
Abstract
Linezolid resistance in Enterococcus spp. is increasingly considered critically important and a public health threat which mandates the need to understand their genomic contents and dissemination patterns. Here, we used whole-genome sequencing to characterize the resistome, virulome and mobile genetic elements of nine linezolid-resistant (LZDR) enterococci (seven optrA-E. faecalis, one poxtA-E. faecium and one optrA-E. casseliflavus) previously obtained from the nares of healthy dogs, pigs, pig farmers and tracheal samples of nestling storks in Spain. Also, the relatedness of the isolates with publicly available genomes was accessed by core-genome single nucleotide polymorphism (SNP) analysis. The optrA gene of the E. faecalis and E. casseliflavus isolates was located downstream of the fexA gene. The optrA gene in the E. casseliflavus isolate was carried in a plasmid (pURX4962), while those in the seven E. faecalis isolates were chromosomally located. The OptrA proteins were mostly variants of wild type (DP-2: Y176D/T481P; RDK: I104R/Y176D/E256K; DD-3: Y176D/G393D; and EDD: K3E/Y176D/G393D), except two that were wild type (one E. faecalis and one E. casseliflavus). The poxtA gene in the E. faecium isolate was found alone within its contig. The cfrD was upstream of ermB gene in the E. casseliflavus isolate and flanked by ISNCY and IS1216. All the LZDR enterococci carried plasmid rep genes (2–3) containing tetracycline, chloramphenicol and aminoglycoside resistance genes. All isolates except E. casseliflavus carried at least one intact prophage, of which E. faecalis-ST330 (X4957) from a pig carried the highest (n = 5). Tn6260 was associated with lnuG in E. faecalis-ST330 while Tn554 was with fexA in E. feaecalis-ST59 isolates. All except E. casseliflavus (n = 0) carried at least two metal resistance genes (MRGs), of which poxtA-carrying E. faecium-ST1739 isolate contained the most (arsA, copA, fief, ziaA, znuA, zosA, zupT, and zur). SNP-based analyses identified closely related optrA-E. faecalis isolates from a pig and a pig farmer on the same farm (SNP = 4). Moreover, optrA- carrying E. faecalis-ST32, -ST59, and -ST474 isolates from pigs were related to those previously described from humans (sick and healthy) and cattle in Spain, Belgium, and Switzerland (SNP range 43–86). These findings strongly suggest the transmission of LZDR-E. faecalis between a pig and a pig farmer and potential inter-country dissemination. These highlight the need to strengthen molecular surveillance of LZDR enterococci in all ecological niches and body parts to direct appropriate control strategies.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Antimicrobial resistance (AMR) constitutes one of the major global health challenges that need a holistic “One Health” and “One World” approach to understand its emergence, evolution, and dissemination pattern [1]. Enterococci are very resilient as they can survive the effect of physicochemical agents for long periods [2]. Enterococcus is one of the suitable bacteria to study AMR across the “One Health” ecosystems, as certain species and genetic lineages could easily acquire or transfer critically important AMR to other species or genera in different hosts or niches via mobile genetic elements [3].
Linezolid (LZD) is one of the most important and “last resort” chemotherapeutic options for severe infections caused by vancomycin-resistant enterococci and multiresistant staphylococci [4]. Linezolid resistance (LZDR) is rare but represents a critically important public health problem that needs to be meticulously studied and reported at all levels. Linezolid is not used in animals but LZDR could be selected by the use of other antibiotics, as is the case of phenicols or lincomycin, among others [5].
Linezolid resistance is mediated either by the acquisition of transferable genes (optrA, poxtA, and cfr), or due to mutations in the V domain of the 23S rRNA or amino acid changes in the ribosomal proteins L3, L4 and/or L22 of the enterococci [6,7,8].
Concerning the transferable LZDR genes, optrA has been frequently reported [6]. At least 69 OptrA variants have been identified so far, with differences of 1–20 amino acid substitutions (i.e., of 97.1–99.8% amino acid identity) [8]. The optrA gene codes for an ATP-binding cassette F ribosomal protection protein that confers cross-resistance to chloramphenicol, linezolid and tedizolid [9]. The poxtA gene was identified and characterized in 2018 from a clinical MRSA in Italy [10]. This gene shares about 32% nucleotide homology with optrA and uses a similar mechanism to confer resistance to linezolid and exists in two variants, poxtA and poxtA2 [10, 11].
The cfr gene encodes a methyltransferase that modifies position A2503 of the 23S rRNA and confers cross-resistance to streptogramin A, lincosamides, pleuromutilins, phenicols and linezolid [8, 12]. There are four variants of the cfr gene, i.e., cfrB, cfrC, cfrD, and cfrE, of which cfrB and cfrD do not confer resistance on their own, except in combination with other linezolid resistance genes [13, 14]. Moreover, the cfrC and cfrE have not been reported in any Enterococcus spp. [6]. It appears that cfr-mediated LZDR is predominantly found in staphylococci whereas the cfr-like gene, cfrD, is common in enterococci [6].
The mutation-mediated LZDR enterococci emerged following linezolid chemotherapy in clinical practice with the predominant being the G2576T or G2505A in 23S rRNA [15]. However, other mutations leading to high-level linezolid resistance such as G2447U, G2576U, and G2504A, among others, have also been reported in enterococci [14]. It is important to remark that LZDR due to target mutations is not transferable as they are not bound to mobile genetic elements. However, the acquired LZDR genes are often carried by plasmids while in some instances, they are chromosomally located [6].
Linezolid-resistant enterococci and the mechanisms of resistance involved have been reported in Spain [7, 16,17,18,19,20,21,22]. However, few reports on their complete genomic characterization have been published [18, 23] and none on healthy human and animal populations. As the incidence of LZDR enterococci is gradually increasing all over the world, it is important to mount a genome-based surveillance system to track their dissemination patterns and relatedness with other reported cases from other countries. The present study characterized the genomic profile in a collection of enterococcal isolates belonging to three species (E. faecalis, E. faecium and E. casseliflavus) carrying LZDR genes previously identified from healthy animals and humans [7], to characterize their resistomes, virulomes, mobile genetic elements and to determine their phylogenetic relatedness with other publicly available strains with similar sequence types and AMR genes.
