
Abstract
Background
The suamc genus Rhus (sensu stricto) includes two subgenera, Lobadium (ca. 25 spp.) and Rhus (ca. 10 spp.). Their members, R. glabra and R. typhina (Rosanae: Sapindales: Anacardiaceae), are two economic important species. Chloroplast genome information is of great significance for the study of plant phylogeny and taxonomy.
Results
The three complete chloroplast genomes from two Rhus glabra and one R. typhina accessions were obtained with a total of each about 159k bp in length including a large single-copy region (LSC, about 88k bp), a small single-copy regions (SSC, about 19k bp) and a pair of inverted repeats regions (IRa/IRb, about 26k bp), to form a canonical quadripartite structure. Each genome contained 88 protein-coding genes, 37 transfer RNA genes, eight ribosomal RNA genes and two pseudogenes. The overall GC content of the three genomes all were same (37.8%), and RSCU values showed that they all had the same codon prefers, i.e., to use codon ended with A/U (93%) except termination codon. Three variable hotspots, i.e., ycf4-cemA, ndhF-rpl32-trnL and ccsA-ndhD, and a total of 152–156 simple sequence repeats (SSR) were identified. The nonsynonymous (Ka)/synonymous (Ks) ratio was calculated, and cemA and ycf2 genes are important indicators of gene evolution. The phylogenetic analyses of the family Anacardiaceae showed that the eight genera were grouped into three clusters, and supported the monophyly of the subfamilies and all the genera. The accessions of five Rhus species formed four clusters, while, one individual of R. typhina grouped with the R. glabra accessions instead of clustering into the two other individuals of R. typhina in the subgenus Rhus, which showed a paraphyletic relationship.
Conclusions
Comparing the complete chloroplast genomes of the Rhus species, it was found that most SSRs were A/T rich and located in the intergenic spacer, and the nucleotide divergence exhibited higher levels in the non-coding region than in the coding region. The Ka/Ks ratio of cemA gene was > 1 for species collected in America, while it was < 1 for other species in China, which dedicated that the Rhus species from North America and East Asia have different evolutionary pressure. The phylogenetic analysis of the complete chloroplast genome clarified the Rhus placement and relationship. The results obtained in this study are expected to provide valuable genetic resources to perform species identification, molecular breeding, and intraspecific diversity of the Rhus species.
Similar content being viewed by others


Background
The sumac genus Rhus L. is the largest and most widespread genus in the family Anacardiaceae, within which two subfamilies Anacardioideae and Spondioideae were recognized, with a disjunct distribution that spans over temperate and subtropical latitudes in the Northern Hemisphere [1,2,3,4,5]. In general, sumac can grow in nonagriculturally viable regions, and various species have been used by indigenous cultures for medicinal and other purposes, suggesting potential for commercializing the bioactivity of these plants [6]. Different parts of Rhus plants can be used in medicine and food, and species of the genus have also been utilized in soil conservation and environmental restoration [7,8,9,10].
The representative species, including R. coriaria (tanner’s sumac), R. copallina (winged or shining sumac), R. glabra (smooth sumac), R. undulate (Kuni bush), and R. verniciflua (Japanese sumac), are native to Mediterranean Basin, Eastern North America, Western North America, South Africa and Asia, respectively [11]. Specially, only the two species R. glabra and R. typhina are the host plant species of the Rhus-gall aphids Melaphis [12]. R. typhina were introduced to China in 1959 [13], that is considered potentially invasive in its non-native habitats [14]. The branches of R. glabra have antimicrobial activity, which is often used in folk medicine by North American native people [7]. As a summary, R. glabra and R. typhina are not only an important species for the landscape of certain open habitats but also ecologically essential as one of the two hosts of the monotypic Rhus-gall aphids Melaphis as well as a medicinal plant.
The genus Rhus (sensu stricto) is classified into two subgenera: subgenus Rhus and subgenus Lobadium [15]. Rhus subgenus includes 10 species and is characterized by the deciduous and imparipinnately compound leaves, the flowers appearing after the leaves, and inflorescences as terminal thyrses, which are subtended by deciduous bracts [15, 16]. The phylogenetic relationship of the genus Rhus was analyzed using a few number of nuclear and chloroplast regions with a limit sampling, and the results based on ITS sequence indicated that the subgenus Rhus was nested into Lobadium to form a paraphyletic group, but R. glabra is not included in this study [4]. The analysis of combined data sets, i.e., ITS, trnL-trnF, ndhF; Nia-i3, trnC-trnD, indicated that subgenus Rhus is monophyletic and R. glabra is sister with R. typhina [17,18,19], and the trnC-trnD region provided slightly more parsimony-informative characters than the ndhF gene and trnL-trnF region [18].
The chloroplast genome is a valuable resource in molecular phylogenetic analysis [20, 21], and contains a pair of inverted repeat (IR) regions separated by a large single copy (LSC) and a small single copy (SSC) region [22, 23]. This quadripartite structure is highly conserved in gene content and genome organization relative to the plant nuclear and mitochondrial genomes [23]. Comparative analyses between chloroplast genomes of plant species revealed structural variations, such as IR or gene loss that are considered as a result of environmental adaptation [23]. Despite the plastid genome generally has a slower evolutionary rate than the nuclear genome, it is frequently used in phylogenetic studies of plants [24, 25]. This is mostly due to the fact of its uniparental inheritance, which provides unique information about the evolutionary history of the group under study [26]. More recently, it is considered that the chloroplast genome data could authenticate evolutionary relationships and confirm phylogenetic classifications for plants at the family and genus level [27]. The chloroplast genome hence has been widely utilized as a good marker for the phylogenetic reconstructions of plants at species, genus and family levels.
In this study, we sequenced three complete chloroplast genomes of Rhus glabra and R. typhina using the high throughout sequencing method on an Illumina HiSeq 4000 platform. We aimed to characterize the structure and organization of the R. glabra and R. typhina chloroplast genomes and the nucleotide divergence in genus Rhus, and conduct an initial chloroplast phylogenomic analysis in the family Anacardiceae, with an emphasis of Rhus subgenus Rhus.
