: plants grown under SD; L1-L3: plants grown under LD
Gene annotation and functional classification
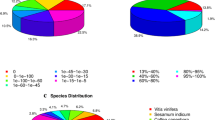
To acquire expression and functional annotation of the assembled unigenes, the assembled Unigene sequences were aligned against the NR, Swiss-Prot, KEGG and COG protein databases and the NT nucleotide database. The number of hits with the NR database was 67,964 (Table 2), and the details of E-value distribution, similarity distribution and species distribution form the result of NR annotation were presented in Fig. 2; while the equivalent numbers for the Swiss-Prot, KEGG, COG and NT databases were, respectively, 46,591, 26,000, 42,512 and 50,337. When subjected to KEGG pathway analysis, 127 pathways were identified, among which the most frequently occurring 30 are detailed in Table 3: the major ones identified were ‘metabolism’, ‘secondary metabolites’, ‘plant-pathogen interaction’, ‘hormone signal transduction’ and ‘spliceosome’. Among the 25 COG categories, the cluster for ‘general function prediction only’ (8974) represented the largest group, followed by ‘transcription’ (4760) and ‘replication, recombination and repair’ (4639). The ‘RNA processing and modification’ (398), ‘Extracellular structure’ (13) and ‘Nuclear structure’ (9) represented the smallest groups (Fig. 3). On the basis of gene ontology (GO), ~50,000 unigenes could be assigned a GO category (Fig. 4). The terms ‘cellular process’, ‘cell’ and ‘catalytic activity’ were dominant in each of these categories.

BLAST outputs of Unigene sequences against the NR database. a The distribution of E-values; b the distribution of similarity scores; c the mix of species contributing sequences to the analysis

Clusters of orthologous groups (COG) classification in Chrysanthemum morifolium ‘Yuuka’. These 25,994 sequences have a COG classification within the 25 categories

GO classification of the unigenes
Identification of TFs and other genes involved in flowering time control
According to unigene annotation, 2000 of the sequences were identified as encoding a member of one of 45 TF families (Additional file 2: Table S2), among which the most frequently occurring were WRKY, followed by G2-like, C3H and MYB. With respect to genes involved in the determination of flowering time, 46 homologs of A. thaliana genes were uncovered (Additional file 3: Table S3). Among those associated with the photoperiod pathway were three homologs of ELF3 (EARLY FLOWERING 3), one of ELF4, four of PIF3, three of FKF1, six of LHY (LATE ELONGATED HYPOCOTYL), six of PRRs (PSEUDO-RESPONSE REGULATORs), one of CCA1 (CIRCADIAN CLOCK ASSOCIATED 1), 17 of GI and 25 of CO. Other flowering time pathways were also well represented, as for example homologs of GA20ox, EMF2, GAI (GIBBERELLIN-INSENSITIVE), GID1 (GA INSENSITIVE DWARF1) and SPY in GA pathway, SVP (SHORT VEGETATIVE PHASE), FCA, FVE, FY, FPA and HOS1 in ambient temperature pathway; LD in autonomous pathway; SPLs (SQUAMOSA PROMOTER BINDING-LIKEs) and JMJ14 in age pathway; FLC (FLOWERING LOCUS C), VIN1 (VERNALIZATION1) and VIN3 in vernalization pathway. A number of flowering integrators were also identified. The details of 46 homologs of flowering time genes were presented in (Additional file 3: Table S3).
The effect of photoperiod on the transcription of floral initiation genes
Genes involved in the determination of photoperiod-induced flowering time in ‘Yuuka’ were identified by initially calculating the transcript abundance of all Unigenes of the above six libraries successively, based on the number of reads per kilobase per million mapped reads (FPKM) method [25]. Subsequently, each Unigene of the five pairwise contrasts CK vs S1 (CKS1), CK vs S2 (CKS2), CK vs L1 (CKL1), CK vs L2 (CKL2) and CK vs L3 (CKL3) was scanned for significant differential transcription, adopting a false discovery rate (FDR) threshold of 0.001 and a |log2ratio| threshold of 1. The resulting set of differentially transcribed genes (DTGs) is given in Additional file 4: Table S4, Additional file 5: Table S5, Additional file 6: Table S6, Additional file 7: Table S7, Additional file 8: Table S8, Additional file 9: Table S9, Additional file 10: Table S10. A greater number of genes were down-regulated in these contrasts than were up-regulated (Fig. 5). More DTGs were identified in CK vs S1 than in CK vs L1, indicating that the response to SD in terms of floral induction and development was more substantial than that to LD; A total of 37 and 38 Unigenes involved the photoperiod pathway with differential transcript abundance were also identified in contrasts L1 vs S1and L2 vs S2 (Additional file 11: Table S11). The details of the key DTGs identified are presented in Tables 4, 5 and Additional file 12: Table S12.

