
Abstract
Background
Within a population, the differences of pharmacogenomic variant frequencies may produce diversities in drug efficacy, safety, and the risk associated with adverse drug reactions. With the development of pharmacogenomics, widespread genetic research on drug metabolism has been conducted on major populations, but less is known about minorities.
Results
In this study, we recruited 100 unrelated, healthy Mongol adults from Xinjiang and genotyped 85 VIP variants from the PharmGKB database. We compared our data with eleven populations listed in 1000 genomes project and HapMap database. We used χ2 tests to identify significantly different loci between these populations. We downloaded SNP allele frequencies from the ALlele FREquency Database to observe the global genetic variation distribution for these specific loci. And then we used Structure software to perform the genetic structure analysis of 12 populations.
Conclusions
Our results demonstrated that different polymorphic allele frequencies exist between different nationalities,and indicated Mongol is most similar to Chinese populations, followed by JPT. This information on the Mongol population complements the existing pharmacogenomic data and provides a theoretical basis for screening and therapy in the different ethnic groups within Xinjiang.
Similar content being viewed by others

Background
It is well known that different individuals have different reactions to the same medications. Pharmacogenomics seeks to identify genetic markers that may influence a person’s response to pharmaceuticals. It will undoubtedly become an indispensable part of medical care in the future [1, 2]. Pharmacogenomic research seeks to identify single nucleotide polymorphisms (SNPs) or multiple gene signatures that are possibly associated with medication responses [3]. The goal of the research is to provide information for personalized medicine, i.e. give to the patient the optimal medication in optimal dose, and promote personalized therapeutics [4–6].
Numerous studies had shown that certain important genes and genetic variations affect critical functions during the drug reaction process. These genetic variations are called very important pharmacogenetic (VIP) variants and listed in the pharmacogenomics databases such as the Pharmacogenomics Knowledge Base (PharmGKB), the Pharmacogenetics of Membrane Transporters (PMT) database, and PharmaADME [6–8]. Currently, PharmGKB (http://www.pharmgkb.org) is the most comprehensive database and dedicates to propagating primary pharmacogenomic data and knowledge. They have extensively annotated the vital drug response genes and presented this information in VIP summaries, pathway diagrams, and curated literature [9].
In China, there are 56 different nationalities. Besides Han, the others account for approximately 100 million people. Due to the different genetic backgrounds and diverse environments of these minor populations, we distinguish them easily from the Han ethnicity. The Mongolian population represents one of the fifteen largest ethnic minorities in China [10]. They primarily live in the Inner Mongolia, Liaoning, Heilongjiang and the Xinjiang Uygur Autonomous Region. The areas are located in the grassland region of Northern China and significantly different with the Central Plains. Special living environments of the Mongol people shaped their unique gene distribution frequencies. An increasing number of studies suggest that genes related to drug response vary between different populations [11], so the pharmacogenomics population genetic studies of different population is valuable.
In this study, we random selected and genotyped 85 VIP variants from the PharmGKB VIP database in 100 Mongols from Xinjiang. We designed primers using MassARRAY Assay Design 3.0 Software [12]. We compared the Mongol’s allele frequencies with 11 populations from 1000 genomes project and the Mongol’s genotype frequencies and haplotype construction with 11 HapMap populations to identify the differences among them. The results will expand the current Mongol pharmacogenomic information and ethnic diversity. We aimed to provide new strategies for medical professionals through use genomic and molecular data to optimize drug administration and therapeutic treatment in the future.
Methods
Ethics statement
Blood samples and signed informed consent forms were obtained from all enrolls. All participants were informed both verbally and in writing of the procedures and purpose of the study, and signed informed consent documents. The clinical protocol was approved by the Clinical Research Ethics of Xizang Minzu University and Northwest University, and it is in compliance with Department of Health and Human Services (DHHS) regulations for human research subject protection.