Results
General characteristics of three chloroplast genome of Rhus species
The two complete chloroplast genomes of Rhus glabra were assembled as a total of 159,984 bp and 159,944 bp, and R. typhina is 159,940 bp in length (Table 1). The nucleotide composition of the genome were calculated to be 31.40% T, 30.80% A, 19.20% C, and 18.60% G, respectively. By BLAST and Finding Repeat Region in Geneious software, the complete chloroplast genomes were divided into a canonical quadripartite structure with a large single-copy region (LSC, 87,904 − 88,002 bp), a small single-copy region (SSC, 18,862 − 18,891 bp) and a pair of inverted repeats regions (IRa/IRb, 26,559 − 26,560 bp). The GC content of the LSC, SSC and IR regions were 35.8%, 32.4% and 42.9%, respectively. The complete chloroplast genomes of two R. glabra individuals (Voucher nos. Ren_P3002 and Ren_P3051) and one R. typhina (Voucher no. Ren_P3053) with gene annotations were submitted to GenBank under the accession numbers OR800752, OR800753 and OR773067, respectively.
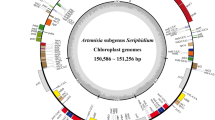
A total of 135 genes in the three chloroplast genomes were both annotated, including 88 protein-coding genes, eight ribosomal RNA genes, 37 transfer RNA genes and two pseudogenes (Table 1). The classifications of the 135 genes in the Rhus glabra complete chloroplast genome were showed in Table 2, which were classified into five categories. Based on the analysis on the protein-coding genes, we found that 88 protein-coding genes were dispersedly located in quadripartite structure, i.e., 60 protein-coding genes in LSC region, 12 in SSC region and eight in IR region (Fig. 1, Fig. S1 and Fig. S2). Four rRNAs, i.e., rrn16, rrn23, rrn4.5 and rrn5, were only located in a pair of inverted repeats regions, and appeared symmetrical distribution. In the 37 tRNA genes, trnL-UAG was only one which was harbored in SSC, while 14 tRNA genes appeared symmetrically in IRs and the remains in LSC.

Chloroplast genome map of Rhus glabra (Accession No. OR800752). Genes encoded in the forward direction are located on the outside of the ring, while those encoded in the reverse direction on the inside of the ring. The gray circles inside represent the GC content
In case of intron-containing genes, there were 17 intron-containing genes including one gene (ndhA) in the SSC, 12 genes in the LSC and four genes (rpl2, ndhB, trnI-GAU, trnA-UGC) duplicated in the IR. The two genes ycf3 and clpP were located in the LSC with possessing two introns (Table 2 and Table S1). The gene trnL-UAA possessed the shortest intron with 469 bp in length, wihle the longest intron was founded in trnK-UUU with 2598 bp in length, in which the the protein-coding gene matK was inserted, which is the general characteristics in plants [28]. The gene rps12 was trans-spliced with the duplicated 3′ end in the IRs and the 5′ end located in the LSC region, as previously reported in other plants [29].
Codon usage of protein-coding genes
The values of RSCU of 88 protein-coding genes of two Rhus glabra chloroplast genomes (accession Nos.OR800752 and OR800753) and three R. typhina accessions (accession Nos. MN866894, MT083895 and OR773067) from GenBank were displayed in Table S2. All the samples had the same condon bias for each amino acid. The protein-coding gene totally contained 26,700 − 26,807 codons (including 88 terminal codons) in different individuals, among which Leu was the most aboudant amino acid, with about 2816 (10.51%) − 2824 (10.53%), while Cys was the least, with 313 (1.18%) − 316 (1.18%), respectively. In all the termination codons, UAA is the most frequently used amino acid. The RSCU values of 30 codons were all > 1 except termination codon, in which codon ended with A/U was 93% and codon ended with C/G was 7%, indicating that these codons tended to end in A/U. The codon usage is extremely conserved in the these species due to as species belong to the genus level conservation, which was consistent with the previous reports on the many land plants’ chloroplast genomes [30, 31].
SSR analysis
SSR loci were detected in three chloroplast genomes, and a total of 152, 153 and 156 SSRs were in one R. typhina individual (accession No. OR773067) and two R. glabra accessions (accession Nos. OR800752 and OR800753), respectively. The SSR distribution in different gene regions and genomic quadripartite structures were shown in Fig. 2. The three samples have the same amounts of repeat types (six dinucleotides, 63 trinucleotides, 10 tetranucleotides, and two pentanucleotides) except mononucleotide type containing 71, 72 and 75, separately (Fig. 2C). Regard to each individuals, majority of SSRs focused on mononucleotide (47 − 48%) and trinucleotide (40 − 41%) in Fig. 2C. Nearly all mononucleotide SSRs were composed of A/T (97%), and trinucleotide repaets of AAT and TTC were the second most common SSRs in these three chloroplast genomes (Fig. 2C).

The type and distribution of SSRs in the three Rhus chloroplast genomes. A: Proportion of SSR distribution in the intergenic spacer, exon and intron; B: Frequency of SSR occurrence in the LSC, SSC, and IR region; C: SSR repeat types and numbers
Contraction and expansion of IR region
Six chloroplast genome sequences of Rhus species in GenBank (Accession Nos. OP326720, MT230556, MN866893, MT230555, MN866894 and MT083895) were compared with three chloroplast genomes present in this study. To analyze the boundary and detect gene contraction and expansion of chloroplast sequences, IRscope software was used to visualize the junction of single copy and the inverted repeat regions, shown in Fig. 3. The length of IR regions ranged from 26,475 to 26,560 bp, and the region of IR/LSC and IR/SSC junction in these Rhus species showed the same boundary genes, i.e., rps19, rpl2, ycf1, ndhF, ycf1, trnN, rpl2 and trnH. The two protein-coding genes ndhF and ycf1 crossed the IRb/SSC and SSC/IRa junction in all the genomes, respectively. Furthermore, ycf1 gene was partially duplicated at the IRb/SSC boundary, resulting in a pseudogene, which can be obversied in nine Rhus genomes. In four accessions (Accession Nos. OP326720, MN866893, MT230556 and MT230555), the JLB boundary was 26 and 40 bp away from rps19, while in the other five accessions species the rps19 gene crossed the JLB boundary. In addition, the pseudogene ycf1 usually located at the end of IRb region crossing SSC with less than 14 bp, while it is very special, that is, in R. typhina (MN866894), ycf1 pseudogene crossed JSB with 512 bp into SSC region, and overlapped with the ndhF gene in SSC region with a stretch of 45 bp. The Rhus chloroplast genomes are generally conserved, but slightly with a variaton of either expansion or contraction of the single copy and IR boundary regions.