DTGs identified in pairwise comparisons between libraries
DTGs involved in the response to floral induction
Many of the DTGs were associated with one of the photoperiod, GA, T6P or sugar signaling pathways (Tables 4 and 5). Unigene11703_All, 38196_All and 29301_All, which are homologs of A. thaliana GI (a key component of the photoperiod pathway) [26, 27], were all up-regulated under both LD and SD, while the abundance of a fourth GI homolog’s (Unigene18727_All) transcript was increased only under LD. Three CO-like homologs (Unigene11436_All, 34062_All and 11502_All) were up-regulated under both LD and SD, CL7046. Contig5_All was only up-regulated under SD and CL11101.Contig1_All under LD, while two additional (Unigene14733_All and CL201.Contig1_All) were down-regulated under both photoperiods. The transcript abundance of one GA20ox homolog (CL9282.Contig2_All) was enhanced under both LD and SD. EMF1 (EMBRYONIC FLOWER 1) and SPY (SPINDLY) were represented by, respectively, CL6401.Contig2_All and CL4303.Contig1_All; the transcript of both of these was less abundant under SD than in CK, while an EMF2 homolog (CL1282.Contig10_All) was induced by LD. Meanwhile, under LD, Unigene28531_All, a PHYB (PHYTOCHROME B) homolog, along with a sucrose synthase homolog (CL2855.Contig4_All) and seven T6P synthase homologs (Unigene10839_All, 35642_All, 10840_All, CL6845.Contig1_All, 24962_All, CL1264.Contig2_All and CL2780.Contig4_All) were all up-regulated. The implication was that the photoperiod and the GA pathways both represent the primary route by which flowering in ‘Yuuka’ is controlled under SD, while under LD, it is the T6P and sucrose signaling pathways which accelerate flowering and the photoperiod pathway which blocks it. No DTG related to other known flowering pathways including ambient temperature, aging, autonomous and vernalization pathway was found. A proposed model for the regulation of floral induction in ‘Yuuka’ was presented in Fig. 6.

A proposed model for the regulation of floral induction in summer-flowering ‘Yuuka’, taking into account the ambient photoperiod
The differential transcription of TF genes
Most of the key regulatory genes involved in floral induction encode a TF. Members of the bHLH, MYB, C3H, BBX, MADS, GATA and WRKY TF families were among the DTGs associated with floral induction in ‘Yuuka’ (Additional file 12: Table S12). Of the bHLH DTGs, 19 encoded Phytochrome-Interacting Factors (PIFs) proteins – two of which (CL10741.Contig1_All and Unigene8480_All) were only up-regulated under SD, while LD induced three other members of this family (CL3244.Contig1_All, Unigene 22647_All and 23299_All) and repressed one (CL11069.Contig3_All); the other 13 were all induced under both photoperiods. With respect to the BBX family, the transcript abundance of CL11101.Contig1_All was increased under LD, and both CL7046.Contig5_All and Unigene11562_All showed increased transcript abundance only under SD. Of the MYB DTGs, three were down- and one was up-regulated by both photoperiod treatments; in four, transcript abundance was reduced under SD, and in another two, it was increased under LD. Certain members of the MADS family (Unigene15098_All and 22350_All) were expressed during the early stages of floral induction, while other members (Unigene12477_All, 36671_All, 56025_All, 8889_All, 33409_All and CL4112.Contig3_All) were expressed throughout floral induction and differentiation.
Quantitative real time PCR (qRT-PCR) validation of differential transcription
To further verify the genes transcript profiles obtained from Illumina RNA-Seq results, a selection of 8 DTGs for their key roles in flowering time control was used for validation by qRT-PCR: the chosen genes were homologs of FLAVINBINDING, KELCH REPEAT, F-BOX 1(FKF1), COL9, COL16, MYB, GATA, GA20ox, APETALA 1(AP1) and AGAMOUS-LIKE 8 (AGL8). The outcome in each case was consistent with the RNA-Seq assay (Fig. 7).

qRT-PCR validation of eight DTGs selected from the RNA-Seq experiment. (a, b) S1 samples; (c, d) L3 samples; (e–h) S2 samples
Discussion
The transcriptome of summer-flowering chrysanthemum
The output of the RNA-Seq-acquired transcriptome of ‘Yuuka’ was a set of some 316 million clean reads, which covered about 28.42 × 109 nt of sequence; the sequences resolved into >115,000 Unigenes, more than 60 % of which could be assigned a putative function. An equivalent transcriptome acquisition program in C. morifolium realized around 91,000 unigenes sequences, of which only ~47 % could be assigned a putative functions [22], while in C. lavandulifolium, nearly 109,000 unigenes were assembled, about 53 % of which were annotated [28]. So far, the total number of Dendranthema spp. unigene sequences deposited in the NCBI EST database is a mere 7371, which means that the present data have provided a >1500 % increase in the representation of the transcriptome of this economically valuable ornamental species.
Regulatory networks controlling the switch to reproductive growth in ‘Yuuka’
Though the differential transcripts represent both developmental as well as condition-dependent differences at the present could not be distinguished, those genes involved in the known flowering pathway and their othologs’ reported to function in flowering time, were further chosed to analyze in detail. Based on the framework of floral induction established in A. thaliana, six regulatory pathways were implicated in the response of ‘Yuuka’ to variation in the photoperiod. In many plants, the photoperiod is the single most important environmental cue affecting the flowering time [6]. The light receptors required for its sensing are the phytochromes (PHYs) and cryptochromes (CRYs), along with phototropin (PHOT) [29]. The transcriptome of ‘Yuuka’ harbored homologs of PHYA, PHYB, PHYC, PHYE, CRY1 and CRY2, but none related to PHOT. Other genes related to the interaction of the photoperiod and flowering time were also among the DTGs, notably ELFs, PIFs, FKF1, LHY, PRRs, CCA1, GI and CO. One FT homolog (Unigene23925_All) was up-regulated under LD, but under SD the otholog of CsFTL3 was not, perhaps because ‘Yuuka’ may be a quantitative rather than a qualitative SD plant. In C. lavandulifolium, genes related to the photoperiod, vernalization, GA and autonomous pathways have all been implicated in the determination of flowering time [28], but this species flowers in the fall (rather than in the summer as ‘Yuuka’ does), so there are probably major differences in the floral switch mechanisms utilized by these members of the chrysanthemum family. In A. thaliana, the gene TPS1 (encoding a T6P synthase 1) is an important controller of the floral transition [13]. Here, three TPS1 homologs (CL1264.Contig2_All, CL2780.Contig4_All and Unigene24962_All) were all up-regulated under LD, implying enhanced activity in the T6P signaling pathway. Sucrose not only acts as a source of energy, but also has a role in signaling through its regulation of the SPLs [30–35]. Here, the transcript abundance of the Unigene CL2855.Contig4_All, a homolog of a gene encoding sucrose synthase, was increased under LD. The conclusion was that both the T6P and sugar sensing/signalling pathways are likely important for the regulation of flowering in ‘Yuuka’ under LD.
