
Abstract
Background
Drug response variability observed amongst patients is caused by the interaction of both genetic and non-genetic factors, and frequencies of functional genetic variants are known to vary amongst populations. Pharmacogenomic research has the potential to help with individualized treatments. We have not found any pharmacogenomics information regarding Uygur ethnic group in northwest China. In the present study, we genotyped 85 very important pharmacogenetic (VIP) variants (selected from the PharmGKB database) in the Uygur population and compared our data with other eleven populations from the HapMap data set.
Results
Through statistical analysis, we found that CYP3A5 rs776746, VKORC1 rs9934438, and VKORC1 rs7294 were most different in Uygur compared with most of the eleven populations from the HapMap data set. Compared with East Asia populations, allele A of rs776746 is less frequent and allele A of rs7294 is more frequent in the Uygur population. The analysis of F-statistics (Fst) and population structure shows that the genetic background of Uygur is relatively close to that of MEX.
Conclusions
Our results show significant differences amongst Chinese populations that will help clinicians triage patients for better individualized treatments.
Similar content being viewed by others

Background
Reactions to the same drug differ significantly among individuals. Thus, analyzing a drug’s safety and efficacy is complicated, causing difficulties in finding new treatments for major diseases. Inherited differences in individual drug-metabolizing enzymes are typically monogenic traits, and their influence on the pharmacokinetics and pharmacologic effects of medications are determined by the importance of the polymorphic enzymes for the activation or inactivation of drug substrates [1]. Pharmacogenetics and pharmacogenomics deal with possible associations of a single genetic polymorphism or multiple gene profiles and responses to drugs [2]. The goal of pharmacogenetic research is to provide information for a patient with the right medicine at the right dose for optimal treatment outcomes. The majority of pharmacogenomic studies have focused on candidate genes thought to be involved in the pharmacokinetics or mechanism of drug action [3, 4].
Recent studies have shown that certain genes have close relationships with the outcomes of drug therapy and that different genotypes may determine how the patient responds to a drug. These gene variants are called very important pharmacogenetic (VIP) variants [5], and are listed in the Pharmacogenomics Knowledge Base (PharmGKB: http://www.pharmgkb.org). In total, there are 126 VIP variants that occur in 44 different genes and variously code for cytochrome P450 oxidases, drug targets, drug receptors, and drug transporters.
Individual responses to medications vary significantly among different populations, and great progress in understanding the molecular basis of drug actions has been made in the past 50 years. The field of pharmacogenomics seeks to elucidate inherited differences in drug disposition and effects. While we know that different populations and ethnic groups are genetically heterogeneous, we have not found any pharmacogenomics information regarding minority groups, such as the Uygur ethnic group in northwest China.
The Uygur is an ethnic group primarily located in the Xinjiang Uygur Autonomous Region of China. The Uygur is one of China’s largest ethnic groups, with a long history in the region and distinct culture and traditions. They were originally a nomadic Turkish people in north and northwestern China. The Uygur language is a Turkic language very similar to Turkish.
In this study, we aimed to identify the allele frequencies of VIP variants in the Uygur and to determine the difference in allele frequencies between the Uygur and 11 populations from the HapMap data set. The results of this study will extend our understanding of ethnic diversity and pharmacogenomics, and enable medical professionals to use genomic and molecular data to effectively implement personalized medicine in the future.
Materials and methods
Study participants
We recruited a random sample of unrelated Uygur adults from the Xinjiang Region of China. The subjects selected were judged to be of good health and had exclusively Uygur ancestry for at least the last three generations. Thus, the subjects were thought to be representative samples of the Uygur population with regard to ancestry and environmental exposures. Blood samples were taken according to the study protocol, which was approved by the Clinical Research Ethics of Northwest University, Tibet University for Nationalities, Xinjiang Medical University, and the people’s hospital of Xinjiang Uygur Autonomous Region. Signed informed consent was also obtained from each participant enrolled in the study. Based on the abovementioned inclusion criteria, 96 randomly-selected, healthy, unrelated Uygur individuals were recruited from the Xinjiang Province.