Materials and Methods
Nine LZDR enterococci were obtained in a previous study [7], and were included in the present work for complete genomic characterization. These isolates were all of nasal origin (except one of tracheal origin in stork), and belonged to three species: a) seven optrA-carrying E. faecalis isolates of pigs (n = 5), pig farmers (n = 1) and dogs (n = 1); b) one optrA/cfrD-carrying E. casseliflavus isolate from a pig; and c) one poxtA-carrying E. faecium isolate of a stork (Table 1). The E. faecalis isolates from the pig farms were selected based on their origins, sequence types (STs) and AMR genes. Details on the strategies of sample collection, processing, isolation, identification of bacteria, their antibiogram and AMR genotyping were presented in our previous study [7].
Whole Genome Sequencing, Assembly, and Phylogenetic Analyses
Whole genome sequencing of the selected LZDR enterococci was carried out on the NextSeq 550 platform (Illumina), while the E. casseliflavius isolate was further sequenced on the MinION platform (Oxford Nanopore Technologies (ONT), Oxford, United Kingdom) as described here. Single colonies were obtained from a fresh over-night blood agar plating and resuspended in enzymatic lysis buffer [Proteinase K (Roche); Lysozyme (Sigma)] and incubated at 37 °C (30 min) and 55 °C (1 h). The final buffer composition was as follows: 1× phosphate buffered Saline (pH 7.2 (ThermoFisher Scientific), 10× solution diluted to 1× in nuclease-free water), 20 mM Tris–HCl (pH 8; ThermoFisher Scientific), 2 mM EDTA (ThermoFisher Scientific), 1.2% Triton X-100 (Merck), 1.7 mg/ml Proteinase K (Roche), 20 mg/ml Lysozyme (Sigma).
The MagNA Pure 96 DNA and Viral NA Small Volume Multi-Sample Kit (Roche) was used to extract genomic DNA according to the manufacturer's instructions. DNA was quantified using the Quant-iT dsDNA BR and HS Assay Kits (Thermo Fisher Scientific, Scoresby, VIC, Australia) and fluorescence was measured on a FLUOstar Omega (BMG LabTech). For short-read sequencing on the Illumina platform, sequencing libraries were prepared using the Illumina Nextera XT DNA Library Preparation Kit (Illumina). The final libraries were analyzed on a TapeStation 4200 (Agilent) before sequencing on the NextSeq 550 platform (Illumina) using a 300-cycle kit to obtain paired-end 150 bp reads, as previously described [24]. For long-read sequencing on the MinION platform, the ONT rapid barcoding kit (SQK-RBK110.96) was used to generate the sequencing library. Four samples were multiplexed and sequenced on an R9.4.1 flow cell. Reads were demultiplexed and base-called with the base-calling model Guppy (v.6.1.5) at super-accurate setting and filtered on quality score > 10. All the genomes analyzed in this study were de novo assembled using SPAdes (v.3.15.5), performing the in silico typing with the settings of a minimum of 90% coverage and 80% identity. Core-genome single nucleotide polymorphisms (SNPs) were detected with the NASP pipeline v.1.0.0 [25]. GATK (v.4.2.2) was used to call SNPs and excluded positions featuring < 90% unambiguous variant calls and < 10 depth. IQ-TREE (v.2.1.2), was used to construct the phylogenetic trees using ModelFinder with 100 bootstraps. The graphical data were added to the phylogenies with iTOL v.6.6 [26].
Genome Annotation, Typing and In Silico Analysis
The STs were determined with MLST (v.2.16, https://cge.food.dtu.dk/services/MLST/). Virulence factors, plasmid replicons, and antimicrobial resistance genes were identified using ABRicate (v.0.9.0) [27], and the respective databases VFDB [28], Plasmidfinder [29], and Resfinder [30] from the Center for Genomic Epidemiology. Mutations associated with AMR were identified using ResFinder (v4.1) [30] and PointFinder [31]. Phaster was used to identify all prophage elements [32]. The genetic environments of optrA, poxtA and cfrD genes were illustrated using the reference strains [E. faecalis (GenBank accession ID KP399637), E. faecium plasmid pGZ8 (GenBank accession ID CP038162), E. faecium (GenBank accession ID MN831413) and E. faecalis (GenBank accession ID CP097040)] in EasyFig Software. The OptrA variants based on amino acid substitutions deduced from the optrA sequences, were analyzed according to a previously described nomenclature [8], using WP_063854496.1 as the wild-type reference.
Phylogenetic Analysis by Core-Genome Single Nucleotide Polymorphism
The SNPs between the genomes of our seven optrA-E. faecalis isolates and those of 12 publicly available optrA-positive E. faecalis isolates was analyzed (GenBank accession numbers: SRR17662732, ERR2008110, ERR2008112, ERR1599987, ERR1599986, ERR2008113, ERR2008114, SRS7549315, SRS7549355, SRS7549357, SRS7549371, SRS7549400). These 12 genomes were selected because they carried the optrA gene and belonged to the same genetic lineages as the seven E. faecalis strains from this study.
Genome Availability
All the raw genome reads of the LZDR enterococci have been deposited at the European Nucleotide Archive under Study Accession number PRJEB62654. The optrA-associated plasmid in E. casseliflavus (pURX4962) was deposited in GenBank with the accession number OR069652.
Results
Genome Features
The genomes of the seven LZDR E. faecalis isolates had sizes in the range of 2.8–3.1 Mb and contigs range from 100 to 164 (Table 1). The LZDR E. faecium isolate had a genome size of 2.7 Mb and 136 contigs, whereas the LZDR E. casseliflavus isolate had a genome size of 3.7 Mb and 247 contigs (Table 1).
Antimicrobial and Metal Resistance
The minimum inhibition concentrations (MICs) of all enterococci to linezolid ranged from 2 to 14 μg/ml (Table 1). The in silico analysis of the optrA sequences of the eight E. faecalis/E.casseliflavus isolates revealed that two of them harbored the gene of the wild-type OptrA (E. faecalis and E. casseliflavus, both of pig origin), while the remaining six E. faecalis isolates carried the gene of four OptrA variants (DP-2: Y176D/T481P; RDK: I104R/Y176D/E256K; DD-3: Y176D/G393D; and EDD: K3E/Y176D/G393D) (Table 1). The OptrA variant DP-2 was detected in three E. faecalis isolates of pig, pig farmer and dog origin (ST330 and ST585), and the OptrA variants RDK, DD-3, and EDD in three E. faecalis isolates recovered from pigs (Table 1). Also, the LZDR-E. faecium isolate carried a poxtA type 1 gene (Table 1). In addition, all the optrA-positive enterococci co-carried the fexA gene; the fexB gene was co-carried by the poxtA-positive E. faecium isolate. Moreover, the E. casseliflavus co-carried the cfrD gene.