The regulatory networks underlying the control of flowering time depend on the photoperiod regime
The photoperiod which induces the switch to reproductive growth in summer-flowering chrysanthemum varieties is longer than that required by conventional varieties [36]. By analysing the transcriptome of ‘Yuuka’ plants exposed to a contrasting photoperiods, it was possible to identify which genes responded to this major environmental cue. Both under SD and LD conditions, a number of the DTGs were found to belong to the photoperiod pathway, some of which (for example a homolog of PHYB, which was up-regulated only under LD) responded in a photoperiod-specific manner. The gene CO (syn. BBX1) was one of the first floral induction genes to be isolated in A. thaliana [37, 38], although in the meantime, additional members of the BBX family have been shown to influence flowering time as well (in particular, the products of BBX4, BBX6, BBX7, BBX24 and BBX32), some positively and others negatively [39]. The rice homolog of BBX14 delays flowering under both LD and SD [40]. CO is transcribed in A. thaliana plants grown under SD, but its product is not accumulated, thereby delaying the onset of flowering [41]. Here, seven Unigenes encoding BBX TFs were represented among the DTGs; their transcription profiling was quite diverse. The implication is that CO-like proteins are likely to contribute in various ways to control the flowering time of ‘Yuuka’.
The phytohormone GA acts as a growth regulator and an accelerator of flowering in A. thaliana [42]. Particularly under SD, any disturbance to the GA pathway, or any reduction in tissue GA content delays flowering. The enzyme GA20ox catalyzes several steps in GA synthesis [43]. GID1 is known to be a receptor for GA, since the triple mutant gid1a/gid1c/gid1a is dwarfed and flowers late [44]. SPY acts upstream of both GAI and RGA to negatively regulate the GA response [45]. Here, a GA20ox homolog (CL9282.Contig2_All) was up-regulated in a photoperiod-independent manner, while a SPY (CL4303.Contig1_All) homolog was down-regulated under SD. The implication was that both the photoperiod and GA pathways promote flowering under SD; meantime, under LD, the T6P and sugar signaling pathways promote flowering, while the photoperiod pathway gene PHYB is up-regulated (the product of this gene blocks flowering).
TF genes involved in floral induction
Transcriptional regulation is an important component of plant growth and development [46]. In A. thaliana, a number of MYB proteins are known to regulate the switch to reproductive growth. An increased abundance of MYB30 in the phloem has been shown to accelerate flowering via its regulation of FT [47]. Another MYB protein (EFM) acts to repress FT transcription in the leaf vasculature [48]. The constitutive expression of CmMYB2 in A. thaliana induces a delay in flowering [49]. Here, ten Unigenes encoding MYB proteins were represented among the set of DTGs. MADS TFs are widely implicated in the control of flower development [50]. The A. thaliana gene SVP encodes one such protein, which, during the plant’s vegetative phase, functions as a flowering repressor [51]. Meanwhile, the constitutive expression in tobacco of a Betula platyphylla AP1 homolog accelerates flowering [52]. AGL15 and AGL18, along with SVP and AGL24, are required to block the initiation of flowering in vegetative organs [53]. Here, nine homologs of various MADS TFs were represented among the DTGs. The implication is that in C. morifolium, as also in a number of other species, MADS TFs are important controllers of floral induction.
In A. thaliana, the bHLH protein PIF4 activates FT with the increase of temperature [54]. A number of PIF proteins (PIF1, PIF3, PIF4 and PIF5) are known to act as constitutive repressors of photomorphogenesis in the dark, and their proteolytic degradation is required to abolish the repression [55]. Members of the TF families WRKY, C3H and GATA are also implicated in the machinery underlying floral induction [56–60]. Here, 44 WRKY members were detected as DTGs. Thus the members of the WRKY family are likely to be important for floral induction in C. morifolium. With one exception, all the GATA homologs were up-regulated, while 16 of the C3H homologs were down-regulated under both SD and LD. The different expression pattern of C3Hs showed its different roles in floral induction. Given that these TFs likely regulate floral induction (either directly or indirectly) differentially depending on the photoperiod, characterization of the DTGs encoding TFs might shed light on the regulation of flowering time.
Conclusions
The present analysis of the ‘Yuuka’ transcriptome has shown that under SD, flowering might be promoted largely though the activity of the photoperiod and GA pathways, while under LD, the T6P and sugar signaling pathways might promote flowering and PHYB might block it. The DTGs identified here as being involved in the process of floral transition provide potential targets for the manipulation of flowering time in chrysanthemum, while their actual functions need to be fully elucidated in future.