Study participants
We recruited 100 random unrelated Mongol adults (50 males and 50 females, average age range 25-40 years) from the Xinjiang Region of China and collected blood samples. The detailed recruitment criteria are the sample have good health body and had at least three generations of exclusive ethnic ancestries. They rarely communicate with other ethnics in Xinjiang because they are still nomads which living on relatively limited pasture. They were determined to be a representative Mongol population sample with regard to both ancestry and environmental exposure.
Variant selection and genotyping
Using the PharmGKB database, we screened published genetic polymorphisms associated with VIP variants, and finally 85 genetic variant loci from 37 genes were randomly selected for our investigation. We extracted genomic DNA from whole blood using a GoldMag-Mini Whole Blood Genomic DNA Purification Kit (GoldMag Ltd. Xi’an, China) according to the manufacturer’s protocol. The genomic DNA concentration was measured by absorbance at 260 nm using a NanoDrop 2000C (Thermo Scientific, Waltham, Massachusetts, USA). We used the Sequenom MassARRAY Assay Design 3.0 software (San Diego, California, USA) to design multiplexed SNP MassEXTEND arrays [12]. We utilized a Sequenom MassARRAY RS1000 (San Diego, California, USA) to genotype the SNPs according to the manufacturer’s instructions. Sequenom Typer 4.0 Software was used for data collection and analysis as described previously [13].
Statistical analyses
We used Microsoft Excel and the SPSS 19.0 statistical package (SPSS, Chicago, IL) to perform a Hardy–Weinberg Equilibrium (HWE) analysis and χ2 tests. All p values calculated were two-sided and Bonferroni’s multiple adjustment was used to correction. The values were considered statistically significant when p < 0.05 and p < 0.05/(85 × 11), respectively [14]. We analyzed each variant frequency in Mongols using an exact test to identify those that departed from HWE. We downloaded the allele frequencies of 85 loci in eleven randomly population of 1000 genomes project, which are a population of African ancestry in the southwestern USA (ASW); a population of Chinese Dai in Xishuangbanna, China (CDX); a Utah residents population (CEPH) with North and Western European Ancestry (CEU); the Chinese Han in Beijing, China (CHB); the Gujarati Indians in Houston, Texas, USA (GIH); the Japanese population in Tokyo, Japan (JPT); the Luhya people in Webuye, Kenya (LWK); people of Mexican ancestry from Los Angeles, USA (MXL); a population of Puerto Ricans from Puerto Rico (PUR); the Tuscan people of Italy (TSI); and the Yoruba in Ibadan, Nigeria (YRI). We downloaded the genotype frequencies of 85 variation loci in eleven populations from the HapMap database that are ASW; a northwestern European population (CEU); CHB; a Chinese population of metropolitan Denver, Colorado, USA (CHD); GIH; JPT; LWK; people of Mexican ancestry living in Los Angeles, California, USA (MEX); the Maasai people in Kinyawa, Kenya (MKK); TSI; and YRI. We first compared the allele frequencies difference between Mongolian and 11 random 1000 genomes project popualtions and calculate the correlation coefficient (R2) among the minor different population, then compared and calculated the selected SNP’s variant frequencies between the Mongol people and eleven HapMap populations (data from the second phase of HapMap: http://hapmap.ncbi.nlm.nih.gov) using a χ2 test. Afterwards, we downloaded the SNP allele frequencies of selected loci from the ALlele FREquency Database (http://alfred.med.yale.edu, ALFRED) and analyzed the global genetic variation patterns. We used Haploview software package (4.2) to perform the linkage disequilibrium (LD) analysis constructed haplotype, and genetic association of significant polymorphism loci.
