
Abstract
Background
The Styracaceae are a woody, dicotyledonous family containing 12 genera and an estimated 160 species. Recent studies have shown that Styrax and Sinojackia are monophyletic, Alniphyllum and Bruinsmia cluster into a clade with an approximately 20-kb inversion in the Large Single-Copy (LSC) region. Halesia and Pterostyrax are not supported as monophyletic, while Melliodendron and Changiostyrax always form sister clades. Perkinsiodendron and Changiostyrax are newly established genera of Styracaceae. However, the phylogenetic relationship of Styracaceae at the generic level needs further research.
Results
We collected 28 complete plastomes of Styracaceae, including 12 sequences newly reported here and 16 publicly available sequences, comprising 11 of the 12 genera of Styracaceae. All species possessed the typical quadripartite structure of angiosperm plastomes, with sequence differences being minor, except for a large 20-kb (14 genes) inversion found in Alniphyllum and Bruinsmia. Seven coding sequences (rps4, rpl23, accD, rpoC1, psaA, rpoA and ndhH) were identified to possess positively selected sites. Phylogenetic reconstructions based on seven data sets (i.e., LSC, SSC, IR, Coding, Non-coding, combination of LSC + SSC and concatenation of LSC + SSC + one IR) produced similar topologies. In our analyses, all genera were strongly supported as monophyletic. Styrax was sister to the remaining genera. Alniphyllum and Bruinsmia form a clade. Halesia diptera does not cluster with Perkinsiodendron, while Perkinsiodendron and Rehderodendron form a clade. Changiostyrax is sister to a clade of Pterostyrax and Sinojackia.
Conclusion
Overall, our results demonstrate the power of plastid phylogenomics in improving estimates of phylogenetic relationships among genera. This study also provides insight into plastome evolution across Styracaceae.
Similar content being viewed by others

Background
The Styracaceae DC. & Spreng (Ericales) comprise an angiosperm clade of 12 genera and over 160 species, mainly distributed in regions of Asia, as well as tropical and temperate America, and the Mediterranean [1]. The family consists of shrubs or trees, usually having stellate pubescent or epidermal scales, simple leaves, with raceme, cyme or panicle inflorescence, and actinomorphic flowers with varying degrees of synsepaly and sympetaly [2]. The fruit of Styracaceae is a drupe or capsule, with persistent calyx, surrounding or united with the fruit. The Styracaceae have been included in a number of morphological studies, analyzing leaf anatomy [3], wood anatomy [4], pollen morphology [5] and floral morphology and anatomy [2], but distinguishing between genera in the family primarily involves variation in fruit morphological characters (e.g. hypanthium at maturity). On one hand the ovary is inferior with a persistent hypanthium combined with the fruit at maturity [(i.e., Changiostyrax C.T. Chen (one species), Halesia J. Ellis ex L (two species), Melliodendron Hand.-Mazz (one species), Parastyrax Siebold & Zucc. (two species), Perkinsiodendron P. W. Fritsch (one species), Pterostyrax W.W. Sm.(four species), Rehderodendron Hu (one species), and Sinojackia Hu (seven species)]. On the other hand, the ovary is superior and a persistent hypanthium forms only at the base of the fruit at maturity [Alniphyllum Matsum (three species), Bruinsmia Boerl. & Koord (two species), and Styrax L (130 species)]. Moreover, the ovary of Huodendron Rehder (four species) is semisuperior with a persistent hypanthium extending from the base to about two-thirds of the fruit length [1, 2], a feature considered to be transitional.
The systematic position of Styracaceae and the genera within have been unstable since the establishment of the family by Dumoritor in 1829 [6]. Early researchers thought Styracaceae was positioned in the order Ebenales, along with the well-known Sapotaceae, Ebenaceae, and Symplocaceae, and the small family Lissocarpaceae [7,8,9,10]. However, Cronquist [10] showed that these families have some original characteristics and some new evolutionary characters, which may have arisen via parallel evolution. Based on embryological and anatomical studies, Herbert [11] suggested that Styracaceae and Theaceae may have originated from a common ancestor, since the two share many common characteristics. According to molecular systematic studies, Styracaceae has been recognized as part of the order Ericales sensu lato [12].
Within the family, phylogenetic resolution generally remains poor. At most 17 genera have been included in Styracaceae, with Symplocos L, Diclidanthera Mart, Afrostyrax Perk et Gil, Foveolaria Ruiz et pav., Pamphilia Mart. ex A. DC, Huapierre et De Wil, and Lissocarpa Benth placed in the Styracaceae by various authors [13]. Symplocos, Diclidanthera, and Lissocarpa were excluded from Styracaceae by Perkins [14]. Symplocos was treated as an independent family (Symplocaceae Desf) [15]. Diclidanthera was placed in Polygalaceae [7, 15], and Lissocarpa was placed in Ebenaceae [16]. Afrostyrax was once included in the genus Styrax [17], but was later reclassified into Huaceae [7, 15, 18]. According to taxonomic revisions, Pamphilia was classified into Styrax [19], while Fritsch [20] combined Foveolaria into Styrax by implementing morphological phylogenetic analyses. In addition, two new genera have been established: (1) Chen [21] segregated Sinojackia dolichocarpa as a new monotypic genus Changiostyrax, and (2) according to morphological and DNA sequences, Halesia macgregorii was removed from Halesia to become a new genus, Perkinsiodendron P.W. Fritsch [22].