Variant selection and genotyping
We selected genetic variants from published polymorphisms associated with VIP variants from the PharmGKB database. We designed assays for the 85 genetically-variant loci in 37 genes that formed the basis for our our analyses. We excluded loci if we could not design an assay. We extracted genomic DNA from peripheral blood obtained from the subjects using the GoldMag-Mini Whole Blood Genomic DNA Purification Kit (GoldMagLtd. Xi’an, China) according to the manufacturer’s protocol. The DNA concentration was measured with a NanoDrop 2000C spectrophotometer (Thermo Scientific, Waltham, MA, USA). The Sequenom MassARRAY Assay Design 3.0 software (San Diego, CA, USA) was used to design multiplexed single nucleotide polymorphism (SNP) MassEXTEND assays [6]. SNP genotyping analysis was performed using the standard protocol recommended by the manufacturer with a Sequenom MassARRAY RS1000. Sequenom Typer 4.0 software was used to manage and analyze the SNP genotyping data as described in a previous report [7].
HapMap genotype data
The genotype data of individuals from eleven populations were downloaded from the International HapMap Project web site (HapMap_release127) at http://hapmap.ncbi.nlm.nih.gov/biomart/martview/e4f42d4d0acde5ea6c35312381c1e461. The eleven populations included those of (1) African ancestry in Southwest USA (ASW); (2) Utah, USA residents with Northern and Western European ancestry from the CEPH collection (CEU); (3) Han Chinese in Beijing, China (CHB); (4) Chinese in metropolitan Denver, CO, USA (CHD); (5) Gujarati Indians in Houston, Texas, USA (GIH); (6) Japanese in Tokyo, Japan (JPT); (7) Luhya in Webuye, Kenya (LWK); (8) Mexican ancestry in Los Angeles, California, USA (MEX); (9) Maasai in Kinyawa, Kenya (MKK); (10) Toscani in Italy (TSI); and (11) Yoruba in Ibadan, Nigeria (YRI).
Statistical analyses
We used Microsoft Excel and SPSS 17.0 statistical packages (SPSS, Chicago, IL, USA) to perform Hardy–Weinberg Equilibrium (HWE) analysis and the χ 2 test. The validity of the frequency of each VIP variant in the Uygur data was tested by assessing the departure from HWE using an exact test. We calculated and compared the genotype frequencies of the variants in the Uygur data with those in the eleven populations separately using the χ 2 test. All p values obtained in this study were two-sided, and Bonferroni’s adjustment for multiple tests was applied to the level of significance, which was set at p < 0.05/(85*11). The purpose of the χ 2 test was to discover sites with significant differences. Afterwards, we obtained the SNP allele frequencies from the ALleleFREquency Database (http://alfred.med.yale.edu), and analyzed the global patterns of genetic variation at specific loci.
Analysis of population genetic structures
Some studies point out that population genetic structure is central to the study of human origins, DNA forensics, and complex diseases [8]. We believe it is also important for pharmacogenomics. Fst and structure analyses are common in population genetic studies. Because of the insights that F-statistics can provide about the processes of differentiation among populations, over the past 50 years they have become the most widely used descriptive statistics in population and evolutionary genetics [9]. Wright’s F-statistics describe the level of heterozygosity in each level of a hierarchically-subdivided population. More specifically, F-statistics relate the departure from panmixia in the total population and within subpopulations to the total homozygosity. The most commonly reported statistic, Fst, measures the differentiation of a subpopulation relative to the total population, and is directly related to the variance in allele frequency between subpopulations. To further investigate variation at the VIP locus in terms of population structure, we used the model-based clustering method implemented in Structure (http://pritchardlab.stanford.edu/structure.html).