Analysis of population genetic structures
There are studies proved that the center of study which research human origins, DNA forensics and complex diseases is population genetic structure. It is also important to our study as a pharmacogenomics population study. Structure analysis is common in population genetic study. To further investigate variation at the VIP locus in terms of population structure we used STRUCTURE ver. 2.3.1 (Pritchard Lab, Stanford University,USA, http://pritchardlab.stanford.edu/structure.html) which based on the Bayesian clustering algorithmto assign the samples within a hypothetical K number of populations hypothesized by Pritchard et al [15]. We performed structure analysis using ancestry model with correlated allele frequencies among clusters. K = 2 to 8 is the range of possible numbers of clusters and 12 trials were run for each K. We performed the MCMC analyses for each structure analysis was run for 10,000 after an initial burn-in period of 10,000 for data collection. △K was calculated to identified the most likely number of clusters by STRUCTURE HARVESTER [16].
Results
We sequenced 85 VIP variants from 100 Mongols. The selected SNP PCR primers were designed using the Sequenom MassARRAY Assay Design 3.0 Software. Information regarding the selected VIP loci and their genotype frequencies is listed in Table 1, including the genes, their positions, the nucleotide change, the amino acid translation, the calculated allele frequencies, and the genotype frequencies for Mongols. Several variants, such as rs698, rs1695, rs5219, rs16974, rs20417, rs890293, rs2740574, and rs3211371, did not meet HWE with a 5 % significance level and were not included in the final 85 loci analyzed. We first compared the allele frequencies differences among the Mongols and the selected 11 groups from 1000 genomes project database (p < 0.05). We found that there are some loci have significantly different between them. In ASW population, there are 22 loci exist different with Mongol. The results of other groups are as follows: CDX, 14; CEU, 19; CHB,15; GIH,15; JPT,15; LWK,18; MXL,18; PUR, 22; TSI, 18; YRI, 18(Table 2), respectively. In Fig. 1, we selected CDX, CHB and JPT which are the minimum difference population compared with Mongol population to calculate the correlation coefficient, R2. From the allele frequencies difference comparison, we figure out one initial conclusion that the Mongolian is relatively close to CDX, followed by CHB and JPT.

Pairwise comparisons of difference in correlation coefficient value R2
We used χ2 analyses to compare differences in the variants’ genotype frequency distributions among the Mongols and eleven HapMap populations (without adjustment, p < 0.05; adjustment, p < 0.05/85 × 11). There were a number of loci had significantly different distribution frequencies among Mongols and the 11 HapMap populations that listed in Table 3 Without adjustment the ASW population had 20 different loci; CEU, 27; CHB, 19; CHD, 10; GIH, 24; JPT, 17; LWK, 24; MEX, 17; MKK, 22; TSI, 14; and YRI, 38. Once the data underwent multiple comparison adjustment, the numbers of significantly different loci variants were revised to 13, 13, 6, 1, 8, 5, 18, 3, 18, 6 and 26, respectively.
When p < 0.05, rs1540339 locus (46489G > A) which located in an intron region of VDR (1, 25- dihydroxyvitamin D3 receptor), showed the greatest number of significant differences between Mongol and 11 HapMap populations; the SNP rs776746 (12083G > A) is a SNP of CYP3A5 which located in an intron region and a significant locus that observed in these populations except TSI. After Bonferroni’s multiple adjustment (p < 0.05/(85 × 11)), the number of HapMap populations with a significantly different rs1540339 locus changed very large which included CEU, CHB, CHD, JPT, MEX and TRI. The rs776746 locus also changed very large which except TSI added CEU, CHD, GIH, JPT and MEX.
Of the 85 variants analyzed, 74 could be classified as part of a superfamily. When the gene superfamily categories were tallied, the number of the associated variants with significantly different frequencies between the Mongols and the eleven HapMap populations were as follows: ASW, 10; CEU, 9; CHB, 5; CHD, 1; GIH, 5; JPT, 4; LWK, 14; MEX, 1; MKK, 14; TSI, 4; and YRI, 21 (Table 4). A number of distinct loci were significantly different and included several pharmacogenomic superfamilies such as the nuclear receptor family, the sodium channel gene family, and the methylenetetrahydrofolate reductase family.