Although the phylogenetic placement of the family has been resolved, relationships between genera remain ambiguous. The phylogeny of Ericales based on the chloroplast gene rbcL [23] suggested that Styracaceae was polyphyletic with Styrax and Clethra Gronov. ex L. (Clethraceae) clustered in a clade, while Halesia, Rehderodendron, and Sinojackia formed a clade that was sister to Diapensia L. and Galax Rafin. (Diapensiaceae). However, the interpretation of polyphyly does not always hold true. Olmstead et al. [24] inferred the phylogeny of Asteridae based on the chloroplast gene ndhF, showing a strongly supported sister relationship between Styrax and Halesia. Albach et al. [25] came to the same conclusion based on the DNA gene sequences atpB, ndhF, rbcL and 18S [24] within the Asterids. In addition, the phylogeny of Styracaceae based on morphology plus three DNA sequences (chloroplast trnL intron/trnL-trnF spacer and rbcL with the nuclear ribosomal DNA region ITS) recovered a monophyletic relationship of Styracaceae [1]. Pterostyrax and Halesia were not supported as monophyletic, since Styrax and Huodendron formed a clade that was sister to a clade of Alniphyllum and Bruinsmia, and a sister relationship was found between Halesia macgregorii and Rehderodendron macrocarpum [1]. Based on ITS, the plastid psbA-trnH intergenic spacer, and microsatellite data, Yao et al. [26] recovered Sinojackia as monophyletic and reported a similar topology as Fritsch et al. [1] with weak support for six genera within Styracaceae. Yan et al. [27] conducted phylogenetic analyses of Styracaceae based on 19 chloroplast genomes. The results showed that Styrax was monophyletic, while Alniphyllum and Bruinsmia clustered in a clade with an approximate 20-kb inversion in the Large Single-Copy (LSC) region. Species of Pterostyrax were not supported as monophyletic, with Halesia carolina L and Pterostyrax hispidus Siebold & Zucc forming a clade.
The chloroplast genomes of most angiosperms are maternally inherited. The rate of evolution of genes in the chloroplast is relatively slow overall, but differences have been observed across different regions of the plastome, which can be applied to phylogenetic studies of various taxonomic scales. Signatures of selection (purifying or positive/adaptive) have been observed in different regions of the plastome, including protein coding regions involved in photosynthesis [28–30]. Several aspects have led to the extensive use of plastomes for phylogenetic inference such as a conserved structure, small effective population size, and lack of recombination due to being predominately uniparentally inherited [31–33]. With the increasing efficiency of next-generation sequencing (NGS) technologies, obtaining whole-plastome sequence data has become cheaper and easier. Whole-plastomes have been used in taxonomically complex groups to generate resolved and well-supported phylogenies, as well as serving as sequence barcodes to identify plant species at the molecular level [34–36].
Despite progress in understanding phylogenetic relationships within Styracaceae, most advances have been based on relatively limited molecular and/or morphological data. Only one study has examined the phylogeny of Styracaceae using plastome-scale data [27], but this study employed only 19 taxa and included only one or two accessions per genus. Here, we increased sampling for some genera, especially Sinojackia (five accessions) and Styrax (seven accessions). We analyzed 28 complete plastomes for resolving the broader phylogeny of Styracaceae. Compared with phylogenetic studies limited to a few complete plastomes or a few plastid loci, plastome phylogenomic studies provide potentially greater resolution and support. The objectives of this study are: (1) infer the plastome structural evolution of Styracaceae, (2) resolve the phylogenetic relationships of Styracaceae, (3) use selective pressure analysis to test for the presence of adaptive evolution in all genes.
Methods
Plant samples, DNA extraction, sequencing and assembly
We collected 28 plastomes of Styracaceae, including 12 newly sequenced and 16 previously sequenced plastomes (Table 1), with representatives from 11 of the 12 genera described by APG IV [37]. We used Symplocos ovatilobata Noot (Symplocaceae), Stewartia monadelpha Siebold et Zucc, and Stewartia sinii (Y. C. Wu) Sealy (Theaceae) as outgroups. A total of 31 sequences were analyzed. Our field collections were permitted by the government following local ethics and laws. Collected plant leaves were put directly into silica gel to dry. The formal identification of the plant material was undertaken by Guowen Xie, and voucher herbarium specimens were deposited at the Institute of Tropical Agriculture and Forestry (HUTB), Hainan University, Haikou, China.