Beside the AMR genes presented in our previous studies [7], the following genes were detected: (a) E. casseliflavus isolate (X4962) carried the dfrG, vanC3, lnuB, aph3’ and lsaE genes; (b) E. faecalis-ST330 (X5386 and X5809) from pigs of farms B-D carried the ant4’, dfrG, lsaA, and aph3’genes; (c) E. faecalis-ST59 (X5799) carried the dfrG, lnuB, lsaE, and lsaA genes; (d) E. faecalis-ST32 (X5445) from a pig of farm B carried the lsaA, ant9’, dfrG, lnuG, and aph3’genes; (e) E. faecalis-ST330 (X5463) from a pig farmer of farm B carried the ant4’, dfrG, lsaA, and aph3’genes; (f) E. faecalis-ST585 (X6347) from a dog carried the lnuB and lsaA genes. There were no additional AMR genes detected in the E. faecium-ST1739 isolate (X3877) from a nesting of stork foraging in landfills (Table 1).
Aside from the AMR genes, chromosomal point mutations leading to ciprofloxacin resistance were detected in E. faecalis-ST330 isolates from pigs of farms B and D, associated with amino acid changes in ParC (S80I) and GyrA proteins (E87G). Moreover, only one amino acid change in ParC (S80I) was detected in the dog E. faecalis-ST475 isolate. In addition, 17 different amino acids substitutions were identified on the penicillin-binding protein 5 (S27G, A68T, A216S, T172A, V24A, 885D, K144Q, A499T, L177I, N496K, G66E, E100Q, D204G, P667S, E525D, T324A, R34Q) of the E. faecium isolate from stork nestling (poxtA-positive), some of them leading to penicillin resistance (Table 1).
All LZDR isolates except E. casseliflavus X4962 carried at least two metal resistance genes (MRGs), of which E. faecium-ST1739 carried most of them (arsA, copA, fief, ziaA, znuA, zosA, zupT, zur) (Table 1). Moreover, E. faecalis X5386 carried only two of the MRGs (cutC and znuA).
Virulence Determinants
Many virulence genes that have been associated with surface adherence, biofilm formation, and cytolysis were detected in the optrA-carrying E. faecalis isolates, most frequently being the ebpA, tpx, elrA, hylA, srtA, gelE, fsrB, ace, cOB1, cCF10, dad, agg, camE, efaAfs, hylB, and cylA genes. In the E. faecium X3877 isolate, only acm and efaAfm genes were found. However, none of these genes were identified in the E. casseliflavus X4962 isolate (Table 1).
Mobile Genetic Elements
All the enterococci carried at least one plasmid replicon gene (1–5 rep genes), however, only some of the plasmid replicons (2–3 per isolate) were associated with AMR genes. The three optrA-positive E. faecalis-ST330 isolates carried different number of plasmid replicons (Table 1). The repUS52 in strain X5386 was found co-located with the aminoglycoside resistance ant4’ gene. Three resistance genes (tet(L), tet(M), and cat) were bound on plasmid rep9a and repUS43 of all E. faecalis isolates from the pigs and pig farmers (Table 1). The aminoglycoside resistance gene, str was found on plasmid rep7 in the E. faecalis isolate from a dog (X6347). Moreover, the agg virulence gene was also co-located on the rep9a contig of E. faecalis-ST330 (X5463) and E. faecalis-ST32 (X5445). Three replicons were identified in the poxtA-carrying E. faecium isolate (rep29, rep1, repUS15) and none was co-located with any other AMR genes. Moreover, five replicons (rep14b, repUS41, repUS1, rep1, and rep40) were identified in the optrA-positive E. casseliflavus X4962 isolate, of which repUS40 was associated with the optrA-fexA genes (Fig. 1), and was 99.83% identical with plasmid pE3954 of E. faecalis (GenBank accession no: KP399637).

Genetic environment of the optrA gene in the eight E. faecalis and E. casseliflavus isolates from healthy pigs, pig farmer, and a dog. Shown in the figure are AMR genes located in the same contigs and frames with their corresponding mobile genetic elements. The percentage of identity and scale bar legends are presented on the right side of the image. The comparison was made with a reference E. faecalis strain E349 (GenBank Accession number: KP399637) (colour figure online)
Two different transposons were identified, viz: Tn6260 and Tn554 associated with lnuG and fexA genes of E. faecalis-ST330 and -ST59 isolates, respectively. Also, the Tn6260 identified in three of our optrA-positive E. faecalis isolates have previously been shown associated with lnuG.
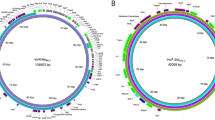
Different insertion sequences were detected among our LZDR isolates: E. faecalis-ST59 (ISS1N and ISEnfa4), E. faecalis-ST330 (ISEnfa1), E. faecium (ISSsu5) and E. casseliflavus (ISEnfa1) (Table 1). The optrA gene was chromosomally located in all our E. faecalis isolates. Nevertheless, the optrA gene of E. casseliflavus strain X4962 was located in a plasmid (37.9 kb, pURX4962) (Fig. 2), that showed 99.98% similarity with the one of an E. faecalis strain from China (GenBank Accession number: KP399637.1). All our LZDR E. faecalis and E. faecium isolates carried at least one prophage, of which E. faecalis -ST330 (X4957) from a pig carried the highest variety (n = 5), viz: phiEf11, phiFL4A, phiFL2A, EFC_1 and LP_101 (Table 1).

Genetic representation of the optrA-carrying plasmid pURX4962. Colors and arrows indicate the represented genes and their orientation (color figure online)
Genetic Environment of the Linezolid Resistance Genes
Five different genetic environments of the optrA gene were detected among the eight isolates that carried this gene: (a) E. casseliflavus X4962 (OptrA wild type); (b) E. faecalis X4957, X5386, and X5463 (OptrA—variants RDK and DP-2); (c) E. faecalis X5445 and X5809 (OptrA -variants DD-3 and EDD); (d) E. faecalis X5799 (OptrA- wild type); and (e) E. faecalis X6347 (OptrA variant DP-2). The fexA gene, which confers resistance to phenicols, was detected upstream of the optrA gene in all the eight E. faecalis and E. casselifavus isolates (Fig. 1). Moreover, an ermA-like gene was detected in the seven optrA-carrying E. faecalis isolates. Of these, the ermA-like gene was found in the environment of the optrA gene in four of isolates (Fig. 1). This ermA-like gene was identical to that detected in Streptococcus suis (GenBank accession number: EU348758). Regarding the cfrD gene, we could identify the presence of a guaA gene encoding a glutamine-hydrolyzing guanosine monophosphate synthase in the downstream region. Upstream of the cfrD gene, we detected the ermB gene flanked by IS1216 and ISNCY (Fig. 3). The genetic environment of the cfrD gene (1074 bp) revealed 100% nucleotide similarity with that of an E. faecium isolate in France (GenBank accession number: NG_067192).