We used the Arlequin ver 3.1 software to calculate the value of Fst to infer the pairwise distance between populations. Pairwise Fst values were calculated on the primary, 84 SNP dataset in Arlequin3.5 [10] using Reynolds’ distance [11] with significance tested using 100 permutations. To further investigate population structure, we used the model-based clustering method implemented in Structure ver. 2.3.1. Fst is directly related to the variance in allele frequency among populations and to the degree of resemblance among individuals within populations. If Fst is small, it means that the allele frequencies within each population are similar; if it is large, it means that the allele frequencies are different.
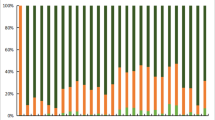
To analyze the genetic structure, the Bayesian clustering algorithm-based program Structure ver. 2.3.1 was used to assign the samples within a hypothetical K number of populations as proposed by Pritchard et al. [12]. Analyses were performed using the ancestry model with correlated allele frequencies in eleven independent runs from K = 2 to K = 7. The MCMC analyses for each structure analysis (from K = 2 to K = 7) was run for 10,000 steps after an initial burn-in period of 10,000 steps. To assess the most likely number of clusters, we calculated △K following Evanno et al. [13]. When the software ran to completion and results were obtained, we constructed bar charts summarizing the results using drawing software.
Results
Basic information about the selected VIP loci in Uygur is listed in Table 1. The 85 VIP loci relate to 37 genes that belong to the cytochrome P450 superfamily, the nuclear receptor family, the G-protein coupled receptor family, the alcohol dehydrogenase family, the adrenergic receptors family, the ATP-binding cassette (ABC) transporters superfamily, and the eag family.
Using the χ 2 test with the Bonferroni correction for multiple hypotheses and multiple comparisons, we found 0, 1, 3, 5, 7, 9, 10, 13, 16, 17, and 25 different loci in the frequency distributions when the Uygur population was compared to the TSI, MEX, GIH, CHD, CEU, CHB, ASW, JPT, MKK, LWK, and YRI populations, respectively. Three loci (rs776746, rs9934438, and rs7294) located in the CYP3A5 and VKORC1 genes were different in the Uygur population when compared with most of the populations (Tables 2 and 3).
For a global analysis, we combined our new data with previously published data, for a total of 66 population samples at rs776746 and rs7294. From Table 4 it can clearly be seen that the frequencies of the A allele of rs776746 were higher in Africa than in Asia and East Asia, but lower in Europe. For the East Asia data, frequencies ranged from 5 % to 50 %, and the frequencies were high in the She and Tujia population and lower in the Uygur and Tu populations. The frequencies of the A allele of rs7294 in East Asia ranged from 1 % to 35 %, and the frequency in the Uygur population was higher than in the other populations from East Asia.
Pairwise Fst values were calculated for all population comparisons across loci. As shown in Table 5, we found that pairwise Fst values for comparisons of the Uygur population with the other 11 populations ranged from 0.49686 to 0.581. Fst is directly related to the variance in allele frequency among populations and to the degree of resemblance among individuals within populations. If Fst is small, it means that the allele frequencies within each population are similar; if it is large, it means that the allele frequencies are different. The value of Fst for the Uygur and MEX populations was the smallest. We therefore conclude that the allele frequencies of the Uygur and MEX are similar. We speculate that the genetic backgrounds of the Uygur and MEX populations are similar.
We used a model-based clustering approach, as implemented in Structure, to infer population structure among the 12 populations. Different values ranging from 2 to 7 were assumed for K in Structure calculations. K = 3, 4, 5 were selected, based on the Estimated Ln Prob of Data and other recommendations of the Structure software manual. As shown in Fig. 1, when the K value was equal to 3, individuals were independently assigned to three affinity groups (subpopulations 1: Uygur, CEU, GIH, MEX, TSI; subpopulations 2: ASW, LWK, MKK, YRI; subpopulations 3: CHB, CHD, JPT) using the relative majority of likelihood to assign individuals to subpopulations. We tested additional values of K and obtained results suggesting that the genetic backgrounds of the Uygur and MEX populations are simila.