Comparison of the borders of the LSC, SSC and IR regions in chloroplast genomes of Rhus. The junction of these parts, i.e., JLB (junction of LSC and IRb), JSB (junction of SSC and IRb), JSA (junction of SSC and IRa) and JLA (junction of LSC and IRa)
Sequence divergence and hotspots
We determined the genome divergence of Rhus genus by genome alignment using the program mVISTA, and the visualization map was shown in Fig. 4. It was showed that the intraspecific variation of each species was low, and the exons were extremely conservative, while the interspecific variation was high, and the divergence regions were mainly from these ones, i.e., trnH-psbA, trnK-rps16, ycf4-cemA, rps19, ndhF-rpl32-trnL, trnS-psbZ and ccsA-ndhD regions, majority of which happened in introns and intergenic spacer.

Visualization map of genome alignment of the chloroplast genomes using Rhus glabra (OR800752) as a reference by mVISTA. X-axis: the coordinates in chloroplast genomes; Y-axis: the average percent identity (50–100%)
We also identified the mutational hotspots of the chloroplast genomes of the Rhus species in Fig. 5. There were three positions (ycf4-cemA, ndhF-rpl32-trnL and ccsA-ndhD) in the nine Rhus species exhibited high nucleotide diversity (Pi values > 0.02) tested by the DNaSP software, among which the average Pi value of the ccsA-ndhD region was the highest (Pi values = 0.03819) in Table 3.

Sliding-window analysis of the chloroplast genomes in Rhus genus. Window length: 800 bp; step size: 200 bp; X-axis: position of the midpoint of a window; Y-axis: nucleotide diversity of each window
Additionally, we calculated the nucleotide diversity between two R. glabra and three R. typhina chloroplast genomes (Table 3). R. glabra and R. typhina chloroplast genomes were aligned with a matrix of 160,022 bp and 161,410 bp, respectively. Twenty three and 430 variable sites were examined, respectively, and intraspecific difference of R. typhina is located on rpl32 gene (Fig. S3). The pair variable sites between the R. glabra and R. typhina individuals indicated that the variation (about 0.27%) of the R. typhina individual (accession No. OR773067) and the other two R. typhina accessions (Nos. MN866894 and MT083895) was much higher than that (about 0.01%) with the two R. glabra accessions (Nos. OR800752, OR800753).
Synonymous (Ks) and nonsynonymous (Ka) substitution rate analyses
We annotated 88 protein-coding genes in our samples of R. typhina and R. glabra, while, there are 86 genes annotated in the Rhus accessions from GenBank. So we used the common 86 protein-coding genes in Rhus genus, and meanwhile excluded the eight repeat genes in the IR region, that is, pairwise comparisons of 78 commom protein-coding genes in all Rhus species were finally employed to calculate Ka and Ks substitution rates as compared with Pistacia chinensis (MK738124). The genes with Ka/Ks values > 0 were shown in Fig. 6, indicating nucleotide substututions of different protein-coding genes are different. Among the genes with Ka/Ks ratio > 1, cemA gene for American species (our samples, accession Nos. OR800752, OR800753 and OR773067) and R. typhina (accession Nos. MN866894 and MT083895) were undergoing positive selection pressure, while Ka/Ks ratio of cemA gene < 1 for other species, which were undergoing purifying selection. Moreovere, the ycf2 gene was different from the above genes with Ka/Ks ratio < 1 in American species, while > 1 in other accessions. The genes with the ratio of Ka/Ks = 0 or NA (Ks = 0) didn’t be displayed in Fig. 6, including 27 protein-coding genes, i.e., atpH, clpP, ndhB, ndhC, ndhE, petB, petD, petG, petN, psbN, psaC, psaJ, psaI, psbA, psbE, psbF, psbI, psbJ, psbL, psbM, psbN, psbZ, rpl36, rps7, rpl23, rps18, rps19, which dedicated that these genes were the most conserved genes and nucleotide substututions were slow.

The Ka/Ks ratios of 78 protein-coding genes in the Rhus genus taking Pistacia chinensis (MK738124) as a reference
Phylogenetic analysis
The phylogenetic tree of the 25 Anacardiaceae species supported the monophyly of each genus, and provided high support for the phylogenetic relationships among genera (Fig. 7). Three well-supported clades were identified: (I) Spondias of Spondiadoideae, (II) Sclerocarya of Spondiadoideae, and the remaining six genera (III) of Anacardioideae. The genus Spondias was sister to the clade of the remaining genera with 100% support. Within subfamily Anacardioideae, Anacardium and Mangifera consitituted a clade; Rhus is sister to the Cotinus + Pistacia clade; and Toxicodendron is then sister to the clade of (Rhus, (Cotinus, Pistacia)). In Rhus genus, R. potaninii was sister to R. punjabensis, then grouped with R. chinensis; the different individuals from the sister species R. glabra and R. typhina formed two groups with paraphyletic relationship: one in two R. typhina individuals (accession Nos. MN866894 and MT083895) from China, and the other one is two R. glabra individuals (accession Nos. OR800752 and OR800753) and one R. typhina individual (accession No. OR773067) from US.