Schematic comparison between the environment of the cfrD gene in the E. casseliflavus isolate X4962 with E. faecalis (GenBank Accession number: CP097040) and E. faecium (GenBank Accession number: MN831413) (color figure online)
Sequence analysis revealed that E. faecium-X3877 harbored the wild-type poxtA gene with 100% nucleotide sequence identity to that of E. faecium plasmid pGZ8 (GenBank accession number: CP038162) (Fig. 4).

Genetic environment of the poxtA gene in the E. faecium isolate X3877 in comparison with the previously described plasmid-bound poxtA gene in the E. faecium strain pGZ8 (GenBank Accession number: CP038162) (color figure online)
Relatedness of the E. faecalis Isolates
First, SNP analyses identified high relatedness (SNP = 4) of a pig isolate (X5386) with that of a pig farmer (X5463) from the same farm in our study (Supplementary Table S1). Both isolates carried the gene associated with the same OptrA variant (DP-2) and the same optrA-genetic environment and were E. faecalis of the lineage ST330.
Then, analyses with other publicly available genomes revealed relatedness of E. faecalis-ST32 (X5445) that was closely related to an isolate from a healthy human (SNP = 86) in Switzerland (SRR17662732). Moreover, the pig isolate (X5799) was related to two isolates from cattle origin (SNP = 44 and 48) in Belgium (id2205 and id2235) (Fig. 5 and Supplementary Table S1). Furthermore, the pig isolate (X5809) was closely related (< 50 SNP) to two isolates previously described from hospitalized patients in Spain (ERR2008113 and ERR2008114).

Phylogenetic tree based on core-genome SNPs analysis of seven optrA-carrying E. faecalis isolates from healthy hosts in this study with twelve publicly available E. faecalis genomes with similar STs and linezolid resistance genes. Colors (in circles) of the AMR genes are as follows: dark purple for optrA while and light purple for fexA and cat (color figure online)
Discussion
Here we present a genomic investigation of nine LZDR enterococci from four different healthy host types (humans, pigs, dogs, and storks). To the best of our knowledge, this is the first genomic comparative study on LZDR enterococci in healthy human and animal populations in Spain.
Since the first detection of LZDR enterococci in 2003 in Spain, two years after the approval of linezolid for clinical use [22], the detection rate of LZDR has risen significantly, especially during and after the post-COVID-19 era [32]. In our previous study [7], LZDR genes were detected after the selective inclusion of chloramphenicol resistance phenotype and characteristic green colonies on CHROMagar LIN by the three Enterococcus species (E. faecalis, E. faecium and E. casseliflavus). This was not expected as the animals and human hosts were healthy and were not on any antibiotic chemotherapy for at least six months before their enrolment into the study. This suggests that LZDR could persist for a long period in enterococci, considering that the bacterial genus is very tolerant to environmental factors such as sunlight and temperature [2].
In a recent study conducted by our research group and others in Spain, LZDR genes were detected from hospital settings with a high frequency of the optrA/poxtA genes among E. faecalis and E. faecium [17, 18]. This partly corroborated our finding as all the E. faecalis carried the optrA gene, but not poxtA, as in the case of Ruiz-Ripa et al. [18].
Worth mentioning is that most of the optrA genes carried by the E. faecalis strains from this current study (6/7) corresponded to variants of the OptrA wild-type (DP-2, RDK, DD-3, and EDD). The OptrA variants may affect the level of MIC value for linezolid [8, 33, 34], in which some strains show low MICs while others are within the breakpoint classified as resistance. In a study that showed a correlation between linezolid MIC and OptrA variants, it was detected that the EDM variant was associated with linezolid MICs ≤ 4 μg/ml, while the variant RDK (also found among our enterococci) with MIC values ≥ 8 μg/ml [34]. In the present study, all the LZDR enterococci had MICs of ≥ 8 µg/ml, except one isolate carrying a wild-type OptrA (X5799) with an MIC of 2 μg/ml. Nevertheless, this unique finding implies that LZDR may go unnoticed by phenotypic assays unless nucleic acid amplification tests are performed. As indicated in our previous study [7], chloramphenicol resistance could be considered as a marker to screen for linezolid resistance genes. Nevertheless, genes that mediate multidrug resistance (MDR) phenotypes identified from our isolates are frequent findings in LZDR enterococci from previous studies [16,17,18,19,20]. Interestingly, none of the isolates was resistant to other last-resort antibiotics such as vancomycin, which could serve as an alternative for the LZDR enterococci in clinical practice.
It is important to remark that the classification of optrA variants and their nomenclature is not uniform. In this regard, two types of classifications are available; one is based on amino acid changes in the wild-type OptrA protein (the one used in this study) while the other is based on numerical classification of the variants as a result of mutations in the nucleotides of the optrA gene [8, 35].
Enterococcus faecium showing ampicillin resistance is an additional problem in the clinical chemotherapy of enterococci [36]. Chromosomal point mutations putatively conferring resistance to linezolid (in 23S rRNA and ribosomal proteins L3/L4/L22) were not detected in our isolates. However, E. faecalis-ST330 from healthy pigs and pig farmer presented ParC (S80I) and GyrA (E87G) anino acid changes associated with fluoroquinolone resistance, which are commonly identified in samples obtained from livestock, foodstuffs, and from human infections [37, 38].
Several MRGs were identified in most of the isolates from this study. Specifically, cutC and znuA encode for copper homeostasis protein and high-affinity zinc uptake binding protein ZnuA, respectively [39]. Other LZDR isolates carried three MRGs, cutC, tcrB, and znuA. It is important to highlight that metal resistance is a matter of public health concern due to its potential hazards in the food chain and co-selection of other AMRs [40]. Specifically, MRGs are common in enterococci strains due to the frequent use of some metals, such as zinc micronutrients in livestock feed supplements [41]. It is important to emphasize that the feed-relate source of these metals could have been deposited in the environment which caused the bacteria to develop resistance to these metals.