Total genomic DNA was extracted from dried leaf tissue using cetyltrimethyl ammonium bromide (CTAB) protocol of Doyle and Doyle [38]. Genomic DNA of each sample was quantified and analyzed with an Agilent BioAnalyzer 2100 (Agilent Technologies). Samples yielding at least 0.8 µg DNA were selected for subsequent library construction and sequencing. Genomic DNA of selected samples was used to build paired‐end libraries with insert sizes of 200–400 bp according to the manufacturer’s instructions [39]. Sequencing of the new 12 accessions was completed using BGISEQ-500 2 × 100 at BGI (Shenzhen, China). This yielded approximately eight Gb of high‐quality data per sample of 100 bp paired‐end reads. Raw reads were trimmed using SOAPfilter v2.2 (BGI-Shenzhen, China) with the following criteria: removal of reads with more than 10 percent base of N, reads with more than 40 percent low quality (phred score less than 10), and reads contaminated by adaptors and PCR duplicates. Approximately six Gb of clean data (high-quality reads > phred score35) were obtained for each sample. For all samples, plastomes were assembled using MITObim v1.8 [40] with default parameters and using plastomes of related species as templates for assembly (Table 2). The assembly was ordered using BLAST and aligned (> 90% similarity and query coverage) according to the reference chloroplast genome (Table 2). To verify sequencing depth and contig overlap, cleaned reads were mapped to reference plastomes in Geneious R11.0.4 [41].
Genome annotation
Plastomes were annotated using Geneious R11.0.4 [41] using the same reference plastomes used for assembly. Start/stop codons and intron/exon boundaries were further corrected using Dual Organellar GenoMe Annotator (DOGMA) [42]. In addition, tRNAscan-SE1.21 was used to further verify all tRNA genes. We also re-annotated the downloaded assembled plastomes from previous studies before using them in our analyses. The 12 newly generated complete plastome sequences were deposited in GenBank (Accession Numbers in Table 2).
Genome comparative and structural analyses
Graphical maps of Styracaceae plastomes were drawn using Organellar Genome DRAW (OGDRAW) [43], with subsequent manual editing. Genome comparisons across the 26 Styracaceae species (selecting one sequence per species) were performed with Shuffle-LAGAN mode in mVISTA [44] using the annotation of Pterostyrax hispidus Siebold & Zucc as a reference. To evaluate whether different chloroplast genome regions have undergone different evolutionary histories and to explore highly variable regions for future population genetic and species identification studies, we sequentially extracted both coding regions and noncoding regions (including intergenic spacers and introns) after aligning with MAFFT v7 [45] under the criteria that the aligned length was > 200 bp and at least one mutation site was present. Finally, nucleotide variability of these regions was evaluated with DNASP v5.10 [46].
Selective pressure analysis
The analyses of selective pressures were conducted along the phylogenetic tree of Styracaceae (see below) for each plastid gene located in the Large Single-Copy (LSC) region, Inverted Repeat (IR) region and Small Single-Copy (SSC) region. Nonsynonymous (dN) and synonymous (dS) substitution rates of each plastid gene were calculated using the yn00 program in PAML v4.9 [47]. In addition, we used the CODEML program in PAML to detect signatures of natural selection among specific lineages. Genes were considered to be under positive/negative selection at a certain clade when its ω value from the two-ratio model was higher/lower than 1 (neutral selection). To avoid potential convergence biases, genes with too few mutations [Pi(nucleotide diversity) < 0.001] were filtered out from selective pressure analysis.
Phylogenetic analyses
Phylogenetic analyses were conducted on the 31 plastomes, using Symplocos ovatilobata, Stewartia sinii, and S. monadelpha as outgroups. Chloroplast sequences were aligned using MAFFT v7.037 [45]. To evaluate possible alternative phylogenetic hypotheses, topologies were constructed by both maximum likelihood (ML) and Bayesian inference (BI) methods using not only the complete genome sequences, but also using seven additional data sets (i.e. LSC, SSC, IR, Coding, Noncoding, combination of LSC + SSC, and concatenation of LSC + SSC + one IR). Data characteristics and the best-fitting models of nucleotide substitutions were determined with Akaike Information Criterion (AIC) in Modeltest v3.7 [48] (Table 3). For the coding data set, PartitionFinder v2.1.1 [49] was used to select the best-fit partitioning scheme of all 79 possible gene-by-codon position partitions (79 genes × 3 codon positions).
Maximum likelihood analyses were conducted using RAXML-HPC v8.2.8 [50] with 1000 bootstrap replicates on the CIPRES Science Gateway website [51] with the GTR + I + G substitution model. Bayesian inference (BI) analyses were performed in MrBayes v3.2 [52] on the CIPRES Science Gateway portal [51] with the following conditions used for the protein-coding dataset: starting from random trees, Markov chain Monte Carlo (MCMC) simulations were ran for 900,000,000 generations with four incrementally heated chains sampling every 1000 generations. BI analyses were set up identically for the remaining data sets, except that 50,000,000 generations were simulated. Convergence of the MCMC chains was determined by examining the average standard deviation of the split frequencies (< 0.01). The first 25% of the trees were discarded as burn-in. The effective sample size (ESS > 200) was determined by using Tracer v 1.7 [53].