Maximum likelihood (ML) tree constructed by protein-coding genes of the Anarcadiceae chloroplast genomes. Stars represent nodes with 100% bootstrap values. Dots represent three Rhus individuals in this study
Discussion
Overall, the Rhus chloroplast genomes were relatively conserved, including the genomes structures, nucleotide composition, gene orders, G + C contents, and codon usages, which are both a canonical quadripartite structure: a large single-copy region (LSC, about 88k bp), a small single-copy regions (SSC, about 19k bp) and a pair of inverted repeats regions (IRa/IRb, about 26k bp). Each genome contained 88 protein-coding genes, 37 transfer RNAs genes, eight ribosomal RNA genes and two pseudogenes. The total GC contents (37.8%) of three Rhus individuals were almost the same as those of other Anacardiaceae species, e.g., R. potaninii (37.9%) [2], R. chinensis (37.8%) [28], R. punjabensis (37.9%) and R. typhina (37.8%). The GC content of IR region in all Rhus genus is the highest, possibly due to the presence of four extremely conserved rRNA genes in IR region, which had high GC contents.
The codon preference in different species is different and has racial specificity [32, 33]. The relative synonymous codon usage (RSCU) is one of the commonly used parameters to measure codon usage bias [33]. The RSCU values of 30 codons were all > 1 except termination codon in two R. glabra and three R. typhina individuals, in which codon ended with A/U was 93% and is similar to the plants of the same genus [30] and other plants, such as Cinnamomum camphora [34], Wurfbainia villosa [35] and Aconitum hemsleyanum [36]. Those were consistent with the analysis that dicotyledons’ codon tend to end in A/T [37].
It was reported that simple sequence repeats (SSRs), or microsatellites, are repeat sequences of typically 1–6 bp that are distributed throughout the genome [34, 36]. Compared with other neutral DNA regions, SSRs usually have a higher mutation rate due to slipped DNA strands and have been used for the study of population genetics, evolutionary and ecological studies [38, 39]. We looked for SSRs of 10 bp or longer, as these have been suggested to be prone to slipped strand mispairing and believed to be the main mutational mechanism for SSR polymorphisms [39]. In Rhus chloroplast genomes, five types were detected and majority of SSRs focused on mononucleotide and trinucleotide, which were consistent with the previous observation that the SSRs of chloroplast genomes are dominated by ‘A’ or ‘T’ mononucleotide repeats, which refects a biased base composition with an overall A-T richness in the chloroplast genomes [34,35,36]. The SSR distribution in different gene regions and genomic quadripartite structures are uneven, and majority of SSRs located on LSC and intergenic spacer, which might provide more information for selecting effective molecular markers for the detection of intra- and interspecifc polymorphisms.
Boundary shifts of the IR region in chloroplast genomes are hypothesized to be one of the primary drivers of overall chloroplast genome size variation [40, 41]. In this study, the longest size of the R. typhina chloroplast genome (accession No. MN866894) was from the contractions or expansion of the gene ndhF or pseudogene ycf1 in IR region into SSC, which was different from other Rhus species. The ycf1 gene in the SSC region was the second largest gene in the plastid genome and encoded a protein of approximately 1,800 amino acids, which was highly variable, while the pseudogene ycf1 located in the IRb region was conserved. Our current results are consistent with the reports from Asaf et al. [38]. Based on a complete chloroplast genome, IR boundary shifts often lead to gene duplication, loss, and large-scale syntenic rearrangement [42]. However, we didn’t detect these events in our samples.
Comparison of the chloroplast genome sequences was considered as an effective strategy to identify the mutation hotspots, which can be used as the specific DNA barcodes [43]. The detection of variability of hotspots as well as universal barcodes (Table 3) showed that the ycf4-cemA has the second highest average nucleotide diversity (0.03528) after ccsA-ndhD region (0.03819), next ndhF-rpl32-trnL (0.03399). Yi et al. found that the trnC-trnD region provided more parsimony-informative characters than the trnL-trnF region and ndhF gene [18], and Pang et al. treated the region trnH-psbA as a universal barcode [44]. However, basing on the whole chloroplast genome, we detected fewer parsimony information sites and lower nucleotide diversity in trnC-trnD and trnH-psbA, whereas found some other more suitable potential molecular markers (ycf4-cemA, ndhF-rpl32-trnL, ccsA-ndhD) than universal barcodes [45]. These potential highly variable chloroplast barcodes will increase, rich and update marker resources, especially for traditional Chinese medicine [46]. Moreover, the divergence of ycf4-cemA, ndhF-rpl32-trnL and ccsA-ndhD gene was high and majority happened in introns and intergentic spacer, which seemly associated with the results that microsatellites major concentrated intergenic spacer (Figs. 4 and 5). All in all, they both reflected that the interspecific variation were greater than the intraspecific variation, and molecular marker could be helpful for species identification.
The non-synonymous (Ka) and synonymous (Ks) nucleotide substitution patterns of gene are important indicators of gene evolution [47]. The Ka/Ks ratio is usually used to assess whether there are selective pressures on protein-coding gene or to evaluate the rate of gene divergence. Ka/Ks ratios indicate that the gene has undergone positive selection (> 1), neutral selection (close to 1), or purifying seletion (< 1). In our study, the Ka/Ks ratios of ycf2 vary dramatically in species, i.e., the ycf2 gene of American individuals (accession Nos. OR800752, OR800753 and OR773067) is undergoing strongly purifying seletion, while for other individuals, the Ka/Ks ratios of ycf2 gene > 1, undergoing positive selection. We will select a broader group of species to analyze Ks and Ka substitution rate, especially R. typhina introduced in China.
Our phylogenetic analyses (Fig. 7) actually supported three major clades, and the broad relationships of Anacardioideae and Spondiadoideae are consistent with the phylogenetic analyses [3, 31, 48]. The genus Rhus had a controversial generic limits with Toxicodendron, sometimes included as part of Rhus [4]. Our results clearly supported that Toxicodendron was distinct from Rhus, and Rhus was more closely related to the Cotinus-Pistacia clade than to Toxicodendron, supporting the narrower generic limit of the genus Rhus [17]. In addition, R. typhina (OR773067) from US didin’t preferentially cluster with other R. typhina individuals from China (MN866894 and MT083895), while grouped with R. glabra collected in North American.