Bayesian clustering of genotypic samples from 12 populations. Each vertical bar denotes an individual, whilst colors denote inferred clusters. Note that colors are not universal between k = 3 and 5
Discussion
The genotype frequencies of VIP variants differs among human populations. In this study, we genotyped the variants related to drug response in the Uygur ethnic group and compared the genotype frequencies with those in eleven populations. From the χ 2 test, we found clear evidence that the allele characteristics of the CYP3A5 rs776746 and VKORC1 (rs9934438 and rs7294) variants in the Uygur population are quite different from that in other ethnic groups. We also found that the genetic backgrounds of the Uygur and MEX populations are similar, via Fst calculations and analysis of population structure.
CYP3A5, localized on chromosome 7q21-q22.1, encodes one of the CYP3A subfamily of enzymes [14]. The most common nonfunctional variant of CYP3A5 is designated as CYP3A5*3. CYP3A5*3 status is determined by the derived allele at rs776746, a change from A to G located in intron 3. This change creates a cryptic splice site that results in altered mRNA splicing, which may alter the reading frame and result in a premature termination codon and hence a nonfunctional protein [14, 15].
Individuals with CYP3A5*1/*1 and *1/*3 expresser genotypes metabolize some CYP3A substrates more rapidly than CYP3A5*3/*3 nonexpressers. One such substrate is tacrolimus, which is used to prevent post-transplantation organ rejection. CYP3A5*1 carriers have a higher rate of tacrolimus clearance than those with the other genotypes, with *1/*1 individuals having a higher clearance than *1/*3 individuals, who have higher clearance than *3/*3 individuals [16]. In ideal situations, the target tacrolimus concentration must be high enough to prevent transplant rejection [17, 18], but low enough to minimize toxicity [19]. Tacrolimus trough concentrations are routinely monitored after transplantation, and the dose is appropriately adjusted.
Carbamazepine (CBZ), a first-line antiepileptic drug, has been widely prescribed for the treatment of partial and generalized tonic-clonic seizures. It has been reported that CYP3A5*3 is associated with CBZ pharmacokinetics in Japanese [20], Korean [21], and Chinese [22] epileptic patients, and that CYP3A5 expressers are more likely to require higher CBZ maintenance doses than nonexpressers (GA + AA vs. GG). The CYP3A5 genotype may also have dose-dependent effects on ABT-773 plasma levels. CYP3A5 expressers have a higher rate of ifosfamide N-demethylation in the liver and kidney and of cyclosporine A metabolism in the kidney [15].
CYP3A5*3 is the most frequent and well-studied variant allele of CYP3A5. Its frequency varies widely across human populations. In white populations, the estimated allele G frequency of CYP3A5*3 is 0.82–0.95, in African American is 0.33, in Japanese is 0.85, in Chinese is 0.65, in Mexicans is 0.75, in Pacific Islanders is 0.65, and in Southwest American Indians is 0.4 [15]. In our study, the frequency of allele G is higher than in other population from China. This suggests that ancestry should be considered when determining dosages for different patients.
The VKORC1 (vitamin K epoxide reductase complex, subunit 1) gene, which encodes vitamin K epoxide reductase complex subunit 1, located on chromosome 16, includes three exons [23]. The 1173C > T (rs9934438) transition in intron 1 and the 3730G > A (rs7294) transition in the 3ʹ untranslated region (UTR), are two common polymorphisms [24].
Several authors have shown that acenocoumarol dose is also influenced by VKORC1 genotype. Reitsma et al. showed in 2005 that Dutch patients carrying one or two variant alleles for the 1173 polymorphism required a 28 % and 47 % lower dose, respectively, when compared with wild types [25]. In Greek acenocoumarol users, heterozygous carriers of a variant allele required a 19 % lower dose and homozygous carriers a 63 % lower dose [26]. Similar percentages were found in a German and Austrian population (25 % and 52 %) [27], in a Serbian population (27 % and 62 %) [28], and amongst Lebanese acenocoumarol users (34 % and 50 %) [29]. Reitsma et al. also investigated the influence of VKORC1 polymorphism on phenprocoumon dose requirements. Patients with a CT genotype at position 1173 had a 10 % lower dose and patients with a TT genotype a 52 % lower dose than wild types (CC) [25]. This effect was also seen in several German and Austrian studies. The dose in phenprocoumon users with one variant VKORC1 allele was 19–31 % lower than in wild type users, and 43–51 % lower in users with two variant alleles [27].