Result
Plastome structure of styracaceae
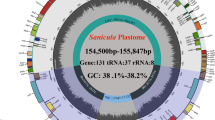
In this study, the plastomes of Styracaceae and outgroups displayed a typical quadripartite structure and similar lengths. Plastome sizes ranged from 155,185 bp (Alniphyllum pterospermu Matsum) to 158,879 bp (Pterostyrax hispidus) with a minimum/maximum read depth of 10 × /40 × for each plastome. The plastomes were composed of a large single-copy (LSC) region (ranging from 83,200 bp to 88,258 bp), a small single-copy (SSC) region (ranging from 17,556 bp to 19,235 bp), and two inverted repeat IR regions (IRa and IRb) (ranging from 24,243 bp to 26,761 bp) (Table 4). Their overall GC content was nearly identical (36.70–37.40%). In all species, the GC content of the LSC and SSC regions (about 35% and 30%) were lower than those of the IR regions (about 43%). The 31 plastomes encoded 113 genes, including 79 protein-coding genes, 30 transfer RNA (tRNA) genes, and four ribosomal RNA (rRNA) genes. Comparison of the genome structures among Styracaceae, revealed an inversion of a large segment spanning trnQ-UUG to rpoB (20-kb) in the LSC region of Alniphyllum fortunei (Hemsl.) Makino (Fig. 1).

Gene map of the Styrax faberi. A The inverted order of genes in Alniphyllum fortunei; B The corresponding region of Styrax faberi
Comparative genomic analysis and divergence hotspot regions
To investigate the levels of sequence divergence, 26 Styracaceae plastomes were plotted using mVISTA with Pterostyrax hispidus as the reference (Fig. 2). The sequence divergence was low among all plastomes. Notably, the proportion of variability in coding regions and inverted repeats (IRs) showed higher conservation than noncoding and small single-copy (SSC) regions. The mutation rate of ycf1 was the highest observed. The variation rates of Styrax and Huodendron in the large and small single copy regions were higher than other species, and the sequence divergence of Huodendron in clpP intron lower than 50%.

Visualization of the alignment of 26 Styracaceae plastome sequences. The plastome of Pterostyrax hispidus was used as the reference. The Y-axis depicts percent identity to the reference genome (50–100%) and the X-axis depicts sequence coordinates within the plastome. Genome regions were color-coded according to coding and noncoding regions
Nucleotide diversity (pi) analyses showed that the proportion of variable sites in noncoding regions were higher than that in coding region, and the greatest diversity change was in the intergenic spacer region (Fig. 3). Among all 209 loci (79 coding genes and 130 noncoding regions), nucleotide diversity values of coding genes ranged from 0.001 (rpl23) to 0.156 (atpH), with four loci greater than 0.1 (psbK, psbI, rpoC2, atpH). Nucleotide diversity of noncoding genes ranged from 0 (rpoC1-rpoB, psaB-psaA, psbF-psbE, rps3-rpl22, rpl2-rpl23, rps7-rps12, trnA (UGC)-rrn23, ndhH-ndhA, orf42-trnA-UGC, ycf2-ycf15) to 0.385 (trnI intron1). Seven loci possessed values > 0.15: e.g. atpF intron (0.151), clpP intron1 (0.151), rps2-rpoC2 (0.151), trnG(GCC)-trnR(UCU) (0.158), rps12-clpP (0.159), atpH-atpI (0.166), trnI(GAU) intron1 (0.385) (Fig. 3).

Comparison of the nucleotide diversity (Pi) values across 28 Styracaceae plastomes. A Protein-coding regions. B Noncoding regions
Selective pressures in plastome evolution of Styracaceae
The results showed that the 79 protein coding genes mainly possessed synonymous substitutions (Fig. 4). In addition, rps12 (0.8874), rps19 (0.5076) and rps11 (0.4466) had the highest synonymous substitution rate. The locus with the highest rate of nonsynonymous substitution was ycf1 (1.016). The rate of nonsynonymous substitutions in other genes was low, in which the rate of nonsynonymous substitution of psb was the lowest, and the nonsynonymous substitution of psbL, psbH, psbN, psbI and psbT was zero. Among the 79 protein coding genes of Styracaceae, there were seven genes with ω value greater than 1: rps4 (1.087), rpl23 (1.126), accD (1.839), rpoC1 (1.990), psaA (2.175), rpoA (1.578) and ndhH (3.459) (Fig. 5).