To further verify the ubiquitous differences between different groups through research the difference of maximum and minimum of two SNPs, we selected two variants, the most significantly different variants -- rs1540339, rs1801131 which is one of the least significantly loci distributed in all 12 populations, and downloaded the population data from the ALFRED database. Combining the new data, we carried out a global analysis. Figure 2 shows the global frequency data of rs1801131 and Fig. 3, the rs1540339 data. From the two figures, we only found that the frequency of Mongol is relatively close to the populations distributed in East Asia.

The global frequency distribution of rs1801131. NA, North America; SA, South America

Rs1540339 frequencies in various global populations. EAsia, East Asia; NA, North America; SA, South America
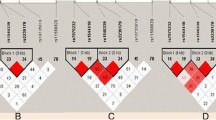
Meanwhile, we focused on rs1540339 to explore the difference of the haplotypes. We performed the LD analysis to define blocks and haplotypes of VDR gene which include rs1540339, rs7975232, rs1544410, rs2239179, rs10735810 and rs11568820 by Haploview. The six SNPs selected from our lists and all of them have the HapMap data. Figure 4 shown that Mongol and CHB has only one block which consisted by rs1540339 and rs2239179, others has obviously different blocks compared with Mongol.

Linkage disequilibrium analysis of the VDR in each of the twelve populations. LD is displayed by standard color schemes with bright red for very strong LD (LOD > 2, D ′ =1), pink red (LOD > 2, D ′ <1), blue (LOD < 2, D ′ = 1) for intermediate LD, and white (LOD < 2, D′ <1) for no LD
For further clarified the genetic structure of Mongol and different populations, we used Structure 2.3.1 performed the population genetic structure comparisons by which works well for 85 loci (K = 2–8). The results are indicated by K = 3–5 (Fig. 5), which based on the Estimated Ln Prob of Data and other recommendations of the STRUCTURE software manual, When k = 3, individuals were divided in three affinity groups (subgroups 1: Mongol, CHD, JPT, CHB; subgroup 2: MEX, TSI, GIH, CEU; subgroup 3: MKK, ASW, LWK, YRI.) which used relative majority of likelihood assignment of individuals to subgroup. Followed by more K value to run STRUCTURE and then displayed the results in bar plots. From the image when k = 4 and 5, we easily found Mongol is closest to CHD, followed by CHB, JPT, and existed significant genetic structure differences with GIH and MEX.

Structure analysis of the genetic relationship between 12 populations. K is the possible numbers of parental population clusters. One color represents one parental population cluster. Each individual is represented by a vertical column partitioned into different color segments. Most suitable K was observed at K = 5, where the proportion of each ancestral component in a single individual is represented by a vertical bar divided into 5 colors
Discussion
Personalized or stratified healthcare is an important goal for medicine in the 21st century. It ensures that the treatments of patients are safe and efficacious [17]. With the rapid development of pharmacogenetics, serious attention has been paid to interethnic or interracial differences in drug responses with the intent to identify the genetic backgrounds of these variations [18]. Our study analyzed the distribution of these VIP variant allele and genotype frequencies to seek out which are altered among the different human populations [19], and found that even the SNP of smallest difference also had significant diversity between different groups. Through the comprehensive analysis, we revealed that Mongol and Chinese populations have the minimum difference.