Warfarin is a commonly prescribed oral anticoagulant, used to prevent thromboembolic diseases in patients with deep vein thrombosis, atrial fibrillation, recurrent stroke, or heart valve prosthesis [30]. Some studies have suggested that carriers of the 1173TT genotype require a dose of warfarin significantly lower than that of carriers with the CC or CT genotypes [24]. On the other hand, the 3730G > A polymorphism was associated with differences in the average dose of warfarin prescribed, with patients carrying the GG genotype being prescribed a significantly lower average daily dose of warfarin [24, 31].
In summary, VKORC1 polymorphisms can significantly alter warfarin pharmacodynamics and maintenance dose requirements. Patients with the 1173T (rs9934438) allele require a lower warfarin dose compared with 35 mg/week for the wild-type carriers [32]. Patients with 3730A (rs7294) need a higher warfarin dose [32, 33]. In our study, the frequency of carriers of the allele T of rs9934438 and allele G of rs7294 are lower than in other Asian populations, and higher than in European and YRI populations, which suggests that the optimal dosage of warfarin should be decided based on the specific genotype in individual Uygur patients.
Conclusion
The genotype frequencies of VIP variants affect a populations’ response to drugs to a great extent. Determination of the genotype distribution and frequencies of VIP variants in a population is necessary to provide a theoretical basis for safer drug administration and an improved curative effect. Our results complement the currently available data on the Uygur ethnic group in the pharmacogenomics database, and furthermore, provide a basis for safer and more effective drug administration in the Uygur. However, our sample size of Uygur is relatively small, and further investigation in a larger cohort of Uygur is necessary to ascertain the generalizability and extrapolation of our results to these and other conditions in the Uygur population.
Abbreviations
- VIP variants:
-
very important pharmacogenetic variants
- SNP:
-
single nucleotide polymorphism
- ASW:
-
African ancestry in Southwest USA
- CEU:
-
Utah residents with Northern and Western European ancestry from the CEPH collection
- CHB:
-
Han Chinese in Beijing, China
- CHD:
-
Chinese in Metropolitan Denver, Colorado
- GIH:
-
Gujarati Indians in Houston, Texas
- JPT:
-
Japanese in Tokyo, Japan
- LWK:
-
Luhya in Webuye, Kenya
- MEX:
-
Mexican ancestry in Los Angeles, California
- MKK:
-
Maasai in Kinyawa, Kenya
- TSI:
-
Toscans in Italy
- YRI:
-
Yoruba in Ibadan, Nigeria (West Africa)
References
Evans WE, Johnson JA. Pharmacogenomics: the inherited basis for interindividual differences in drug response. Annu Rev Genomics Hum Genet. 2001;2:9–39.
Szekanecz Z, Mesko B, Poliska S, Vancsa A, Szamosi S, Vegh E, et al. Pharmacogenetics and pharmacogenomics in rheumatology. Immunol Res. 2013;56(2–3):325–33.
Evans WE, McLeod HL. Pharmacogenomics–drug disposition, drug targets, and side effects. N Engl J Med. 2003;348(6):538–49.
Peet NP, Bey P. Pharmacogenomics: challenges and opportunities. Drug Discov Today. 2001;6(10):495–8.
Peters EJ, McLeod HL. Ability of whole-genome SNP arrays to capture ‘must have’ pharmacogenomic variants. Pharmacogenomics. 2008;9(11):1573–7.
Gabriel S, Ziaugra L, Tabbaa D. SNP genotyping using the Sequenom MassARRAY iPLEX platform. Curr Protoc Hum Genet. 2009:2.12. 11–12.12. 16. doi:10.1002/0471142905.hg0212s60
Thomas RK, Baker AC, Debiasi RM, Winckler W, Laframboise T, Lin WM, Wang M, Feng W, Zander T, MacConaill L et al. High-throughput oncogene mutation profiling in human cancer. Nat Genet. 2007;39(3):347–51.