Methods
Plant materials and growing conditions
C. morifolium cultivar ‘Yuuka’ cuttings were obtained from Jiangsu Junma Park Technology Co. Ltd (Zhangjiagang, China). Uniformly-sized cuttings were planted into a 2:1 mixture of peat and perlite at the end of December and grown in a greenhouse under natural light, with additional lighting between 22:00 and 02:00 provided by high pressure sodium lamps. Rooted cuttings were transplanted into garden soil and maintained in a greenhouse running at 5–7 °C, ventilated when the temperature increased above 10 °C, under the same lighting conditions as described above. By April, the plants had reached a height of around 60 cm, and they were exposed to 1 week at 15 °C in advance to induce the plants to enter the photo-sensitive phase. Some plants were then subjected to either a SD (11.5 h photoperiod) or to LD (15.5 h photoperiod) for a further 60 days. Visible flower buds first appeared on the SD plants after 24 days, and on the LD ones after 44 days. The fourth fully expanded leaves and stem apical meristem were sampled on days 3, 5, 7 and 10 after the initiation of the photoperiod treatment and bulked to form sample S1, while S2 was a bulk of material harvested on days 16 and 20; similarly, the L1 sample was a bulk of material harvested on days 3, 5, 7 and 10, L2 on days 16 and 20, and L3 on days 28, 32 and 36. The CK sample was harvested immediately prior to the imposition of the photoperiod treatments. Three plants were sampled on each occasion and maintained as three biological replicates. Immediately after harvest, the tissue was snap-frozen in liquid nitrogen and stored at −80 °C until required.
RNA extraction, cDNA library construction and Illumina sequencing
RNA was extracted using a Total RNA Isolation System (Takara, Kyoto, Japan) following the manufacturer’s protocol. Its quality and quantity were assessed with either a 2100 Bioanalyzer RNA Nano chip device (Agilent, Santa Clara, CA, USA) or a Nanodrop ND-430 1000 spectrophotometer (Agilent, Santa Clara, CA, USA), and only samples delivering a λ260/280 ratio of 1.8–2.1, a λ260/230 ratio of 2.0–2.5 and an RNA integrity number >8.0 were retained. A 30 μg pool of RNA formed by combining 10 μg from each biological replicate was subjected to RNase-free DNase I (Takara, Dalian, China) treatment to remove contaminating DNA, and mixed with oligo (dT) coated magnetic beads to separate the poly A fraction. Fragmentation buffer was added to break the mRNA into the short fragments required as template for the synthesis of the first cDNA strand, which was achieved by random hexamer priming. The second cDNA strand was synthesized by a reaction driven by DNA polymerase I (Takara, Dalian, China). The resulting dsDNA fragments were purified using a QiaQuick PCR purification kit (Qiagen, Valencia, CA, USA) and resuspended in EB buffer for end repair and the addition of an adenine. Sequencing adapters were ligated onto the dsDNAs. Suitable fragments were selected for PCR amplification following agarose gel electrophoretic separation. The resulting libraries were then submitted for sequencing by a HiSeqTM 2000 device (Illumina, San Diego, CA, USA) at the Beijing Genomics Institute (Shenzhen, China; http://www.genomics.cn/index.php).
Transcriptome assembly and and gene annotation
The raw sequence data acquired from each of the libraries was stored in fastq format. Reads including either adapter sequence and/or more than five unknown nucleotides were removed, as were those having a quality value <10. The remaining clean reads were taken forward for bioinformatic analysis. A transcriptome assembly of the filtered sequences was carried out using the Trinity program (−−seqType fq --min_contig_length 100; −-min_glue 3 --group_pairs_distance 250; −-path_reinforcement_distance 85 --min_kmer_cov 3) [61]. Trinity includes three independent software modules: Inchworm, Chrysalis, and Butterfly. In brief, the Inchworm module was applied to assemble the sequences into the unique sequences of transcripts, and a full length transcript for the predominant isoform was generated; the Inchworm-derived contigs were then clustered and a complete de Bruijn graph for each cluster constructed. The Chrysalis module was then applied to partition the full read set among these disjoint graphs, and finally, the Butterfly module was used to process the individual graphs in parallel to derive full length transcripts for alternatively spliced isoforms, and to identify transcripts produced by paralogous genes. The resulting outputs were termed “Unigenes”. When multiple samples from the same species were sequenced, the Unigene set from each sample was subjected to further process of sequence splicing and redundancy removing with sequence clustering software to acquire non-redundant Unigenes as long as possible. The Unigenes were divided into clusters and singletons. Each cluster gathered several Unigene sequences sharing a similarity of >70 % and was prefixed by “CL”, while the singlets were prefixed by “Unigene”.
For Unigene annotation, a BLASTX alignment between Unigenes sequences and various protein databases (an E-value < 1E-5), namely the NCBI non-redundant protein databases (NR), Swiss-Prot, Kyoto Encyclopedia of Genes and Genomes (KEGG) and COG (Cluster of Orthologous Groups) was performed, as well as a BLASTN alignment (E-value <1E-5) of the sequences against the NCBI non-redundant nucleotide database (NT). Where conflicting results arose, the priority order was NR, Swiss-Prot, KEGG and COG. For the NR annotation, GO (gene ontology) annotation was achieved using the Blast2GO program [62], and GO functional classifications were acquired using the WEGO software [63].