More worrisome is that E. faecium isolate X3877 carried diverse MRGs including those that represent higher environmental health challenges such as arsenic (arsA). For this reason, there is great debate in the EU about the banning of metals in the feed of pig farming to avoid these processes of co-selection to clinically relevant antibiotics [42]. The presence of these MGRs in X3877 of a nestling of a parent stork that forages in a landfill is an indication that metal pollution in the environment is linked to high anthropogenic activities [42, 43].
Virulence factors are very relevant in the pathogenesis of enterococci. Many of these genes such as the ebpA seem to be related to clinical enterococcal strains [18, 44]. Other virulence genes that contribute to the colonization and persistence of enterococcal infections were also identified in most of the isolates [45, 46]. Also, the optrA-carrying E. faecalis isolates recovered from dog, pigs and pig farmers carried ace, gelE and agg genes, previously described in hospital-associated E. faecalis isolates [35, 47].
Regarding virulence factors of the poxtA-positive E. faecium X3877 isolate, it carried the acm and efaA genes that encode collagen-binding, although it seems the efaA is widely distributed in most E. faecium isolates, regardless of whether they are clinical or commensal strains [13].
Generally, the E. faecalis isolates analyzed carried plasmids belonging to many of the known replicon families in enterococci [35, 48]. The variability of plasmid content found illustrates the diverse nature of MGE in enterococci and their potential to facilitate the dissemination of some critical AMR genes, such as the optrA, as in the case of the E. casseliflavus isolate. Importantly, four plasmid replicons were found bound to genes that mediate resistance to tetracyclines, aminoglycosides and chloramphenicol. This could explain the reason why these AMR genes (tet(M), tet(L), cat and ant4’) persist in the enterococci and other related bacteria (such as staphylococci) from pigs and pig farmers and the farm environments [35, 49].
Oftentimes, the fexA gene is associated with Tn554, as previously reported in LZDR-E. faecalis [35, 50]. However, only one E. faecalis isolate carried this transposon in our study. The lnuG gene identified confers resistance to lincomycin through nucleotidylation in enterococci [51, 52]. The detection of lnuG in the LZDR E. faecalis-ST330 isolates from two pigs could lead to increased dissemination of lincosamide resistance in the food chain through Tn6260 [52].
Concerning phages in enterococci, when detected, most intact prophage-associated sequences are found in clinical enterococci in humans and in animal populations [53], but their distribution and involvement in the pathogenesis of enterococcal infection are poorly characterized [47]. Nonetheless, it is important to mention that the therapeutic potential of some of the identified intact prophages has previously been evaluated [54, 55].
Contrary to most other poxtA-carrying E. faecium isolates, the fexB gene in our isolate was in entirely a different contig of the genome, which is often downstream of poxtA [23, 56]. Perhaps, the termination of the contig carrying the poxtA in our E. faecium isolate precludes the co-location of the fexB on the same contig.
The phylogenomic analyses from this study strongly suggests the zoonotic transmission of LZDR-E. faecalis isolates and highlight the impact of pigs’ gut bacteria (such as multidrug-resistant E. faecalis) contaminating the farm environment and finding its way to the farmer’s nostrils and other body parts [57, 58]. This is a situation of occupational biohazard on the side of the pig farmer [57, 58]. Moreover, the SNP-based phylogeny illustrates the potential flow and transfer of LZDR-E. faecalis isolates from multiple sources and countries.
Conclusions
The data presented in this study comprise one of the few available comprehensive genomic datasets of LZDR enterococci in healthy hosts, including key AMR genes, virulence, prophages, and the plasmidome. The phylogenetic relatedness of our optrA-positive E. faecalis with those of publicly available genomes from discrete lineages but varied sources and geographical locations revealed close relatedness with most strains from Spain, and other European, and American countries. Additionally, our study detected an interesting variability of optrA in some E. faecalis isolates of the same lineage (ST330). Findings from this study demonstrated the transmission of LZDR-E. faecalis between a pig and a pig farmer (zoonosis) and highlight the need to strengthen molecular surveillance of LZDR enterococci in all ecological niches to direct appropriate control strategies. A larger number of isolates could have provided more credence to the findings obtained. However, as linezolid resistance is a rare trait in healthy humans and animals, the data from this study enhances our comprehension of the molecular epidemiology of this critical resistance in healthy hosts.
References
Aslam B, Khurshid M, Arshad MI, Muzammil S, Rasool M, Yasmeen N, Shah T, Chaudhry TH, Rasool MH, Shahid A, Xueshan X, Baloch Z (2021) Antibiotic resistance: one health one world outlook. Front Cell Infect Microbiol 11:771510. https://doi.org/10.3389/fcimb.2021.771510
Dubin K, Pamer EG (2014) Enterococci and their interactions with the intestinal microbiome. Microbiol Spectr. 5(6). https://doi.org/10.1128/microbiolspec.BAD-0014-2016
Monteiro Marques J, Coelho M, Santana AR, Pinto D, Semedo-Lemsaddek T (2023) Dissemination of Enterococcal genetic lineages: a one health perspective. Antibiotics 12(7):1140. https://doi.org/10.3390/antibiotics12071140
Santimaleeworagun W, Changpradub D, Hemapanpairoa J, Thunyaharn S (2021) Optimization of linezolid dosing regimens for treatment of vancomycin-resistant Enterococci infection. Infect Chemother 53(3):503–511. https://doi.org/10.3947/ic.2021.0034