Elhaik E. Empirical distributions of FST from large-scale human polymorphism data. PLoS One. 2012;7(11), e49837.
Holsinger KE, Weir BS. Genetics in geographically structured populations: defining, estimating and interpreting FST. Nat Rev Genet. 2009;10(9):639–50.
Excoffier L, Laval G, Schneider S. Arlequin (version 3.0): an integrated software package for population genetics data analysis. Evol Bioinformatics Online. 2005;1:47.
Reynolds J, Weir B, Cockerham CC. Estimation of the coancestry coefficient: basis for a short-term genetic distance. Genetics. 1983;105(3):767–79.
Pritchard JK, Stephens M, Donnelly P. Inference of population structure using multilocus genotype data. Genetics. 2000;155(2):945–59.
Evanno G, Regnaut S, Goudet J. Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol. 2005;14(8):2611–20.
Suarez-Kurtz G, Vargens DD, Santoro AB, Hutz MH, de Moraes ME, Pena SD, Ribeiro-dos-Santos Â, Romano-Silva MA, Struchiner CJ. Global Pharmacogenomics: Distribution of CYP3A5 Polymorphisms and Phenotypes in the Brazilian Population. PLoS One. 2014;9(1), e83472.
Lamba J, Hebert JM, Schuetz EG, Klein TE, Altman RB. PharmGKB summary: very important pharmacogene information for CYP3A5. Pharmacogenet Genomics. 2012;22(7):555.
Passey C, Birnbaum AK, Brundage RC, Oetting WS, Israni AK, Jacobson PA: Dosing equation for tacrolimus using genetic variants and clinical factors. Br J Clin Pharmacol. 2011;72(6):948–57.
Borobia AM, Romero I, Jimenez C, Gil F, Ramirez E, De Gracia R, Escuin F, Gonzalez E, Sansuán AJC: Trough tacrolimus concentrations in the first week after kidney transplantation are related to acute rejection. Ther Drug Monit. 2009;31(4):436–42.
O’Seaghdha C, McQuillan R, Moran A, Lavin P, Dorman A, O’Kelly P, Mohan D, Little P, Hickey D, Conlon P: Higher tacrolimus trough levels on days 2–5 post‐renal transplant are associated with reduced rates of acute rejection. Clin Transpl. 2009;23(4):462–8.
Laskow DA, Vincenti F, Neylan JF, Mendez R, Matas AJ. AN OPEN-LABEL, CONCENTRATION-RANGING TRIAL OF FK506 IN PRIMARY KIDNEY TRANSPLANTATION: A Report Of The United States Multicenter FK506 Kidney Transplant Group1. Transplantation. 1996;62(7):900–5.
Seo T, Nakada N, Ueda N, Hagiwara T, Hashimoto N, Nakagawa K, Ishitsu T: Effect of CYP3A5*3 on carbamazepine pharmacokinetics in Japanese patients with epilepsy. Clin Pharmacol Ther. 2006;79(5):509–10.
Park PW, Seo Y, Ahn J, Kim KA, Park JY. Effect of CYP3A5* 3 genotype on serum carbamazepine concentrations at steady‐state in Korean epileptic patients. J Clin Pharm Ther. 2009;34(5):569–74.
Zhu X, Yun W, Sun X, Qiu F, Zhao L, Guo Y. Effects of major transporter and metabolizing enzyme gene polymorphisms on carbamazepine metabolism in Chinese patients with epilepsy. Pharmacogenomics. 2014;15(15):1867–79.
Rost S, Fregin A, Ivaskevicius V, Conzelmann E, Hortnagel K, Pelz HJ, Lappegard K, Seifried E, Scharrer I, Tuddenham EG et al. Mutations in VKORC1 cause warfarin resistance and multiple coagulation factor deficiency type 2. Nature. 2004;427(6974):537–41.