Clarifying the phylogenetic positions of R. glabra and R. typhina are important for understanding the evolution of Rhus and the biodiversity assembly in the context of the co-evolution of the Rhus-gall aphids and its host plants. Two phylogenetic studies [17, 18] have focused on the genus Rhus with a broad sampling, and the results showed incongruence between the nuclear and chloroplast phylogenies, i.e., R. glabra appears as sister to R. typhina in a clade, sistering to R. michauxii according to the nuclear ITS region, whereas R. glabra and R. michauxii constitute a clade, sistering to R. typhina by the chloroplast DNA sequences, and they interpreted such incongruence as evidence for hybridization. One individual of R. typhina (OR773067) clustered with R. glabra indivuduals might be from the natural hybrids, which has been detected between R. glabra and R. typhina [49]. Also, Natural hybridization has been reported frequently for Rhus species [50]. We will expand the taxon sampling at the genome scale incluing R. michauxii and further analyze its evolutionary process on the association with the Rhus-gall aphid Melaphis.
The present results may provide valuable sequence information for molecular phylogenetics and aid in the development of molecular markers for genus Rhus and evolutionary analyses of the biological interactions of Rhus and the Rhus-gall aphids. It will also offer a theoretical basis for resource utilization and conservation of the Rhus germplasms.
Conclusion
In this study, we assemblyed three chloroplast genomes of Rhus and identified their gene content and general features, which are both a canonical quadripartite structure. Compared with other Rhus species, the interspecific variation were greater than the intraspecific variation, and molecular marker could be helpful for species identification. We detected three sequence variation hotspots (ycf4-cemA, ndhF-rpl32-trnL, ccsA-ndhD) which have more parsimony information sites and nucleotide diversity than universal barcodes (trnC-trnD, trnL-trnF), which will potentially provide chloroplast markers for further taxonomic, phylogenetic, and population genetic studies in the Rhus genus. In Rhus genus, the average Ka/Ks ratio was 0.267, which dedicated protein-coding genes of Rhus genus was undergoing purifying seletion pressure. And cemA and ycf2 gene are important indicators of gene evolution. The phylogenomic analyses supported R. glabra was sister to R. typhina. The present results may provide valuable sequence information for molecular phylogenetics and aid in the development of molecular markers for genus Rhus and evolutionary analyses of the biological interactions of Rhus and the Rhus-gall aphids.
Methods
Sample, DNA extraction and sequencing
We collected the fresh leaves of Rhus glabra and R. typhina in USA in August, 2015. Two R. glabra accessions (Voucher nos. Ren_P3002 and Ren_P3051) were from Cincinnati in Ohio and Mansfield in Georgia, respectively, and the R. typhina sample (Voucher no. Ren_P3053) from New York. The leaf samples were put in silica gel prior to DNA extraction and the specimen was stored at the Herbarium of School of Life Science, Shanxi University, China. We extracted the total genomic DNA from the leaves by the modified CTAB method [51], and sent the DNAs to the Genomic Sequencing and Analysis Facility (GSAF), (Shanghai, China) for library construction and sequencing on an Illumina HiSeq 4000 platform [52]. The paired-end (PE) reads of 2 × 150 bp (insert size of 400 bp) were generated and the sequencing reads were trimmed for obtained high quality reads [53].
Chloroplast genome assembly and annotation
After quality trimming, high-quality clean reads were reference-based assembled by the programs GetOrganelle [54] with kmers 21, 45, 65, 85, and 105. The genome annotation was conducted by using Plastid Genome Annotator (PGA) [55], with manual correction using Geneious software (version 11.0.3) [56]. For unannotated or ambiguous region, BLAST in GenBank searched homologous sequences to attempt annotate these regions.
General characteristics of the chloroplast genome of Rhus
The total chloroplast genome lengths, gene numbers, gene sizes, nucleotide compositions and the lengths of exons and introns were calculate with Geneious (version 11.0.3). The chloroplast physical mappings were drawn using OGDraw (http://ogdraw.mpimp-golm.mpg.de/index.shtml). In MEGA7.0 [57], we calculated the situation frequency of the relative synonymous codon usage by selecting plant plastid. MIcroSAtellite (MISA) (http://pgrc.ipk-gatersleben.de/misa/) software was employed to identify the simple sequence repeats (SSR), and tandem repeats of 1–6 nucleotides was considered as microsatellites [58]. The parameters were set as follows: >10 for mononucleotides, > 6 for dinucleotides, > 3 for trinucleotides, > 3 for tetranucleotides, > 3 for pentanucleotides, and > 3 for hexanucleotides.
Comparasion of chloroplast genomes in Rhus
24 complete chloroplast genomes representing eight genera of Anacardiaceae from GenBank and our current chloroplast genomes were used to analyze the chloroplast genomic variation of Anacardiaceae species. We re-annotated the chloroplast genomes of Rhus potaninii (accession Nos. MN866893 and MN866893), R. punjabensis (No. MT230555) and R. typhina (Nos. MT083895 and MN866894).
The differences between complete chloroplast genome sequences were analyzed, and chloroplast genome structure of these species compared using mVISTA for further analysis [59]. And LSC/ IRb/ SSC/ IRa junctions can be visualized in IRscope online software (http://irscope.shinapps.io/irapp/).
We used the complete chloroplast genomes of Rhus genus to detect the hotspots of species divergence, and analyze the nucleotide diversity (Pi) using the DnaSP v5 software using the sliding window method with a step size of 200 bp and the window length of 800 bp [60] and we considered the outlier values as mutational hotspots.
Synonymous (Ks) and nonsynonymous (Ka) substitution rate analyses
The DnaSP v5 software [60] was used to estimate substitution rates and Ka/Ks ratios of all 78 protein-coding genes across Rhus species (Nos. OP326730, MT230555, MT230556, MN866893, MN866894, MT083895, OR800752, OR800753 and OR7730677). To do this, each protein-coding gene was aligned respectively together with Pistacia chinensis (MK738124) as a reference using MAFFT Alignment in Geneious (11.0.3). The parameter settings were as following: Genetic Code: Nuclear Universal. The indication for Ka/Ks “NA” which appears when Ks = 0 (in cases with no substitutions in the alignment, or 100% match) was replaced in all cases with 0.