Lees P, Pelligand L, Giraud E, Toutain PL (2021) A history of antimicrobial drugs in animals: evolution and revolution. J Vet Pharmacol Ther 44(2):137–171. https://doi.org/10.1111/jvp.12895
Brenciani A, Morroni G, Schwarz S, Giovanetti E (2022) Oxazolidinones: mechanisms of resistance and mobile genetic elements involved. J Antimicrob Chemother 77(10):2596–2621. https://doi.org/10.1093/jac/dkac263
Abdullahi IN, Lozano C, Juárez-Fernández G, Höfle U, Simón C, Rueda S, Martínez A, Álvarez-Martínez S, Eguizábal P, Martínez-Cámara B, Zarazaga M, Torres C (2023) Nasotracheal enterococcal carriage and resistomes: detection of optrA, poxtA- and cfrD-carrying strains in migratory birds, livestock, pets, and in-contact humans in Spain. Eur J Clin Microbiol Infect Dis. 42(5):569–581. https://doi.org/10.1007/s10096-023-04579-9
Schwarz S, Zhang W, Du XD, Krüger H, Feßler AT, Ma S, Zhu Y, Wu C, Shen J, Wang Y (2021) Mobile oxazolidinone resistance genes in Gram-positive and Gram-negative bacteria. Clin Microbiol Rev 34(3):e0018820. https://doi.org/10.1128/CMR.00188-20
Wang Y, Lv Y, Cai J, Schwarz S, Cui L, Hu Z, Zhang R, Li J, Zhao Q, He T, Wang D, Wang Z, Shen Y, Li Y, Feßler AT, Wu C, Yu H, Deng X, Xia X, Shen J (2015) A novel gene, optrA, that confers transferable resistance to oxazolidinones and phenicols and its presence in Enterococcus faecalis and Enterococcus faecium of human and animal origin. J Antimicrob Chemother 70:2182–2190. https://doi.org/10.1093/jac/dkv116
Antonelli A, D’Andrea MM, Brenciani A, Galeotti CL, Morroni G, Pollini S et al (2018) Characterization of poxtA, a novel phenicol–oxazolidinone–tetracycline resistance gene from an MRSA of clinical origin. J Antimicrob Chemother 73:1763–1769. https://doi.org/10.1093/jac/dky088
Baccani I, Antonelli A, Di Pilato V, Coppi M, Di Maggio T, Spinicci M, Villagran AL, Revollo C, Bartoloni A, Rossolini GM (2021) Detection of poxtA2, a presumptive poxtA ancestor, in a plasmid from a linezolid-resistant Enterococcus gallinarum isolate. Antimicrob Agents Chemotherap 65(8):e0069521. https://doi.org/10.1128/AAC.00695-21
Schwarz S, Werckenthin C, Kehrenberg C (2000) Identification of a plasmidborne chloramphenicol-florfenicol resistance gene in Staphylococcus sciuri. Antimicrob Agents Chemother 44:2530–2533. https://doi.org/10.1128/AAC.44.9.2530-2533.2000
Bender JK, Fleige C, Klare I, Fiedler S, Mischnik A, Mutters NT, Dingle KE, Werner G (2016) Detection of a cfr(B) variant in german Enterococcus faecium clinical isolates and the impact on linezolid resistance in Enterococcus spp. PLoS ONE 11:e0167042
Hu Y, Won D, Nguyen LP, Osei KM, Seo Y, Kim J, Lee Y, Lee H, Yong D, Choi JR, Lee K (2022) Prevalence and genetic analysis of resistance mechanisms of linezolid-nonsusceptible Enterococci in a tertiary care hospital examined via whole-genome sequencing. Antibiotics (Basel, Switzerland) 11(11):1624. https://doi.org/10.3390/antibiotics11111624
Bi R, Qin T, Fan W, Ma P, Gu B (2018) The emerging problem of linezolid-resistant Enterococci. J Glob Antimicrob Resist 13:11–19. https://doi.org/10.1016/j.jgar.2017.10.018
Rodríguez-Lucas C, Fernández J, Vázquez X, de Toro M, Ladero V, Fuster C, Rodicio R, Rodicio MR (2022) Detection of the optrA gene among polyclonal linezolid-susceptible isolates of Enterococcus faecalis recovered from community patients. Microb Drug Resist 28(7):773–779. https://doi.org/10.1089/mdr.2021.0402
Moure Z, Lara N, Marín M, Sola-Campoy PJ, Bautista V, Gómez-Bertomeu F, Gómez-Dominguez C, Pérez-Vázquez M, Aracil B, Campos J, Cercenado E, Oteo-Iglesias J (2020) Interregional spread in Spain of linezolid-resistant Enterococcus spp. isolates carrying the optrA and poxtA genes. Int J Antimicrob Agents 55:105977. https://doi.org/10.1016/j.ijantimicag.2020.105977.
Ruiz-Ripa L, Feßler AT, Hanke D, Eichhorn I, Azcona-Gutiérrez JM, Pérez-Moreno MO, Seral C, Aspiroz C, Alonso CA, Torres L, Alós JI, Schwarz S, Torres C (2020) Mechanisms of linezolid resistance among enterococci of clinical origin in Spain-detection of optrA- and cfr(D)-carrying E. faecalis. Microorganisms. 8(8): 1155. https://doi.org/10.3390/microorganisms8081155
Càmara J, Camoez M, Tubau F, Pujol M, Ayats J, Ardanuy C, Domínguez MÁ (2019) Detection of the novel optrA Gene Among linezolid-resistant Enterococci in Barcelona Spain. Microb Drug Resist 25(1):87–93. https://doi.org/10.1089/mdr.2018.0028
García-Gil V, Gómez-Gil MR, Escosa-García L, Hierro-Llanillo L (2015) Linezolid and vancomycin-resistant Enterococcus faecium peritonitis in a child after liver transplantation. Enferm Infecc Microbiol Clin 33(1):66. https://doi.org/10.1016/j.eimc.2014.05.008
Gómez-Gil R, Romero-Gómez MP, García-Arias A, Ubeda MG, Busselo MS, Cisterna R, Gutiérrez-Altés A, Mingorance J (2009) Nosocomial outbreak of linezolid-resistant Enterococcus faecalis infection in a tertiary care hospital. Diagn Microbiol Infect Dis 65(2):175–179. https://doi.org/10.1016/j.diagmicrobio.2009.06.010
Sánchez-Gómez JC, Fraile-Malmierca F, Valverdé-Romero ED, Sánchez M, García-Rodríguez JA, García-Sánchez JE (2006) Linezolid-resistant Enterococcus faecalis: first report in Spain. J Chemother (Florence, Italy) 18(4):440–442. https://doi.org/10.1179/joc.2006.18.4.440
Ruiz-Ripa L, Feßler AT, Hanke D, Sanz S, Olarte C, Eichhorn I, Schwarz S, Torres C (2020) Detection of poxtA- and optrA-carrying E. faecium isolates in air samples of a Spanish swine farm. J Glob Antimicrob Resist 22:28–31. https://doi.org/10.1016/j.jgar.2019.12.012
Abdullahi IN, Lozano C, Zarazaga M, Saidenberg ABS, Stegger M, Torres C (2023) Clonal relatedness of coagulase-positive staphylococci among healthy dogs and dog-owners in Spain. Detection of multidrug-resistant-MSSA-CC398 and novel linezolid-resistant-MRSA-CC5. Front Microbiol. 14:1121564. https://doi.org/10.3389/fmicb.2023.1121564