Identification and functional assignment of DTGs
The transcript abundance of a given Unigene was estimated using the FPKM (fragments per Kbp per million reads) method [25], designed to eliminate any bias generated by variation in sequence length and sequencing quality. Following Audic et al. (1997), an algorithm was developed to identify differential transcript abundance between pairs of samples [64]. FDR (false discovery rate) control, a statistical method, was applied to correct for p-value in multiple tests for calculating the expression between two samples [65]. The smaller FDR and the larger ratio represented the larger difference of the expression level between the two samples. In the present analysis, an FDR threshold of less than 0.001 and a |log2ratio| threshold of at least 1 were applied [64]. Here, DTGs were identified in pairwise contrasts CK vs S1 (CKS1), CK vs S2 (CKS2), CK vs L1 (CKL1), CK vs L2 (CKL2) and CK vs L3 (CKL3). Then, DTGs identified in this way were subjected to GO functional analysis and KEGG Pathway analysis on the base of a hypergeometric test.
qRT-PCR analysis
To validate the ability of RNA-Seq to detect differential transcription, a subset of the DTGs was tested using qRT-PCR. Primer5 software was used to design the appropriate primers (Table 6), and the reactions were performed in a Mastercycler®ep realplex 2 S device (Eppendorf, Hamburg, Germany) using a SYBR Premix Ex Taq™ Kit (Takara, Dalian, China), following the manufacturers’ protocols. Each sample was represented by three biological replicates. The reference gene was chrysanthemum EF-1α (GenBank ID, KF305681). Each 25 μL reaction contained 10 ng cDNA, 0.2 μM of each primer and 10 μl SYBR Green PCR master mix. The reactions were exposed to an initial denaturation (95 °C/2 min), followed by 40 cycles of 95 °C/15 s, 60 °C/15 s, 72 °C/15 s. After amplication, a melting curve analysis was conducted to verify the specificity of the reaction. Relative transcript abundances were calculated using the 2-ΔΔC T method [66].
Abbreviations
- COG:
-
Clusters of orthologous groups
- DTG:
-
Differentially transcribed gene
- FDR:
-
False discovery rate
- FPKM:
-
Fragments per Kbp per million reads
- GO:
-
Gene ontology
- KEGG:
-
Kyoto encyclopedia of genes and genomes
- NR:
-
Non-redundant database
- NT:
-
Nucleotide database
- qRT-PCR:
-
Quantitative Real-Time PCR
References
Amasino RM, Michaels SD. The timing of flowering. Plant Physiol. 2010;154(2):516–20.
Bäurle I, Dean C. The timing of developmental transitions in plants. Cell. 2006;125(4):655–64.
Jung C, Müller AE. Flowering time control and applications in plant breeding. Trends Plant Sci. 2009;14(10):563–73.
Fornara F, de Montaigu A, Coupland G. SnapShot: control of flowering in Arabidopsis. Cell. 2010;141(3):550. 550. e552.
Simpson GG, Dean C. Arabidopsis, the Rosetta stone of flowering time? Science. 2002;296(5566):285–9.
Song YH, Ito S, Imaizumi T. Flowering time regulation: photoperiod-and temperature-sensing in leaves. Trends Plant Sci. 2013;18(10):575–83.
Yanovsky MJ, Kay SA. Molecular basis of seasonal time measurement in Arabidopsis. Nature. 2002;419(6904):308–12.
An H, Roussot C, Suárez-López P, Corbesier L, Vincent C, Piñeiro M, Hepworth S, Mouradov A, Justin S, Turnbull C. CONSTANS acts in the phloem to regulate a systemic signal that induces photoperiodic flowering of Arabidopsis. Development. 2004;131(15):3615–26.
Tiwari SB, Shen Y, Chang HC, Hou Y, Harris A, Ma SF, McPartland M, Hymus GJ, Adam L, Marion C. The flowering time regulator CONSTANS is recruited to the FLOWERING LOCUS T promoter via a unique cis-element. New Phytol. 2010;187(1):57–66.
Choi K, Kim J, Hwang HJ, Kim S, Park C, Sang YK, Lee I. The FRIGIDA complex activates transcription of FLC, a strong flowering repressor in Arabidopsis, by recruiting chromatin modification factors. Plant Cell. 2011;23(1):289–303.
Moon J, Suh SS, Lee H, Choi KR, Hong CB, Paek NC, Kim SG, Lee I. The SOC1 MADS-box gene integrates vernalization and gibberellin signals for flowering in Arabidopsis. Plant J. 2003;35(5):613–23.
Balasubramanian S, Sureshkumar S, Lempe J, Weigel D. Potent induction of Arabidopsis thaliana flowering by elevated growth temperature. Plos Genet. 2006;2(7):e106.
Wahl V, Ponnu J, Schlereth A, Arrivault S, Langenecker T, Franke A, Feil R, Lunn JE, Stitt M, Schmid M. Regulation of flowerin CONSTANS acts in the phloem to regulate a systemic signal that g by trehalose-6-phosphate signaling in Arabidopsis thaliana. Science. 2013;339(6120):704–7.
Yang L, Xu M, Koo Y, He J, Poethig RS. Sugar promotes vegetative phase change in Arabidopsis thaliana by repressing the expression of MIR156A and MIR156C. Elife. 2013;2:e00260.