Synonymous (dS) and nonsynonymous (dN) substitution rates of the protein coding genes

ω (dN/dS) values of genes in plastomes of the Styracaceae. The red line represents neutral selection, while values above one represents positive/adaptative selection, and values below one represents negative/purifying selection
Phylogenetic analyses
The optimal partitioning scheme identified under the Akaike information criterion with correction (AICc) using relaxed clustering analysis in PartitionFinder (lnL = − 18247.90; AICc = 379952.05) contained 64 partitions (Additional file 7: Table S1). BI analyses and ML analyses using the unpartitioned and partitioned schemes produced identical topologies (Fig. 6). Genera within Styracaceae were all recovered as monophyletic with strong support (BS/PP = 100/1). All species of Styrax form a clade sister to the rest of the family (BS/PP = 100/1). The second branch is Huodendron, followed by two genera with the 20-Kb inversion, Alniphyllum and Bruinsmia. Halesia diptera did not cluster with Perkinsiodendron but was sister to the remaining genera (BS/PP = 100/1), while Perkinsiodendron and Rehderodendron form a clade (BS/PP = 100/1). The position of Melliodendron does change based on the data partition analyzed. In most analysis Melliodendron is sister to a clade of Perkinsiodendron, Rehderodendron, Changiostyrax, Pterostyrax, and Sinojackia (BS/PP = 100/1) except for in LSC, which Melliodendron is sister to Changiostyrax form a clade (BS/PP = 56/1). Changiostyrax is sister to a clade composed of Pterostyrax and Sinojackia (BS/PP = 65/0.67). Pterostyrax and Sinojackia are sister with strong support (BS/PP = 85/1). To test for conflicting signals across different data, we used six data sets for analyses (Additional files 1, 2, 3, 4, 5, 6: Fig. S1–S6). The ML and BI analyses produced similar topologies over all data sets except for the different positions of Sinojackia sarcocarpa (L.) Q. Luo, Changiostyrax dolichocarpus (C. J. Qi) Tao Chen and Pterostyrax hispidus in the IR regions (Additional file 1: Fig S1). In trees inferred from the IR regions, Sinojackia and Pterostyrax were not monophyletic. Characteristics of all data sets are shown in Table 2.

Optimal phylogenetic tree resulting from analyses of 79 protein-coding genes using Maximum Likelihood (ML). Bayesian inference (BI) topology is the same as ML. Support values next to the nodes are maximum likelihood bootstrap support/Bayesian posterior probability; asterisks indicate 100%/1.0 support values. The genera of Styracaceae are indicated by different branch colors. The inset shows the same tree as a phylogram
Discussion
Plastome structure comparisons and sequence divergence hotspots
This study included 31 plastomes, 28 representative taxa from 11 genera of Styracaceae, and three outgroups. Plastomes displayed a typical quadripartite structure and similar size, containing a pair of inverted repeat IR regions (IRa and IRb), one large single-copy (LSC) region, and one small single-copy (SSC) region. The plastome size of Styracaceae is within the normal range of angiosperms (120–190 kb), and the size, structure, gene sequence and content of the whole family are highly conserved (155,185 bp–158,879 bp), with a typical tetragonal structure [54]. The plastome of Alniphyllum fortunei, which was first reported in this study, contained a 20-kb inversion which includes 14 coding genes from trnQ-UUG to rpoB. The presence of this inversion has previously been verified using PCR and Sanger sequencing by Yan et al. [55]. The inversion has also been observed in plastomes of A. eberhardtii Guill, A. pterospermum Matsum, Bruinsmia polysperma (C. B. Clarke) Steenis and B. styracoides Boerl. & Koord, suggesting that the inversion is common to Bruinsmia and Alniphyllum. The large 20-kb inversion has the same gene composition and relative position as the normal plastome structure and is not due solely to the genome assembly method [55]. Plastid structure is usually conserved in most angiosperms, but large inversions have been detected in many taxa. For example, a 4-kb inverted fragment in the LSC between rpoB-trnT was found in Myriophyllum spicatum [56], and a large gene inversion has also found in Lotus japonicas, Arabidopsis thaliana [57] and members of Oleaceae [58]. Because of their scarcity, plastid inversions are of great value to the study of genome evolution [59, 60]. Previous studies have suggested that gene inversions are closely related to repetitive sequences, and dispersed repetitive sequences promote inversions through intermolecular recombination [61–63].
In the sequence divergence analysis, the variation in loci of noncoding regions is higher than those of coding regions, which is similar to previous results of most angiosperms [64–66]. The results also show that the degree of evolution in the noncoding regions is greater than that of coding regions, and highly variable noncoding regions are of great value for the study of plant phylogenetics [67, 68]. In addition, the rate of variation in the IR region was lower than the two single copy regions. Previous studies have shown that the accumulation of point mutations in the inverted repeat region is slower than the single copy region [69–71].