Two of the variants were identified, rs1801133 (C677T) and rs1801131 (A1298C), included one of the least significant locus in our data, they are located in the same gene -- methylenetetrahydrofolate reductase (MTHFR) gene. MTHFR is located on chromosome 1p36.3 in human which is an important regulatory enzyme that involved in the folate pathway. It catalyzes the conversion of 5,10-methylenetetrahydrofolate to 5-methyltetrahydrofolate [20, 21]. Thymidylate synthesis required a lower 5,10-methylenetetrahydrofolate levels which leading to misincorporation of uracil into DNA, increasing chromosome damage frequency. A lower levels of 5-methyltetrahydrofolate may decrease the methylation process of homocysteine to methionine which could lead to hyperhomocysteinemia and DNA hypomethylation. Severe MTHFR enzyme deficiency is the most common inherited folate metabolism disorder which leads to hyperhomocysteinemia and homocystinuria that eventually destroy the central nervous system and vascular system [22]. Several studies revealed that the C677T and A1298C mutations reduce MTHFR enzyme activity [20–25]. In Caucasians, the C677T of TT and CT carriers had 70 % and 35 % reduced MTHFR enzyme activity, respectively, compared to CC carriers [26]. In Mongolians, CT and TT carriers had a frequency about 0.39 and 0.09. We should pay more attention on capecitabine, cisplatin, pemetrexed, cyanocobalamin and related agents in the Mongolian. Research of this mutation in other populations had not been performed. The enzyme activity reduction extent of different A1298C carriers had not been researched, but the study would play a large role in clinical treatment when one medication cure different patient who carriers different A1298C genotype.
We randomly selected one of the middle significantly different variants in Mongols -- the non-synonymous SNP rs1805124 (A1673G-H558R), which is located in exon 12 of SCN5A [27]. SCN5A encodes the integral membrane protein, voltage-dependent sodium channel α-subunit. It primarily traffics sodium in human heart muscle cells [28, 29]. SCN5A can cause fast depolarization during the upstroke phase of cardiac action potentials, that is the reason as a molecular antiarrhythmic drug target [30]. Amounts of Studies reveals SCN5A is associated with various cardiac diseases including long-QT syndrome (LQTS), Brugada syndrome (Brs), progressive cardiac conduction defect, atrial fibrillation (AF), dilated cardiomyopathy, and overlapping syndromes [27–31]. SCN5A-H558R has been shown to generate moderate electrophysiological functions that can regulate the phenotypic expression of cardiac conduction. It is associated with the mechanism of atrial fibrillation [30, 32] and can modify QTc duration in people with LQTS [33]. Studies of different genotype frequencies in various populations related to SCN5A-H558R function have not yet been performed, but SY Nikulina.et.al already found that AG genotype of the H558R (rs1805124) polymorphism of the SCN5A gene is a genetic predictor of idiopathic disorders of atrioventricular and intraventricular conduction [34] We can carry out the prevention and early treatment of these diseases by gene sequencing.
Among Mongols and others global populations, numerous important genetic variants play critical roles in drug response and this information should directly applied to clinical guidelines. For instance rs1540339 (46489G > A), the most significant locus in our data, is associated with bronchodilator responsiveness [35]. Studies have been performed on the correlation between asthma and rs1540339; however, evaluation of this polymorphism in a clinical setting is not yet routine [36, 37].
Beyond the genetic factor, we also determined that long-term survival in different environments affects genetic adaption. Environmental pressures shape genotype distributions towards specific functions, particularly in pharmacogenetic genes. Studies by Janha et al., Sabbagh et al., and Fuselli et al. directly demonstrated that the different genotype frequencies of CYP2C19, NAT2, and CYP2D6 significantly differed between populations race, subsistence modes, and dietary habits also play a role in the evolutionary trajectory [38–40].
Conclusions
Different populations exists different genetic distribute frequencies. The drug dosage and usage of different genotype carriers is difference. Identifying genotype distribution and VIP variant frequencies in different populations to determine what medications might be most effective may provide a theoretical foundation for safe drug administration and improved curative effects. Besides, we figured out the minimum allele difference between Mongol and CDX. We also preliminary supplemented the pharmacogenomic data on the Mongol ethnic group and illustrated the differences between Mongols and other populations, and finally found Mongol and Chinese populations have the minimum difference. To the study, the sample size is relatively small and further investigation using a larger cohort of Mongols is needed to verify the generalizability of our results, and would be help us to establish a more reasonable and effective individualized treatment plan.