Ren L, Sun J, Chen S, Gao J, Dong B, Liu Y, Xia X, Wang Y, Liao Y, Teng N. A transcriptomic analysis of Chrysanthemum nankingense provides insights into the basis of low temperature tolerance. BMC Genomics. 2014;15(1):844.
da Silva JA T, Shinoyama H, Aida R, Matsushita Y, Raj SK, Chen F. Chrysanthemum biotechnology: quo vadis? Crit Rev Plant Sci. 2013;32(1):21–52.
Fu J, Wang L, Wang Y, Yang L, Yang Y, Dai S. Photoperiodic control of FT- like gene ClFT initiates flowering in Chrysanthemum lavandulifolium. Plant Physiol Biochem. 2014;74:230–8.
Ren H, Zhu F, Zheng C, Sun X, Wang W, Shu H. Transcriptome analysis reveals genes related to floral development in chrysanthemum responsive to photoperiods. Biochem Genet. 2013;51(1):20–32.
Oda A, Narumi T, Li T, Kando T, Higuchi Y, Sumitomo K, Fukai S, Hisamatsu T. CsFTL3, a chrysanthemum FLOWERING LOCUS T-like gene, is a key regulator of photoperiodic flowering in chrysanthemums. J Exp Bot. 2012;63(3):1461–77.
Chan A. Some factors affecting flower bud development of chrysanthemums. In: Reports of the 14th Int horticultural congress: 1955. 1955. p. 1023–39.
Seeley J, Weise A. Photoperiodic response of garden and greenhouse chrysanthemums. In: Proceedings of the American Society for Horticultural Science: 1965: Amer Soc Horticultural Science 701 North Saint Asaph Street, Alexandria, VA 22314–1998; 1965. p. 464
Liu H, Sun M, Pan H, Cheng T, Wang J, Zhang Q. Whole-transcriptome analysis of differentially expressed genes in the vegetative buds, floral buds and buds of Chrysanthemum morifolium. PLoS One. 2015;10(5):e0128009.
Higuchi Y, Narumi T, Oda A, Nakano Y, Sumitomo K, Fukai S, Hisamatsu T. The gated induction system of a systemic floral inhibitor, antiflorigen, determines obligate short-day flowering in chrysanthemums. Proc Natl Acad Sci. 2013;110(42):17137–42.
Yang Y, Ma C, Xu Y, Wei Q, Imtiaz M, Lan H, Gao S, Cheng L, Wang M, Fei Z. A zinc finger protein regulates flowering time and abiotic stress tolerance in chrysanthemum by modulating gibberellin biosynthesis. Plant Cell. 2014;26(5):2038–54.
Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods. 2008;5(7):621–8.
Berns MC, Nordström K, Cremer F, Tóth R, Hartke M, Simon S, Klasen JR, Bürstel I, Coupland G. Evening expression of Arabidopsis GIGANTEA is controlled by combinatorial interactions among evolutionarily conserved regulatory motifs. Plant Cell. 2014;26(10):3999–4018.
Song YH, Estrada DA, Johnson RS, Kim SK, Lee SY, MacCoss MJ, Imaizumi T. Distinct roles of FKF1, GIGANTEA, and ZEITLUPE proteins in the regulation of CONSTANS stability in Arabidopsis photoperiodic flowering. Proc Natl Acad Sci. 2014;111(49):17672–7.
Wang Y, Huang H, Ma Y, Fu J, Wang L, Dai S. Construction and de novo characterization of a transcriptome of Chrysanthemum lavandulifolium: analysis of gene expression patterns in floral bud emergence. Plant Cell Tissue Organ Cult. 2014;116(3):297–309.
Schäfer E, Nagy F. Photomorphogenesis in plants and bacteria: function and signal transduction mechanisms. Springer Science & Business Media; Heidelberg, Germany. 2006.
Teotia S, Tang G. To bloom or not to bloom: role of microRNAs in plant flowering. Mol Plant. 2015;8(3):359–77.
Wang JW. Regulation of flowering time by the miR156-mediated age pathway. J Exp Bot. 2014;65(17):4723–30.
Smeekens S, Hellmann HA. Sugar sensing and signaling in plants. Front Plant Sci. 2014;5:113.
Yu S, Cao L, Zhou CM, Zhang TQ, Lian H, Sun Y, Wu J, Huang J, Wang G, Wang JW. Sugar is an endogenous cue for juvenile-to-adult phase transition in plants. Elife. 2013;2:e00269.
Moghaddam MRB, Van den Ende W. Sugars, the clock and transition to flowering. Front Plant Sci. 2013;4:22.
Rolland F, Sheen J. Sugar sensing and signalling networks in plants. Biochem Soc Trans. 2005;33(1):269–71.
Kinter A, Catanzaro A, Monaco J, Ruiz M, Justement J, Moir S, Arthos J, Oliva A, Ehler L, Mizell S, Jackson R, Ostrowski M, Hoxie J, Offord R, Fauci AS. The phasic development of chrysanthemum as a basis for the regulation of vegetative growth and flowering in Japan. Acta Hortic. 1987;95(95):11880–5.
Putterill J, Robson F, Lee K, Simon R, Coupland G. The CONSTANS gene of Arabidopsis promotes flowering and encodes a protein showing similarities to zinc finger transcription factors. Cell. 1995;80(6):847–57.
Robson F, Costa MMR, Hepworth SR, Vizir I, Reeves PH, Putterill J, Coupland G. Functional importance of conserved domains in the flowering-time gene CONSTANS demonstrated by analysis of mutant alleles and transgenic plants. Plant J. 2001;28(6):619–31.