Phylogenetic analysis
The phylogenetic position of R. glabra and R. typhina were assessed using all the protein-coding genes from the complete chloroplast genome sequence of 24 species in the family Anarcadiceae with two species Boswellia sacra and Commiphora gleadensis from the family Burseraceae as outgroups. Each protein-coding gene was aligned using MAFFT version 7 [61, 62] with Translation Align implemented and then the 78 protein-coding genes formed a dataset by Concatenate Sequence or Alignments in Geneious 10.2.4 with default settings. Maximum-likelihood (ML) analysis was run by RAxML program under the GTRGAMMA model with 1000 bootstrap replicates [63], and the best-fit model for each partition was GTR + G. The phylogenetic tree was visualized by Figtree v1.4.2 (http://tree.bio.ed.ac.uk/software/figtree/).
Data availability
The identified Rhus glabra (Voucher nos. Ren_P3002 and Ren_P3051) and R. typhina (Voucher no. Ren_P3053) sequences in this study have been submitted to NCBI, and the accession numbers are OR800752, OR800753 and OR773067, respectively. The SRA numbers are SRR26711334, SRR26712902 and SRR26713523, respectively.
References
Muellner-Riehl AN, Weeks A, Clayton JW, et al. Molecular phylogenetics and molecular clock dating of Sapindales based on plastid rbcL, atpB and trnL-trnF. DNA Sequences Taxon. 2016;65(18):1019–36. https://doi.org/10.12705/655.5
Pell SK. Molecular systematics of the cashew family (Anacardiaceae). La State Univ. 2004. https://doi.org/10.31390/gradschool_dissertations.1472
Pell SK, Mitchell JD, Miller AJ et al. Anacardiaceae. In Kubitzki, K, editor, The families and genera of vascular plants. Flowering plants. Eudicots. Sapindales, Cucurbitales, Myrtaceae. (Springer-Verlag: Berlin). 2011; 10: 7–50.
Miller AJ, Young DA, Wen J. Phylogeny and biogeography of Rhus (Anacardiaceae) based on its sequence data. Int J Plant Sci. 2001;162:1401–7. https://doi.org/10.1086/322948
Lee WK, Kim MJ, Heo K. Phylogeny of Korean Rhus spp. based on ITS and rbcL sequences. J Korean Med Sci. 2004;12:60–66.
Rayne S, Mazza G. Biological activities of extracts from sumac (Rhus spp.): a review. Nat Preceding. 2007;62(4):165–75. https://doi.org/10.1007/s11130-007-0058-4
Saxena G, Mccutcheon AR, Farmer S, et al. Antimicrobial constituents of Rhus glabra. J Ethnopharmacol. 1994;42:95–9. https://doi.org/10.1016/0378-8741(94)90102-3
Ustunda Z. Polarized energy dispersive X-ray fluorescence applications of spice samples. Spectrosc Lett. 2009;42:7–11. https://doi.org/10.1080/00387010802428450
Khalil M, Bazzi A, Zeineddine D, et al. Repressive effect of Rhus coriaria L. fruit extracts on microglial cells-mediated inflammatory and oxidative stress responses. J Ethnopharmacol. 2021;269:113748. https://doi.org/10.1016/j.jep.2020.113748
Yan MF, Li TH, Li XR, et al. Microbial biomass and activity restrict soil function recovery of a post-mining land in eastern Loess Plateau. CATENA. 2021;199:105107. https://doi.org/10.1016/j.catena.2020.105107
Wang S, Zhu F. Chemical composition and biological activity of staghorn sumac (Rhus typhina). Food Chem. 2017;237:431–43. https://doi.org/10.1016/j.foodchem.2017.05.111
Ren ZM, von Dohlen CD, Harris AJ, et al. Congruent phylogenetic relationships of Melaphidina aphids (Aphididae: Eriosomatinae: Fordini) according to nuclear and mitochondrial DNA data with taxonomic implications on generic limits. PLoS ONE. 2019;14(2):e0213181. https://doi.org/10.1371/journal.pone.0213181
Pan ZG, You YT. Growing exotic trees in China. Beijing: Beijing Science and Technology Press; 1994. (in Chinese).