Positive selection analysis
In the selection pressure analysis, Styracaceae is dominated by synonymous substitutions. A previous study indicated that the rate of nonsynonymous substitutions is positively correlated with the degree of variation in the genome, while the rate of synonymous substitution exhibits a weak correlation with the degree of variation in the genome [72]. There are seven coding genes under positive selection, including five gene types: NADH dehydrogenase gene (ndhH), ribosomal protein coding gene (rps4 & rpl23), RNA polymerase gene (rpoC1 & rpoA), a photosynthetic gene (psaA) and one additional protein gene (accD). The chloroplast NADH dehydrogenase (NDH) complex participates in the circular electron transport and chlorine respiration around the light system [73]. However, due to NDH complex existing in low abundance and being of a fragile nature, it is difficult to analyze its function [74]. The plants of Styracaceae are mainly distributed in the tropics and subtropics, which are subjected to growing conditions of high light and high temperature. Ribosomal proteins are a part of the ribosomal complex, which is a translation mechanism, and is essential for the correct production of proteins required for normal cell function. The selection of ribosomal proteins may increase the stability of ribosomal complexes under high light conditions, as well as high temperature, which is similar to the selection of ndh proteins under high light conditions [75]. However, whether these ribosomal proteins have increased stability over those of the original proteins under strong light or related conditions has not been determined, and further experimental verification is still needed. The rpoC gene is in the same operon as rpoA, which encodes the β subunit of RNA polymerase. Increasing the rpoA & rpoC mutations may lead to alterations in cell wall metabolism, possibly as a result of altered transcription [76]
Phylogenetic analyses
We constructed data matrices from seven different partitions, and analyzed the phylogeny of the different matrices to maximize the resolution of phylogenetic relationships and to test for conflicting signals. Overall, the phylogenetic relationships constructed by the different data matrices show consistent topologies with moderate support. The phylogeny based on the complete plastome is consistent with the inferred phylogenies of the other six data sets with the exception of the IR region. According to Fritsch et al.’s [1] analysis of morphology and three DNA sequence data sets, Styrax is monophyletic, forming a clade with Huodendron. However, our analyses show that Styrax is monophyletic with high support (BS/PP = 100/1) but is sister to the remainder of the family, which is consistent with the conclusions of Yan et al. [27]. Alniphyllum and Bruinsmia formed a clade that has the longest branches in the phylogram, which may be due to higher rates of substitution in these two genera.
Fritsch et al. [1] and Yao et al. [26] consistently showed that Melliodendron formed a clade with Changiostyrax, whereas in all our data sets, except in the LSC data set, Melliodendron and Changiostyrax do not form a clade. Changiostyrax is weakly supported as sister to a clade composed of Pterostyrax and Sinojackia (BS/PP = 65/0.67). Halesia and Pterostyrax have not previously been fully resolved [1, 26, 27]. Here, we collected four accessions of Pterostyrax to analyze and Pterostyrax was recovered as monophyletic in all analyses except when P. hispidus was observed as being excluded from the other two species with a relatively low support value (BS/PP = 56/1) in the IR data set. Our study only included one species of Halesia, and its systematic relationship needs to be further verified by increasing the sample size or combining with nuclear gene analysis. Perkinsiodendron and Rehderodendron form a clade in our all data sets, with Perkinsiodendron being established as a new genus from Halesia macgregorii Chun based on molecular data and morphological characters [22]. Furthermore, our study strongly supports the monophyly of Sinojackia based on plastid data, as has been detected in previous studies [26], except in the IR data set where Sinojackia sarcocarpa is separated from the other species (BS/PP = 71/1). The different topological structure of the IR data set may be the result of a slower mutation and evolution rate compared to that of the single copy region [69–71, 77]. There are many possible reasons for differences between data sets in inferring phylogenetic trees, including taxonomic sampling and biological factors such as hybridization/introgression, incomplete lineage sorting, gene duplication and/or loss, and horizontal gene transfer [78–80]. However, most of these reasons do not explain differences observed between different partitions of complete plastome sequences. The conflicting signal from different partitions of the chloroplast may be caused by homoplasy rather than hybridization [1].
Conclusions
Our results presented here utilize a phylogenomic data set to investigate phylogenetic relationships among the genera of Styracaceae. Based on 28 complete plastomes, our results show that the plastome structure of Styracaceae have small differences except for Alniphyllum and Bruinsmia, which have an approximately 20-kb inversion. Based on our almost complete species sampling for all genera except Styrax, all genera of Styracaceae are monophyletic, and the establishment of Perkinsiodendron and Changiostyrax are supported. Nevertheless, the lack of sequence data for species of Parastyrax necessitates that our results need to be further verified by increasing taxon sampling or population level sampling. With the increased sampling of taxa we can more effectively use the characteristics of faster evolving loci for phylogenetic inference [81, 82].
Availability of data and materials
All sequences used in this study are available from the National Center for Biotechnology Information (NCBI) (Accession Numbers: MT700470-MT700481; see Additional file 7: Table S1) .
Abbreviations
- BI:
-
Bayesian Inference
- CTAB:
-
Cetyltrimethylammonium bromide
- dN:
-
Nonsynonymous
- DnaSP:
-
DNA Sequences Polymorphism
- dS:
-
Synonymous
- IR:
-
Inverted repeat
- LSC:
-
Large single copy
- GTR:
-
General time reversible
- ML:
-
Maximum Likelihood
- PI:
-
Phylogenetic informativeness
- rRNA:
-
Ribosomal RNA
- SSC:
-
Small single copy
- tRNA:
-
Transfer RNA
References
Fritsch PW, Morton CM, Chen T, Meldrum C. Phylogeny and biogeography of the Styracaceae. Int J Plant Sci. 2001;162:S95–116.
Dickison WC. Floral anatomy of the Styracaceae, including observations on intraovarian trichomes. Bot J Linn Soc. 1993;112:223–55.
Dickison WC. A note on the wood anatomy of Dillenia (Dilleniaceae). IA WA Bull. 1979:2 3:57–60.
Dickison WC, Phend KD. Wood anatomy of the styracaceae: evolutionary and ecological considerations. IAWA J. 1985;6(1):3–22.