Wang CQ, Guthrie C, Sarmast MK, Dehesh K. BBX19 interacts with CONSTANS to repress FLOWERING LOCUS T transcription, defining a flowering time checkpoint in Arabidopsis. Plant Cell Online. 2014;26(9):3589–602.
Bai B, Zhao J, Li Y, Zhang F, Zhou J, Chen F, Xie X. OsBBX14 delays heading date by repressing florigen gene expression under long and short-day conditions in rice. Plant Sci. 2016;247:25–34.
Wong A, Hecht V, Picard K, Diwadkar P, Laurie RE, Wen J, Mysore K, Macknight RC, Weller JL. Isolation and functional analysis of CONSTANS-LIKE genes suggests that a central role for CONSTANS in flowering time control is not evolutionarily conserved in Medicago truncatula. Front Plant Sci. 2014;5(17):486.
Srikanth A, Schmid M. Regulation of flowering time: all roads lead to Rome. Cell Mol Life Sci. 2011;68(12):2013–37.
Olszewski N, Sun T-p, Gubler F. Gibberellin signaling biosynthesis, catabolism, and response pathways. Plant Cell. 2002;14(1):S61–80.
Griffiths J, Murase K, Rieu I, Zentella R, Zhang Z-L, Powers SJ, Gong F, Phillips AL, Hedden P, Sun TP, Thomas SG. Genetic characterization and functional analysis of the GID1 gibberellin receptors in Arabidopsis. Plant Cell. 2006;18(12):3399–414.
Xu H, Liu Q, Yao T, Fu X. Shedding light on integrative GA signaling. Curr Opin Plant Biol. 2014;21:89–95.
Ramachandran S, Hiratsuka K, Chua NH. Transcription factors in plant growth and development. Curr Opin Genet Dev. 1994;4(5):642–6.
Liu L, Zhang J, Adrian J, Gissot L, Coupland G, Yu D, Turck F. Elevated levels of MYB30 in the phloem accelerate flowering in Arabidopsis through the regulation of FLOWERING LOCUS T. PLoS One. 2014;9(2):e89799.
Yan Y, Shen L, Chen Y, Bao S, Thong Z, Yu H. A MYB-Domain protein EFM mediates flowering responses to environmental cues in Arabidopsis. Dev Cell. 2014;30(4):437–48.
Shan H, Chen S, Jiang J, Chen F, Chen Y, Gu C, Li P, Song A, Zhu X, Gao H, Zhou G, Li T, Yang X. Heterologous expression of the chrysanthemum R2R3-MYB transcription factor CmMYB2 enhances drought and salinity tolerance, increases hypersensitivity to ABA and delays flowering in Arabidopsis thaliana. Mol Biotechnol. 2012;51(2):160–73.
Smaczniak C, Immink RG, Muiño JM, Blanvillain R, Busscher M, Busscher-Lange J, Dinh QP, Liu S, Westphal AH, Boeren S, Parcy F, Xu L, Carles CC, Angenent GC, Kaufmann K. Characterization of MADS-domain transcription factor complexes in Arabidopsis flower development. Proc Natl Acad Sci. 2012;109(5):1560–5.
Gregis V, Andrés F, Sessa A, Guerra RF, Simonini S, Mateos JL, Torti S, Zambelli F, Prazzoli GM, Bjerkan KN, Grini PE, Pavesi G, Colombo L, Coupland G, Kater MM. Identification of pathways directly regulated by SHORT VEGETATIVE PHASE during vegetative and reproductive development in Arabidopsis. Genome Biol. 2013;14(6):R56.
Qu GZ, Zheng T, Liu G, Wang W, Zang L, Liu H, Yang C. Overexpression of a MADS-box gene from Birch (Betula platyphylla) promotes flowering and enhances chloroplast development in transgenic tobacco. PLoS One. 2013;8(5):e63398.
Fernandez DE, Wang CT, Zheng Y, Adamczyk B, Singhal R, Hall PK, Perry SE. The MADS-domain factors AGAMOUS-LIKE15 and AGAMOUS-LIKE18, along with SHORT VEGETATIVE PHASE and AGAMOUS-LIKE24, are necessary to block floral gene expression during the vegetative phase. Plant Physiol. 2014;165(4):1591–603.
Kumar SV, Lucyshyn D, Jaeger KE, Alós E, Alvey E, Harberd NP, Wigge PA. Transcription factor PIF4 controls the thermosensory activation of flowering. Nature. 2012;484(7393):242–5.
Leivar P, Monte E, Oka Y, Liu T, Carle C, Castillon A, Huq E, Quail PH. Multiple phytochrome-interacting bHLH transcription factors repress premature seedling photomorphogenesis in darkness. Curr Biol. 2008;18(23):1815–23.
Fu J, Yang L, Dai S. Conservation of Arabidopsis thaliana circadian clock genes in Chrysanthemum lavandulifolium. Plant Physiol Biochem. 2014;80:337–47.
Chao Y, Zhang T, Yang Q, Kang J, Sun Y, Gruber MY, Qin Z. Expression of the alfalfa CCCH-type zinc finger protein gene MsZFN delays flowering time in transgenic Arabidopsis thaliana. Plant Sci. 2014;215:92–9.
Cai Y, Chen X, Xie K, Xing Q, Wu Y, Li J, Du C, Sun Z, Guo Z. Dlf1, a WRKY transcription factor, is involved in the control of flowering time and plant height in rice. PLoS One. 2014;9(7):e102529.