D'Andrea G, D'Ambrosio RL, Di Perna P, Chetta M, Santacroce R, Brancaccio V, Grandone E, Margaglione M: A polymorphism in the VKORC1 gene is associated with an interindividual variability in the dose-anticoagulant effect of warfarin. Blood. 2005;105(2):645–9.
Reitsma PH, Van Der Heijden JF, Groot AP, Rosendaal FR, Büller HR. A C1173T dimorphism in the VKORC1 gene determines coumarin sensitivity and bleeding risk. PLoS Med. 2005;2(10), e312.
Markatos CN, Grouzi E, Politou M, Gialeraki A, Merkouri E, Panagou I, Spiliotopoulou I, Travlou A: VKORC1 and CYP2C9 allelic variants influence acenocoumarol dose requirements in Greek patients. Pharmacogenomics. 2008;9(11):1631–8.
Cadamuro J, Dieplinger B, Felder T, Kedenko I, Mueller T, Haltmayer M, Patsch W, Oberkofler H: Genetic determinants of acenocoumarol and phenprocoumon maintenance dose requirements. Eur J Clin Pharmacol. 2010;66(3):253–60.
Kovac MK, Maslac AR, Rakicevic LB, Radojkovic DP. The c.-1639G> A polymorphism of the VKORC1 gene in Serbian population: retrospective study of the variability in response to oral anticoagulant therapy. Blood Coagul Fibrinolysis. 2010;21(6):558–63.
Esmerian MO, Mitri Z, Habbal MZ, Geryess E, Zaatari G, Alam S, Skouri HN, Mahfouz RA, Taher A, Zgheib NK: Influence of CYP2C9 and VKORC1 polymorphisms on warfarin and acenocoumarol in a sample of Lebanese people. J Clin Pharmacol. 2011;51(10):1418–28.
Rettie AE, Tai G. The pharmocogenomics of warfarin: closing in on personalized medicine. Mol Interv. 2006;6(4):223–7.
Limdi NA, Wadelius M, Cavallari L, Eriksson N, Crawford DC, Lee MT, Chen CH, Motsinger-Reif A, Sagreiya H, Liu N et al. Warfarin pharmacogenetics: a single VKORC1 polymorphism is predictive of dose across 3 racial groups. Blood. 2010;115(18):3827–34.
Wadelius M, Chen LY, Downes K, Ghori J, Hunt S, Eriksson N, Wallerman O, Melhus H, Wadelius C, Bentley D et al. Common VKORC1 and GGCX polymorphisms associated with warfarin dose. Pharmacogenomics J. 2005;5(4):262–70.
Herman D, Peternel P, Stegnar M, Breskvar K, Dolzan V. The influence of sequence variations in factor VII, gamma-glutamyl carboxylase and vitamin K epoxide reductase complex genes on warfarin dose requirement. Thromb Haemostasis-Stuttg. 2006;95(5):782.
Acknowledgments
This work is supported by The Social Science Foundation of Chinese Ministry of Education (No. 12YJA850011), Natural Science Foundation of Xizang (Tibet) Autonomous Region (20152R-13-11), and Major Training Program of Tibet University for Nationalities (No. 13myZP06). We would also like to thank BioScience Writers for the assistance in the preparation of this manuscript.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Competing interests
The authors declare that they have no competing interests, and the manuscript is approved by all authors for publication.
Authors’ contributions
LW and AA drafted the manuscript. LW, AY, and SD performed the statistical analyses and AY helped edit the manuscript. BY and YZ performed the genotyping. TG and AA reviewed all the genotypes. TJ assisted in the study design. TJ and JY conceived the study and co-supervised the work. All the authors have read and approved the final manuscript.
Li Wang and Ainiwaer Aikemu joint first authors.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Wang, L., Aikemu, A., Yibulayin, A. et al. Genetic polymorphisms of pharmacogenomic VIP variants in the Uygur population from northwestern China. BMC Genet 16, 66 (2015). https://doi.org/10.1186/s12863-015-0232-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12863-015-0232-x