Sahl JW, Lemmer D, Travis J, Schupp JM, Gillece JD, Aziz M, Driebe EM, Drees KP, Hicks ND, Williamson CHD, Hepp CM, Smith DE, Roe C, Engelthaler DM, Wagner DM, Keim P (2016) NASP: an accurate, rapid method for the identification of SNPs in WGS datasets that supports flexible input and output formats. Microb Genom 2:e000074. https://doi.org/10.1099/mgen.0.000074
Letunic I, Bork P (2021) Interactive Tree Of Life (iTOL) v5: an online tool for phylogenetic tree display and annotation. Nucl Acids Res 49:W293–W296. https://doi.org/10.1093/nar/gkab301
Seemann T, Abricate, 2019. Github https://github.com/tseemann/abricate
Liu B, Zheng D, Zhou S, Chen L, Yang J (2022) VFDB 2022: a general classification scheme for bacterial virulence factors. Nucl Acids Res 50(D1):D912–D917. https://doi.org/10.1093/nar/gkab1107
Clausen PTLC, Aarestrup FM, Lund O (2018) Rapid and precise alignment of raw reads against redundant databases with KMA. BMC Bioinformatics 19(1):307. https://doi.org/10.1186/s12859-018-2336-6
Bortolaia V, Kaas RS, Ruppe E, Roberts MC, Schwarz S, Cattoir V et al (2020) ResFinder 4.0 for predictions of phenotypes from genotypes. J Antimicrob Chemother 75:3491–3500. https://doi.org/10.1093/jac/dkaa345
Zankari E, Allesøe R, Joensen KG, Cavaco LM, Lund O, Aarestrup FM (2017) PointFinder: a novel web tool for WGS-based detection of antimicrobial resistance associated with chromosomal point mutations in bacterial pathogens. J Antimicrob Chemother 72:2764–2768. https://doi.org/10.1093/jac/dkx217
Arndt D, Grant JR, Marcu A, Sajed T, Pon A, Liang Y, Wishart DS (2016) PHASTER: a better, faster version of the PHAST phage search tool. Nucleic Acids Res 44(W1):W16–W21. https://doi.org/10.1093/nar/gkw387
Li P, Yang Y, Ding L, Xu X, Lin D (2020) Molecular investigations of linezolid resistance in Enterococci OptrA variants from a hospital in Shanghai. Infect Drug Resist 13:2711–2716. https://doi.org/10.2147/IDR.S251490
Cai J, Schwarz S, Chi D, Wang Z, Zhang R, Wang Y (2019) Faecal carriage of optrA-positive enterococci in asymptomatic healthy humans in Hangzhou, China. Clin Microb Infect 25(5):630.e1-630.e6. https://doi.org/10.1016/j.cmi.2018.07.025
Freitas AR, Tedim AP, Novais C, Lanza VF, Peixe L (2020) Comparative genomics of global optrA-carrying Enterococcus faecalis uncovers a common chromosomal hotspot for optrA acquisition within a diversity of core and accessory genomes. Microbial Genomics 6(6):e000350. https://doi.org/10.1099/mgen.0.000350
Darehkordi H, Saffari F, Mollaei HR, Ahmadrajabi R (2019) Amino acid substitution mutations and mRNA expression levels of the pbp5 gene in clinical Enterococcus faecium isolates conferring high level ampicillin resistance. APMIS 127(3):115–122. https://doi.org/10.1111/apm.12922
Kim YB, Seo HJ, Seo KW, Jeon HY, Kim DK, Kim SW, Lim SK, Lee YJ (2018) Characteristics of high-level ciprofloxacin-resistant Enterococcus faecalis and Enterococcus faecium from retail chicken meat in Korea. J Food Prot 2018(81):1357–1363. https://doi.org/10.4315/0362-028X.JFP-18-046
Werner G, Fleige C, Ewert B, Laverde-Gomez JA, Klare I, Witte W (2010) High-Level ciprofloxacin resistance among hospital-adapted Enterococcus faecium (CC17). Int J Antimicrob Agents 35(2):119–125. https://doi.org/10.1016/j.ijantimicag.2009.10.012
Belloso Daza MV, Milani G, Cortimiglia C, Pietta E, Bassi D, Cocconcelli PS (2022) Genomic insights of Enterococcus faecium UC7251, a multi-drug resistant strain from ready-to-eat food, highlight the risk of antimicrobial resistance in the food chain. Front Microbiol 13:894241. https://doi.org/10.3389/fmicb.2022.894241
Vats P, Kaur UJ, Rishi P (2022) Heavy metal-induced selection and proliferation of antibiotic resistance: a review. J Appl Microbiol 132(6):4058–4076. https://doi.org/10.1111/jam.15492
European Commission (2017) Commission implementing decision of 26.6.2017 concerning, in the framework of Article 35 of Directive 2001/82/EC of the European Parliament and of the Council, the marketing authorizations for veterinary medicinal products containing “zinc oxide” to be administered orally to food-producing species. https://ec.europa.eu/health/documents/community-register/2017/20170626136754/dec_136754_en.pdf. Accessed 25 Dec 2023
Rebelo A, Mourão J, Freitas AR, Duarte B, Silveira E, Sanchez-Valenzuela A, Almeida A, Baquero F, Coque TM, Peixe L, Antunes P, Novais C (2021) Diversity of metal and antibiotic resistance genes in Enterococcus spp. from the last century reflects multiple pollution and genetic exchange among phyla from overlapping ecosystems. Sci Total Environ 787:147548. https://doi.org/10.1016/j.scitotenv.2021.147548
Rebelo A, Almeida A, Peixe L, Antunes P, Novais C (2023) Unraveling the role of metals and organic acids in bacterial antimicrobial resistance in the food chain. Antibiotics (Basel, Switzerland) 12(9):1474. https://doi.org/10.3390/antibiotics12091474
López M, Alvarez-Martínez MJ, Marco F, Torres C (2013) Glycopeptide-resistant Enterococcus faecium. analysis of the resistance genotype, virulence and genetic lines. Enferm Infecc Microbiol Clin 31:10–14. https://doi.org/10.1016/j.eimc.2012.05.010
Zaheer R, Cook SR, Barbieri R, Goji N, Cameron A, Petkau A, Polo RO, Tymensen L, Stamm C, Song J, Hannon S, Jones T, Church D, Booker CW, Amoako K, Van Domselaar G, Read RR, McAllister TA (2020) Surveillance of Enterococcus spp. reveals distinct species and antimicrobial resistance diversity across a one-health continuum. Sci Rep 10(1):3937. https://doi.org/10.1038/s41598-020-61002-5
Raven KE, Reuter S, Gouliouris T, Reynolds R, Russell JE, Brown NM, Török ME, Parkhill J, Peacock SJ (2016) Genome-based characterization of hospital-adapted Enterococcus faecalis lineages. Nat Microbiol 1:15033. https://doi.org/10.1038/nmicrobiol.2015.33.