Luo X, Sun X, Liu B, Zhu D, Bai X, Cai H, Ji W, Cao L, Wu J, Wang M, Ding X, Zhu Y. Ectopic expression of a WRKY homolog from Glycine soja alters flowering time in Arabidopsis. PLoS One. 2013;8(8):e73295.
Zhao Y, Medrano L, Ohashi K, Fletcher JC, Yu H, Sakai H, Meyerowitz EM. HANABA TARANU is a GATA transcription factor that regulates shoot apical meristem and flower development in Arabidopsis. Plant Cell. 2004;16(10):2586–600.
Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I, Adiconis X, Fan L, Raychowdhury R, Zeng Q. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol. 2011;29(7):644–52.
Conesa A, Götz S, García-Gómez JM, Terol J, Talón M, Robles M. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics. 2005;21(18):3674–6.
Ye J, Fang L, Zheng H, Zhang Y, Chen J, Zhang Z, Wang J, Li S, Li R, Bolund L. WEGO: a web tool for plotting GO annotations. Nucleic Acids Res. 2006;34(Web Server issue):293–7.
Audic S, Claverie JM. The significance of digital gene expression profiles. Genome Res. 1997;7(10):986–95.
Benjamini Y, Yekutieli D. The control of the false discovery rate in multiple testing under dependency. Ann Stat. 2001;29(4):1165–88.
Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2− ΔΔCT method. Methods. 2001;25(4):402–8.
Acknowledgments
This research was financially supported by grants awarded by the National Natural Science Foundation of China (31372100; 31572159), the Priority Academic Program Development of Jiangsu Higher Education Institutions, the Program for Natural science of Anhui Province Education Department (2014KJ019) and “Programs of Innovation and Entrepreneurship Talents” of Jiangsu Province.
Availability of data and material
All data analysed during this study are included within the article and its additional files, and the raw sequencing data have been deposited at the Sequence Read Archive (SRA) of the National Center for Biotechnology under accession number SRP076366.
Authors’ contributions
LR and TL contributed to bioinformatics analysis and writing of the manuscript. JJ, FC and SC conceived the study, participated in its design and contributed to revisions of the manuscript. YC and JS participated in experiment materials preparation. JG and BD helped with RNA extraction and qRT-PCR. All authors have read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable.
Author information
Authors and Affiliations
Corresponding author
Additional files
Additional file 1: Table S1.
Statistics of assembly quality. Total Consensus Sequences: the full set of assembled Unigenes; Distinct Clusters: clustered Unigenes – each cluster harbors a set of highly similar (>70 % similarity) Unigenes; Distinct Singletons: a Unigene derived from a single gene. The length of sequences assembled provides a measure of assembly success. (XLSX 11 kb)
Additional file 2: Table S2.
The representation of transcription factors in the set of Unigenes. (XLSX 10 kb)
Additional file 3: Table S3.
Homologs of genes from other species known to be involved in the regulation of flowering time. (XLSX 12 kb)
Additional file 4: Table S4.
Genes differentially transcribed in the comparison CK vs S1. The criteria applied for assigning significance were: P-value < 0.05, FDR ≤ 0.001, and |log2ratio| ≥ 1. The genes are listed in descending order of |log2ratio|. The “GeneLength” column gives the length of exon sequence. CK- and S1- transcript abundance derived from the frequency measured in reads per Kbp per million reads (FKPM). |log2ratio|: the ratio between the FPKM measured in CK and S1. The up- or down-regulation (S1/CK), P-value and FDR of each gene are also shown. KEGG: annotation according to the KEGG database, BLAST NR: identification of homologs in GenBank. GO Component, GO Function and Go Process: ontology information relating to “Cellular Components”, “Molecular Function” and “Biological Processes”. “-”: no hit. (XLS 2434 kb)
Additional file 5: Table S5.
Genes differentially transcribed in the comparison CK vs S2. Criteria as given in Additional file 4: Table S4. (XLSX 1287 kb)
Additional file 6: Table S6.
Genes differentially transcribed in the comparison in the comparison CK vs L1. Criteria as given in Additional file 4: Table S4. (XLS 1282 kb)
Additional file 7: Table S7.
Genes differentially transcribed in the comparison in the comparison CK vs L2. Criteria as given in Additional file 4: Table S4. (XLS 3076 kb)
Additional file 8: Table S8.
Genes differentially transcribed in the comparison in the comparison CK vs L3. Criteria as given in Additional file 4: Table S4. (XLS 1026 kb)
Additional file 9: Table S9.
Genes differentially transcribed in the comparison in the comparison L1 vs S1. Criteria as given in Additional file 4: Table S4. (XLS 333 kb)
Additional file 10: Table S10.
Genes differentially transcribed in the comparison in the comparison L2 vs S2. Criteria as given in Additional file 4: Table S4. (XLS 1772 kb)
Additional file 11: Table S11.
Differential transcription genes involved in the photoperiod pathway in the comparisons L1 vs S1 and L2 vs S2. (XLS 52 kb)
Additional file 12: Table S2.
Transcription behavior of unigenes encoding C3H, Dof, GATA, GRAS, bHLH, BBX, bZIP, WRKY, MYB and MADS transcription factors. (XLSX 39 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Ren, L., Liu, T., Cheng, Y. et al. Transcriptomic analysis of differentially expressed genes in the floral transition of the summer flowering chrysanthemum. BMC Genomics 17, 673 (2016). https://doi.org/10.1186/s12864-016-3024-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-016-3024-4