Morton CM, Dickison WC. Comparative pollen morphology of the Styracaceae. Grana 1992:31: 1–15. 19n. Odensc. ISSN 0017–3134
Dumortier B-C. Analyse des familles des plantes :avec l'indication des principaux genres quis'y rattachent (French Edition). Tournay Impr de J Casterman, Ainé, 1829.
Takhtajan A. Diversity and classification of flowering plants. New York: Columbia University Press; 1997.
Thorne RF. Classification and geography of the flowering plants. Bot Rev. 1992;58(3):225–348.
Voss EG. An integrated system of classification of flowering plants-Cronquist, A. Econ Botany. 1983;37(4):498–498.
Cronquist A. An integratcd system of classification of flowering plants. NewYork: CDlumbia Univ Pesrs; 1981. p. 492–506.
Copeland HF. The kingdom of organisms. Q R Biol. 1938;13:383.
APG. An ordinal classification for the families of flowering plants. Ann Mo Bot Gard. 1998:85(4): 531–553. https://doi.org/10.2307/2992015.
Bentham G, JD Hooker. Genera plantarum, vol 2. LovellReeve, London. 1279 pp. 1873.
Perkins J. Styracaceae. In: Engler A, ed. Pflanzenreich IV, 241 (Heft 30). Engelmann, Leipzig. 1907.
Thorne RF. The classification and geography of the flowering plants: dicotyledons of the class angiospermae (subclasses Magnoliidae, Ranunculidae, Caryophylliidae, Dilleniidae, Rosidae, Asteridae, and Lamiidae). Bot Rev. 2000;66:441–647.
Franceschi, D. de. Phylogenie des Ebenales: analyse de l'ordre et origine biogeographique des especes indiennes. Pub. Dept. Ecol. Institut Franais de Pondicherry. 1993:33: 1–153.
Hutehinson J. The genera of flowering plant. Oxford, 1967:2:34–39
Baas P. Anatomical contributions to plant taxonomy 2. The affinities of Hua Pierre and Afrostyrax Perkins et Gilg. Blumea 1972:20:161–192.
Wallnöfer B. A revision of Styrax L. section Pamphilia (Mart. ex A.DC.) B.Walln.(Styracaceae). Annalen des Naturhistorischen Museums in Wien. Serie B für Botanikund Zoologie, 1997:99B: 681–720.
Fritsch PW. Phylogeny of Styrax based on morphological characters, with implications for biogeography and infrageneric classification. Syst Bot. 1999;24(3):356–78.
Chen CT. Changiostyrax, a new genus of Styracaceae from China. Guihaia. 1995;15:289–92.
Fritsch PW, Yao X, Simison WB, Cruz BC, Chen T. Perkinsiodendron, a new genus in the Styracaceae based on morphology and DNA sequences. J Bot Res I Tex. 2016;10:109–17.
Morton CM, Chase MW, Swensen KAKM. A molecular evaluation of the monophyly of the order ebenales based upon rbcl sequence data. Syst Bot. 1996;21(4):567–86.
Olmstead RG, Kim KJ, Jansen RK, Wagstaff SJ. The phylogeny of the Asteridae sensu lato based on chloroplast ndhF gene sequences. Mol Phylogenet Evol. 2000;16:96–112.
Albach DC, Soltis PS, Soltis DE, Olmstead RG. Phylogenetic analysis of Asterids based on sequences of four genes. Ann Mo Bot Gard. 2001;88:163–212. https://doi.org/10.2307/2666224.
Yao XH, Ye Q, Fritsch PW, Cruz BC, Huang H. Phylogeny of Sinojackia (Styracaceae) based on DNA sequence and microsatellite data: implications for taxonomy and conservation. Ann Bot. 2008;101:651–9.
Yan M, Fritsch PW, Moore MJ, et al. Plastid phylogenomics resolves infrafamilial relationships of the Styracaceae and sheds light on the backbone relationships of the Ericales. Mol Phylogenet Evol. 2018;121:198–211.
Allen JF, de Paula WB, Puthiyaveetil S, Nield J. A structural phylogenetic map for chloroplast photosynthesis. Trends Plant Sci. 2011;16:645–55.
Carbonell-Caballero J, Alonso R, Iba VE, Terol J, Talon M, Dopazo J. A phylogenetic analysis of 34 chloroplast genomes elucidates the relationships between wild and domestic species within the genus Citrus. Mol Biol Evol. 2015;32:2015–35.
Hu SL, Sablok G, Wang B, Qu D, Barbaro E, Viola R, Li MA, Varotto C. Plastome organization and evolution of chloroplast genes in Cardamine species adapted to contrasting habitats. BMC Genom. 2015;16:306.
Shaw J, Shafer HL, Leonard OR, Kovach MJ, Schorr M, Morris AB. Chloroplast DNA sequence utility for the lowest phylogenetic and phylogeographic inferences in angiosperms: the tortoise and the hare IV. Am J Bot. 2014;101:1987–2004.
Moore MJ, Soltis PS, Bell CD, Burleigh JG, Soltis DE. Phylogenetic analysis of 83 plastid genes further resolves the early diversification of eudicots Proc. Natl Acad Sci USA. 2010;107:4623–8.