Zischka M, Künne CT, Blom J, Wobser D, Sakιnç T, Schmidt-Hohagen K, Dabrowski PW, Nitsche A, Hübner J, Hain T, Chakraborty T, Linke B, Goesmann A, Voget S, Daniel R, Schomburg D, Hauck R, Hafez HM, Tielen P, Jahn D, Werne G (2015) Comprehensive molecular, genomic and phenotypic analysis of a major clone of Enterococcus faecalis MLST ST40. BMC Genomics 16(1):175. https://doi.org/10.1186/s12864-015-1367-x
McHugh MP, Parcell BJ, Pettigrew KA, Toner G, Khatamzas E, El Sakka N, Karcher AM, Walker J, Weir R, Meunier D, Hopkins KL, Woodford N, Templeton KE, Gillespie SH, Holden MTG (2022) Presence of optrA-mediated linezolid resistance in multiple lineages and plasmids of Enterococcus faecalis revealed by long read sequencing. Microbiology (Reading, England) 168(2):001137. https://doi.org/10.1099/mic.0.001137
Abdullahi IN, Lozano C, Zarazaga M, Simón C, Höfle U, Sieber RN, Latorre-Fernández J, Stegger M, Torres C (2024) Comparative genomics of Staphylococcus aureus strains from wild birds and pig farms elucidates levels of mobilomes, antibiotic pressure and host adaptation. J Glob Antimicrob Resist 36:142–150. https://doi.org/10.1016/j.jgar.2023.12.003
Clewell DB, Weaver KE, Dunny GM, Coque TM, Francia MV (2014) Extrachromosomal and mobile elements in enterococci: transmission, maintenance, and epidemiology. Boston: Massachusetts eye and ear infirmary. https://pubmed.ncbi.nlm.nih.gov/24649505/. Accessed 2 Feb 2023
Cabal A, Rab G, Daza-Prieto B, Stöger A, Peischl N, Chakeri A, Mo SS, Bock H, Fuchs K, Sucher J (2022) Characterizing antimicrobial resistance in clinically relevant bacteria isolated at the human/animal/environment interface using whole-genome sequencing in Austria. Int J Mol Sci 23:11276. https://doi.org/10.3390/ijms231911276
Zhu XQ, Wang XM, Li H, Shang YH, Pan YS, Wu CM, Wang Y, Du XD, Shen JZ (2017) Novel lnu(G) gene conferring resistance to lincomycin by nucleotidylation, located on Tn6260 from Enterococcus faecalis E531. J Antimicrob Chemother 72(4):993–997. https://doi.org/10.1093/jac/dkw549
Matos RC, Lapaque N, Rigottier-Gois L, Debarbieux L, Meylheuc T, Gonzalez-Zorn B, Repoila F, de Lopes MF, Serror P (2013) Enterococcus faecalis prophage dynamics and contributions to pathogenic traits. PLoS Genet 9:e1003539. https://doi.org/10.1371/journal.pgen.1003539.
Song M, Wu D, Hu Y, Luo H, Li G (2021) Characterization of an Enterococcus faecalis bacteriophage vB_EfaM_LG1 and its synergistic effect with antibiotic. Frontiers Cell Infect Microb 11:698807. https://doi.org/10.3389/fcimb.2021.698807
Bolocan AS, Upadrasta A, Bettio PHA, Clooney AG, Draper LA, Ross RP, Hill C (2019) Evaluation of phage therapy in the context of Enterococcus faecalis and its associated diseases. Viruses 11(4):366. https://doi.org/10.3390/v11040366
Cinthi M, Coccitto SN, Morroni G, D’Achille G, Brenciani A, Giovanetti E (2022) Detection of an Enterococcus faecium carrying a double copy of the poxtA Gene from freshwater river, Italy. Antibiotics (Basel, Switzerland) 11(11):1618. https://doi.org/10.3390/antibiotics11111618
Tan SC, Chong CW, Teh CSJ, Ooi PT, Thong KL (2018) Occurrence of virulent multidrug-resistant Enterococcus faecalis and Enterococcus faecium in the pigs, farmers and farm environments in Malaysia. PeerJ 6:e5353. https://doi.org/10.7717/peerj.5353
Badul S, Abia ALK, Amoako DG, Perrett K, Bester LA, Essack SY (2021) From the farms to the dining table: the distribution and molecular characteristics of antibiotic-resistant enterococcus spp. in intensive pig farming in South Africa. Microorganisms 9(5):882. https://doi.org/10.3390/microorganisms9050882
Acknowledgements
We cordially thank Andre Becker Saidenberg for their assistance in some of the genome analyses. Also, the authors appreciate the support of Ursula Hofle, Carmen Simon and other veterinarians in the animal samples collection.
Funding
Open Access funding provided thanks to the CRUE-CSIC agreement with Springer Nature. This work was finnanced by MICIU/AEI/10.13039/501100011033 of Spain (project PID2019-106158RB-100). Also, it received funding from the European Union’s H2020 research and innovation programme under the Marie Sklodowska-Curie grant agreement N° 801586.
Author information
Authors and Affiliations
Contributions
Conceptualization: I.N.A., C.T.; methodology: I.N.A., M.S.; laboratory experiments: I.N.A., S.H.; software analysis: I.N.A., M.S., S.H., A.R., J.L.F.; validation: C.T., I.N.A., J.L.F., S.H., A.R., M.S., M.Z. and C.L.; formal analysis: I.N.A., C.T., S.H., A.R., M.S.; data curation: C.T., I.N.A., M.S.; writing—original draft preparation, I.N.A., C.T.; writing—review and editing: C.T., I.N.A., J.L.F., M.Z., S.H., A.R., M.S., and C.L.; supervision: C.T., C.L., project administration: C.T., funding acquisition: C.T., M.Z., I.N.A. All authors have revised and agreed to the published version of the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
None declared by the authors.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Abdullahi, I.N., Lozano, C., Zarazaga, M. et al. Genomic Characterization and Phylogenetic Analysis of Linezolid-Resistant Enterococcus from the Nostrils of Healthy Hosts Identifies Zoonotic Transmission. Curr Microbiol 81, 225 (2024). https://doi.org/10.1007/s00284-024-03737-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00284-024-03737-2