Abbreviations
ALFRED, the ALlele FREquency Database; ASW, a population of African ancestry in the southwestern USA; CEU, a northwestern European population; CHB, the Han Chinese in Beijing, China; CHD, the population of metropolitan Denver, Colorado, USA; GIH, the Gujarati Indians in Houston, Texas, USA; HWE, Hardy–Weinberg Equilibrium; JPT, the Japanese population in Tokyo, Japan; LWK, the Chinese living in Luhya in Webuye, Kenya; MEX, people of Mexican ancestry living in Los Angeles, California, USA; MKK, the Maasai people in Kinyawa, Kenya; MTHFR, methylenetetrahydrofolate reductase; PharmGKB, the Pharmacogenomics Knowledge Base; PMT, the Pharmacogenetics of Membrane Transporters database; PUR, a population of Puerto Ricans from Puerto Rico; TSI, the Tuscan people of Italy; VIP, very important pharmacogenetic; YRI, the Yoruba in Ibadan, Nigeria
References
Daly AK. Genome-wide association studies in pharmacogenomics. Nat Rev Genet. 2010;11(4):241–6.
Roden D, Tyndale R. Pharmacogenomics at the tipping point: challenges and opportunities. Clin Pharmacol Ther. 2011;89(3):323–7.
Szekanecz Z, Meskó B, Poliska S, Váncsa A, Szamosi S, Végh E, Simkovics E, Laki J, Kurkó J, Besenyei T. Pharmacogenetics and pharmacogenomics in rheumatology. Immunol Res. 2013;56(2-3):325–33.
Food U, Administration D. FDA clears genetic test that advances personalized medicine test helps determine safety of drug therapy. FDA News P05-53 Rockville, MD: FDA 2005.
Scott SA. Personalizing medicine with clinical pharmacogenetics. Genet Med. 2011;13(12):987–95.
Whirl-Carrillo M, McDonagh E, Hebert J, Gong L, Sangkuhl K, Thorn C, Altman R, Klein TE. Pharmacogenomics knowledge for personalized medicine. Clin Pharmacol Ther. 2012;92(4):414–7.
Sim SC, Altman RB, Ingelman‐Sundberg M. Databases in the area of pharmacogenetics. Hum Mutat. 2011;32(5):526–31.
Poon AH, Gong L, Brasch-Andersen C, Litonjua AA, Raby BA, Hamid Q, Laprise C, Weiss ST, Altman RB, Klein TE. Very important pharmacogene summary for VDR. Pharmacogenet Genomics. 2012;22(10):758–63.
Sangkuhl K, Berlin DS, Altman RB, Klein TE. PharmGKB: understanding the effects of individual genetic variants. Drug Metab Rev. 2008;40(4):539–51.
Yang ZF, Cui HW, Hasi T, Jia SQ, Gong ML, Su XL. Genetic polymorphisms of cytochrome P450 enzymes 2C9 and 2C19 in a healthy Mongolian population in China. Genet Mol Res. 2010;9(3):1844–51.
Takahashi H, Wilkinson GR, Nutescu EA, Morita T, Ritchie MD, Scordo MG, Pengo V, Barban M, Padrini R, Ieiri I. Different contributions of polymorphisms in VKORC1 and CYP2C9 to intra-and inter-population differences in maintenance dose of warfarin in Japanese, Caucasians and African-Americans. Pharmacogenet Genomics. 2006;16(2):101–10.
Gabriel S, Ziaugra L, Tabbaa D. SNP genotyping using the Sequenom MassARRAY iPLEX platform. Current protocols in human genetics/editorial board, Jonathan L Haines [et al] 2009, Chapter 2:Unit 2 12.
Thomas RK, Baker AC, Debiasi RM, Winckler W, Laframboise T, Lin WM, Wang M, Feng W, Zander T, MacConaill L, et al. High-throughput oncogene mutation profiling in human cancer. Nat Genet. 2007;39(3):347–51.
Song MK, Lin FC, Ward SE, Fine JP. Composite variables: when and how. Nurs Res. 2013;62(1):45–9.