Zhang ZJ, Jiang CD, Zhang JZ, et al. Ecophysiological evaluation of the potential invasiveness of Rhus typhina in its non-native habitats. Tree Physiol. 2009;29(11):1307–16. https://doi.org/10.1093/treephys/tpp065
Young DA. Re-evaluation of the section of Rhus L. subgenus Lobadium (Raf.) T. and G. (Anacardiaceae). Brittonia. 1978;30:411–415. https://doi.org/10.2307/2806344
Barkley FA. A monographic study of Rhus and its immediate allies in North and Central America, including the West Indies. Ann Mo Bot Gard. 1937;24(3):265–498. https://doi.org/10.2307/2394183
Yi TS, Miller AJ, Wen J. Phylogenetic and biogeographic diversification of Rhus (Anacardiaceae) in the Northern Hemisphere. Mol Phylogenet Evol. 2004;33:861–79. https://doi.org/10.1016/j.ympev.2004.07.006
Yi TS, Mille AJ, Wen J. Phylogeny of Rhus (Anacardiaceae) based on sequences of nuclear Nia-i3 intron and chloroplast trnC-trnD. Syst Bot. 2007;32:379–91. https://doi.org/10.1600/036364407781179635
Andres-Hernandez AR, Terrazas T, Salazar G, et al. Phylogenetic analysis based on structural and combined analyses of Rhus s.s. (Anacardiaceae). Bot J Linn Soc. 2014;176:452–68. https://doi.org/10.1111/boj.12222
Li XW, Yang Y, Henry RJ, et al. Plant DNA barcoding: from gene to genome. Biol Rev. 2015;9:157–66. https://doi.org/10.1111/brv.12104
Li XJ, Baskin JM, Baskin CC. Pericarp ontogeny and anatomy in Rhus aromatica Ait. And R-glabra L-(Anacardiaceae). J Torrey Bot Soc. 1999;126:279–88. https://doi.org/10.2307/2997311
Ravi V, Khurana JP, Tyagi AK, et al. An update on chloroplast genomes. Plant Syst Evol. 2008;271:101–22. https://doi.org/10.1007/s00606-007-0608-0
Gualberto JM, Mileshina D, Wallet C, et al. The plant mitochondrial genome: dynamics and maintenance. Biochimie. 2014;100:107–20. https://doi.org/10.1016/j.biochi.2013.09.016
Wang YB, Liu BB, Nie ZL, et al. Major clades and a revised classification of Magnolia and Magnoliaceae based on whole plastid genome sequences via genome skimming. J Syst Evol. 2020;58(5):673–95. https://doi.org/10.1111/jse.12588
Welker CA, McKain MR, Estep MC, et al. Phylogenomics enables biogeographic analysis and a new subtribal classification of the Andropogoneae (Poaceae-Panicoideae). J Syst Evol. 2020;58(6):1003–30. https://doi.org/10.1111/jse.12691
Saina JK, Li ZZ, Gichira AW, et al. The complete chloroplast genome sequence of tree of heaven (Ailanthus altissima (Mill.) (Sapindales: Simaroubaceae), an important Pantropical tree. Int J Mol Sci. 2018;19:929. https://doi.org/10.3390/ijms19040929
Kim Y, Shin J, OH DR, et al. Comparative analysis of complete chloroplast genome sequences and insertion-deletion (Indel) polymorphisms to distinguish five Vaccinium species. Forests. 2020;11:927. https://doi.org/10.3390/f11090927
Xu YJ, Wen J, Su X, et al. Variation among the complete chloroplast genomes of the Sumac species Rhus chinensis: reannotation and comparative analysis. Genes. 2022;13:1936. https://doi.org/10.3390/genes13111936
Lew KA, Manhart JR. The rps12 gene in Spirogyra maxima (Chlorophyta): its evolutionary significance. J Phycol. 1993;29:500–5. https://doi.org/10.1111/j.1529-8817.1993.tb00151.x
Zhu B, Qian F, Hou YF, et al. Complete chloroplast genome features and phylogenetic analysis of Eruca sativa (Brassicaceae). PLoS ONE. 2021;16:e0248556. https://doi.org/10.1371/journal.pone.0248556
Redwan RM, Saidin A, Kumar SV. Complete chloroplast genome sequence of MD-2 pineapple and its comparative analysis among nine other plants from the subclass Commelinidae. BMC Plant Biol. 2015;15:294. https://doi.org/10.1186/s12870-015-0619-x
Wang WB, Yu H, Qiu XP. Analysis of repeat sequence and codon bias of chloroplast genome in Scutellaria baicalensis. Mol Plant Breed. 2018;16:2445–52.
Liu H, Hu HB, Zhang SH, et al. The complete chloroplast genome of the rare species Epimedium tianmenshanensis and comparative analysis with related species. Physiol Mol Biol Plants. 2020;26(10):2075–83.
Chen CH, Zheng YJ, Liu SA, et al. The complete chloroplast genome of Cinnamomum camphora and its comparison with related Lauraceae species. PeerJ. 2017;5:e3820.
An WL, Li J, Yang ZR, et al. Characteristics analysis of the complete Wurfbainia villosa chloroplast genome. Physiol Mol Biol Plants. 2020;26:747–58.
Meng J, Li XP, Li HT, et al. Comparative analysis of the complete chloroplast genomes of four Aconitum medicinal species. Molecules. 2018;23:1015–7.
Kawabe A, Miyashita NT. Patterns of codon usage bias in three dicot and four monocot plant species. Genes Genet Syst. 2003;78:343–52.
Asaf S, Khan AL, Khan MA, et al. Chloroplast genomes of Arabidopsis halleri ssp. gemmifera and Arabidopsis lyrata ssp. petraea: structures and comparative analysis. Sci Rep. 2017;7(1):7556. https://doi.org/10.1038/s41598-017-07891-5
Provan J, Powell W, Hollingsworth PM. Chloroplast microsatellites: new tools for studies in plant ecology and evolution. Trends Ecol Evol. 2001;16:142–7. https://doi.org/10.1016/S0169-5347(00)02097-8
Wicke S, Schneeweiss GM, DePamphilis CW, et al. The evolution of the plastid chromosome in land plants: gene content, gene order, gene function. Plant Mol Biol. 2011;76(3–5):273–97. https://doi.org/10.1007/s11103-011-9762-4
Ruhlman TA, Jansen RK. The plastid genomes of flowering plants. In: Maliga P, editor. Chloroplast biotechnology: methods and protocols—methods in molecular biology. Totowa: Humana Press; 2014. pp. 3–38.
Zhu A, Guo W, Gupta S, et al. Evolutionary dynamics of the plastid inverted repeat: the effects of expansion, contraction, and loss on substitution rates. New Phytol. 2016;209(4):1747–56. https://doi.org/10.1111/nph.13743
Dong W, Liu J, Yu J, et al. Highly variable chloroplast markers for evaluating plant phylogeny at low taxonomic levels and for DNA barcoding. PLoS ONE. 2012;7(4):e35071.
Pang X, Liu C, Shi L, et al. Utility of the trnH–psbA intergenic Spacer Region and its combinations as plant DNA barcodes: a meta-analysis. PLoS ONE. 2012;7(11):e48833. https://doi.org/10.1371/journal.pone.0048833
Hao DC, Huang BL, Chen SL, et al. Evolution of the chloroplast trnL-trnF region in the gymnosperm lineages Taxaceae and Cephalotaxaceae. Biochem Genet. 2009;47(5–6). https://doi.org/10.1007/s10528-009-9233-7. 351– 69.