Pritchard JK, Stephens M, Donnelly P. Inference of population structure using multilocus genotype data. Genetics. 2000;155(2):945–59.
Evanno G, Regnaut S, Goudet J. Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol. 2005;14(8):2611–20.
Everett JR, Loo RL, Pullen FS. Pharmacometabonomics and personalized medicine. Ann Clin Biochem. 2013;50(Pt 6):523–45.
Zand N, Tajik N, Moghaddam AS, Milanian I. Genetic polymorphisms of cytochrome P450 enzymes 2C9 and 2C19 in a healthy Iranian population. Clin Exp Pharmacol Physiol. 2007;34(1‐2):102–5.
Li J, Zhang L, Zhou H, Stoneking M, Tang K. Global patterns of genetic diversity and signals of natural selection for human ADME genes. Hum Mol Genet. 2011;20(3):528–40.
Yu L, Chen J. Association of MHTFR Ala222Val (rs1801133) polymorphism and breast cancer susceptibility: an update meta-analysis based on 51 research studies. Diagn Pathol. 2012;7:171.
Hider S, Bruce I, Thomson W. The pharmacogenetics of methotrexate. Rheumatology. 2007;46(10):1520–4.
Schwahn B, Rozen R. Polymorphisms in the methylenetetrahydrofolate reductase gene. Am J Pharmacogenomics. 2001;1(3):189–201.
Zhou B-S, Bu G-Y, Li M, Chang B-G, Zhou Y-P. Tagging SNPs in the MTHFR gene and risk of ischemic stroke in a Chinese population. Int J Mol Sci. 2014;15(5):8931–40.
van der Put NM, Gabreëls F, Stevens E, Smeitink JA, Trijbels FJ, Eskes TK, van den Heuvel LP, Blom HJ. A second common mutation in the methylenetetrahydrofolate reductase gene: an additional risk factor for neural-tube defects? Am J Hum Genet. 1998;62(5):1044–51.
Gra O, Mityaeva O, Berdichevets I, Kozhekbaeva Z, Fesenko D, Kurbatova O, Goldenkova-Pavlova I, Nasedkina T. Microarray-Based Detection of CYP1A1, CYP2C9, CYP2C19, CYP2D6, GSTT1, GSTM1, MTHFR, MTRR, NQO1, NAT2, HLA-DQA1, and AB0 Allele Frequencies in Native Russians. Genet Test Mol Biomarkers. 2010;14(3):329–42.
Frosst P, Blom HJ, Milos R, Goyette P, Sheppard CA, Matthews RG, Boers GJ, den Heijer M, Kluijtmans LA, van den Heuvel LP, et al. A candidate genetic risk factor for vascular disease: a common mutation in methylenetetrahydrofolate reductase. Nat Genet. 1995;10(1):111–3.
Park HS, Kim YN, Lee YS, Jung BC, Lee SH, Shin DG, Cho Y, Bae MH, Han SM, Lee MH. Genetic analysis of SCN5A in Korean patients associated with atrioventricular conduction block. Genomics Inform. 2012;10(2):110–6.
Ye B, Valdivia CR, Ackerman MJ, Makielski JC. A common human SCN5A polymorphism modifies expression of an arrhythmia causing mutation. Physiol Genomics. 2003;12(3):187–93.
Makielski JC, Ye B, Valdivia CR, Pagel MD, Pu J, Tester DJ, Ackerman MJ. A ubiquitous splice variant and a common polymorphism affect heterologous expression of recombinant human SCN5A heart sodium channels. Circ Res. 2003;93(9):821–8.
Chen L, Zhang W, Fang C, Jiang S, Shu C, Cheng H, Li F, Li H. Polymorphism H558R in the human cardiac sodium channel SCN5A gene is associated with atrial fibrillation. J Int Med Res. 2011;39(5):1908–16.