Zhu S, Liu Q, Qiu S, et al. DNA barcoding: an efficient technology to authenticate plant species of traditional Chinese medicine and recent advances. Chin Med. 2022;17(1):112. https://doi.org/10.1186/s13020-022-00655-y
Ivanova Z, Sablok G, Daskalova E, et al. Chloroplast genome analysis of resurrection tertiary relict Haberlea rhodopensis highlights genes important for desiccation stress response. Front Plant Sci. 2017;8:204. https://doi.org/10.3389/fpls.2017.00204
Weeks A, Zapata F, Pell SK, et al. To move or to evolve: contrasting patterns of intercontinental connectivity and climatic niche evolution in Terebinthaceae (Anacardiaceae and Burseraceae). Front Genet. 2014;5:409. https://doi.org/10.3389/fgene.2014.00409
Hardin JW, Phillips LL. Hybridization in the eastern north American Rhus (Anacardiaceae). Assoc Southeast Biol Bull. 1985;32:99–106.
Burke JM, Hamrick JL. Genetic variation and evidence of hybridization in the genus Rhus (Anacardiaceae). J Hered. 2003;93(1):37–41. https://doi.org/10.1093/jhered/93.1.37
Li L, Peng JY, Bai RX. Effect of different methods on Chinensis jujube DNA extraction from leaves. J Fruit Sci. 2007;24:389–92.
Zimmer EA, Wen J. Using nuclear gene data for plant phylogenetics: progress and prospects II next-gen approaches. J Syst Evol. 2015;53:371–9. https://doi.org/10.1111/jse.12174
Chen SF, Zhou YQ, Chen YR, et al. Fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics. 2018;34(17):i884–90. https://doi.org/10.1093/bioinformatics/bty560
Jin JJ, Yu WB, Yan JB, et al. GetOrganelle: a fast and versatile toolkit for accurate de novo assembly of organelle genomes. Genome Biol. 2020;21:241. https://doi.org/10.1186/s13059-020-02154-5
Qu XJ, Moore MJ, Li DZ, et al. PGA: a software package for rapid, accurate, and flexible batch annotation of plastomes. Plant Methods. 2019;15:50. https://doi.org/10.1186/s13007-019-0435-7
Kearse M, Moir R, Wilson A, et al. Geneious basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics. 2012;28(12):1647–9. https://doi.org/10.1093/bioinformatics/bts199
Kumar S, Stecher G, Tamura K. MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol. 2016;33:1870–4. https://doi.org/10.1093/molbev/msw054
Beier S, Thiel T, Mu¨nch T, et al. MISA-web: a web server for microsatellite prediction. Bioinformatics. 2017;33:2583–5. https://doi.org/10.1093/bioinformatics/btx198
Brudno M, Do CB, Cooper GM, et al. NISC comparative sequencing progra. LAGAN and Multi-LAGAN: efficient tools for large-scale multiple alignment of genomic DNA. Genome research. 2003;13(4):721–731. https://doi.org/10.1101/gr.926603.
Librado P, Rozas J. DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics. 2009;25:1451–2. https://doi.org/10.1093/bioinformatics/btp187
Shigehiro K, Zmasek CM, Osamu N, et al. Aleaves facilitates ondemand exploration of metazoan gene family trees on MAFFT sequence alignment server with enhanced interactivity. Nucleic Acids Res. 2013;41:W22–8. https://doi.org/10.1093/nar/gkt389
Katoh K, Rozewicki J, Yamada KD. Mafft online service: multiple sequence alignment, interactive sequence choice and visualization. Brief Bioinform. 2017;20(4):1160–6. https://doi.org/10.1093/bib/bbx108
Stamatakis A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 2014;30:1312–3. https://doi.org/10.1093/bioinformatics/btu033
Acknowledgements
The authors would like to thank Mr. Joe Boggs and Mr. Karl Hilling, who helped for collecting the samples.
Funding
This work was supported by the National Natural Science Foundation of China (31870366), Shanxi International Science and Technology Cooperation Project (201803D421051), Research Project Supported by Shanxi Scholarship Council of China (2020-018), the National High Technology Research and Development “863” Program (2014AA021802).
Author information
Authors and Affiliations
Contributions
ZR conceived this research and collected plant materials. LH and YX conducted experiments, assembled and annotated the chloroplast genome. LH analyzed the data, prepared figures and tables and wrote the original draft. ZR, VV, SL and JW revised the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
In the current investigation, we collected the three samples of Rhus species in 2015, and the samples with Voucher nos. Ren_P3002 and Ren_P3053 were from the property of Mr. Joe Boggs and Mr. Karl Hilling, who helped us for collecting the samples. The sample with Voucher no. Ren_P3051 was from publicly accessible area, and the leaf collection of this wild species for research purposes does not pose a threat to the local ecology. All the voucher specimens of Rhus accessions were meticulously preserved at the Herbarium of School of Life Science, Shanxi University, China. These specimens were taxonomically identified by Prof. Jun Wen, botanist from Smithsonian Institution, US.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Supplementary Material 1: Fig. S1
Chloroplast genome map of Rhus glabra (Accession No. OR800753)
Supplementary Material 2: Fig. S2
Chloroplast genome map of Rhus typhina (Accession No. OR773067)
Supplementary Material 3: Fig. S3
Sliding-window analysis on the three chloroplast genomes of Rhus typhina. Window length: 800 bp; step size: 200 bp; X-axis: position of the midpoint of a window; Y-axis: nucleotide diversity of each window
Supplementary Material 4: Table S1
Introns and exons of protein coding genes of three Rhus chloroplast genomes
Supplementary Material 5: Table S2
Relative synonymous codon usage (RSCU) of chloroplast genomes of three Rhus individuals
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

{kind=link}
Cite this article
Huang, L., Xu, Y., Valcárcel, V. et al. Three complete chloroplast genomes from two north American Rhus species and phylogenomics of Anacardiaceae. BMC Genom Data 25, 30 (2024). https://doi.org/10.1186/s12863-024-01200-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12863-024-01200-6