Daneshjou R, Zappala Z, Kukurba K, Boyle SM, Ormond K, Klein T, Snyder M, Bustamante C, Altman R, Montgomery S. Path-scan: a reporting tool for identifying clinically actionable variants. In: Pacific Symposium on Biocomputing Pacific Symposium on Biocomputing: 2013. World Scientific: 229-240.
Gouas L, Nicaud V, Berthet M, Forhan A, Tiret L, Balkau B, Guicheney P. Association of KCNQ1, KCNE1, KCNH2 and SCN5A polymorphisms with QTc interval length in a healthy population. Eur J Hum Genet. 2005;13(11):1213–22.
Giudicessi JR, Ackerman MJ. Arrhythmia risk in long QT syndrome beyond the disease-causative mutation. Circ Cardiovasc Genet. 2013;6(4):313–6.
Nikulina SY, Chernova AA, Shulman VA, Maksimov VN, Gavrilyuk OA, Tretyakova SS, et al. An investigation of the association of the H558R polymorphism of the SCN5A gene with idiopathic cardiac conduction disorders. Genet Test Mol Biomarkers. 2015.
Baig S, Moatter T. Vitamin D is not just about healthy bones anymore. J Coll Physicians Surg Pak. 2010;20(6):355–6.
Michel S, Liang L, Depner M, Klopp N, Ruether A, Kumar A, Schedel M, Vogelberg C, von Mutius E, von Berg A. Unifying candidate gene and GWAS approaches in asthma. PLoS One. 2010;5(11), e13894.
Pillai DK, Iqbal SF, Benton AS, Lerner J, Wiles A, Foerster M, Ozedirne T, Holbrook HP, Payne Jr PW, Gordish-Dressman H. Associations between genetic variants in vitamin D metabolism and asthma characteristics in young African Americans: a pilot study. J Investig Med. 2011;59(6):938.
Janha RE, Worwui A, Linton KJ, Shaheen SO, Sisay-Joof F, Walton RT. Inactive alleles of cytochrome P450 2C19 may be positively selected in human evolution. BMC Evol Biol. 2014;14(1):71.
Sabbagh A, Darlu P, Crouau-Roy B, Poloni ES. Arylamine N-acetyltransferase 2 (NAT2) genetic diversity and traditional subsistence: a worldwide population survey. PLoS One. 2011;6(4), e18507.
Fuselli S, de Filippo C, Mona S, Sistonen J, Fariselli P, Destro-Bisol G, Barbujani G, Bertorelle G, Sajantila A. Evolution of detoxifying systems: the role of environment and population history in shaping genetic diversity at human CYP2D6 locus. Pharmacogenet Genomics. 2010;20(8):485–99.
Acknowledgments
Authors thank BioScience Writers for assistance in the manuscript preparation. We also appreciate the reviewers and editors for their patience to the work.
Funding
The work is supported by Natural Science Foundation of Xizang (Tibet) Autonomous Region (20152R-13-11), Major science and technology research projects of Xizang (Tibet) Autonomous Region (2015XZ01G23) and National Natural Science Foundation of China (No. 81560516).
Availability of data and material
All relevant data are available within the manuscript.
Authors’ contributions
TJ and XS drafted the manuscript. TJ, TF, and XS performed the statistical analyses and LW helped edit the manuscript. LW and HW performed the genotyping. TF and XS reviewed all the genotypes. TJ assisted in the study design. TJ and LK conceived the study and co-supervised the work. All the authors have read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interest.
Consent for publication
Not applicable.
Ethics approval and consent to participate
This study was approved by the Clinical Research Ethics of Xizang Minzu University and Northwest University, and it is in compliance with Department of Health and Human Services (DHHS) regulations for human research subject protection. Written informed consent was received from all of the study participants.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Tianbo Jin and Xugang Shi are joint first authors.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Jin, T., Shi, X., Wang, L. et al. Genetic polymorphisms of pharmacogenomic VIP variants in the Mongol of Northwestern China. BMC Genet 17, 70 (2016). https://doi.org/10.1186/s12863-016-0379-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12863-016-0379-0