Jansen RK, Cai Z, Raubeson LA, Daniell H, Depamphilis CW, Leebensmack J, Müller KF, Guisinger-Bellian M, Haberle RC, Hansen AK, Chumley TW, Lee SB, Peery R, McNeal JR, Kuehl JV, Boore JL. Analysis of 81 genes from 64 plastomes resolves relationships in angiosperms and identifies genome-scale evolutionary patterns. Proc Natl Acad Sci USA. 2007;104:19369–74.
Barrett CF, Specht CD, Leebens-Mack J, Stevenson DW, Zomlefer WB, Davis JI. Resolving ancient radiations: can complete plastid gene sets elucidate deep relationships among the tropical gingers (Zingiberales)? Ann Bot. 2014;113:119–33.
Malé PG, Bardon L, Besnard G, Coissac E, Delsuc F, Engel J, Lhuillier E, Scotti-Saintagne C, Tinaut A, Chave J. Genome skimming by shotgun sequencing helps resolve the phylogeny of a pantropical tree family. Mol Ecol Res. 2014;14:966–75.
Yu XQ, Gao LM, Soltis DE, Soltis PS, Yang JB, Fang L, Yang SX, Li DZ. Insights into the historical assembly of East Asian subtropical evergreen broadleaved forests revealed by the temporal history of the tea family. New Phytol. 2017;215:1235–48.
Iv APG. An update of the Angiosperm Phylogeny Group classification for the orders and families of flowering plants: APG IV. Bot J Linn Soc. 2016;181:1–20. https://doi.org/10.1111/boj.12385.
Doyle JJ, Doyle JL. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochemical Bull. 1987;19:11–5.
Gao S, Mu F, Yang Z, Liu X, Jiang H, Liao S, et al. BGISEQ-500 WGS library construction. Protocols. 2018. https://doi.org/10.17504/protocols.io.ps5dng6.
Hahn C, Bachmann L, Chevreux B. Reconstructing mitochondrial genomes directly from genomic next-generation sequencing reads—a baiting and iterative mapping approach. Nucleic Acids Res. 2013;41:e129.
Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, Buxton S, Cooper A, Markowitz S, Duran C, Thierer T, Ashton B, Mentjies P, Drummond A. Geneious basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics. 2012;28:1647–9.
Wyman SK, Jansen RK, Boore JL. Automatic annotation of organellar genomes with DOGMA. Bioinformatics. 2004;20:3252–5.
Lohse M, Drechsel O, Kahlau S, Bock R. Organellar GenomeDRAW-a suite of tools for generating physical maps of plastid and mitochondrial genomes and visualizing expression data sets. Nucleic Acids Res. 2013;41:W575.
Frazer KA, Pachter L, Poliakov A, Rubin EM, Dubchak I. Vista: computational tools for comparative genomics. Nucleic Acids Res. 2004;32:273–9.
Katoh K, Standley DM. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 2013;30:772–80.
Librado P, Rozas J. DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics. 2009;25:1451–2.
Yang ZH. PAML 4: phylogenetic analysis by maximum likelihood. Mol Biol Evol. 2007;24:1586–91.
Posada D, Crandall KA. Modeltest: testing the model of DNA substitution. Bioinformatics. 1998;14(9):817–8.
Lanfear R, Frandsen PB, Wright AM, Senfeld T, Calcott B. PartitionFinder 2: new methods for selecting partitioned models of evolution for molecular and morphological phylogenetic analyses. Mol Biol Evol. 2016;34:772–3.
Stamatakis A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 2014;30:1312–3.
Miller MA, Pfeiffer W, Schwartz T. Creating the CIPRES Science Gateway for inference of large phylogenetic trees. Gateway Comput Environ Workshop (GCE). 2010;14:1–8.
Ronquist F, Huelsenbeck JP. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics. 2003;19:1572–4.
Rambaut A, Drummond AJ, Xie D, Baele G, Suchard MA. Posterior summarisation in Bayesian phylogenetics using Tracer 1.7. Syst Biol. 2018;67:901–4.
Jansen RK, Ruhlman TA, Plastomes of seed plants. Sharkey TD, eds. Genomics of chloroplasts and Mitochondria, Advances in Photosynthesis and Respiration. 2012; 35:103–126.
Yan MH, Moore MJ, Meng AP, Yao XH, Wang HC. The first complete plastome sequence of the basal asterid family Styracaceae (Ericales) reveals a large inversion. Plant Syst Evol. 2017;303:61–70. https://doi.org/10.1007/s00606-016-1352-0.
Liao Y, Liu Y, Liu X, et al. The complete chloroplast genome of Myriophyllum spicatum reveals a 4-kb inversion and new insights regarding plastome evolution in Haloragaceae. Ecol Evol. 2020;10(6):3090.
Tomohiko K, Takakazu K, Shusei S, et al. Complete structure of the chloroplast genome of a legume. Lotus japonicus Dna Res. 2000;6:6.
Lee HL, Jansen RK, Chumley TW, Kim KJ. Gene relocations within chloroplast genomes of Jasminum and Menodora (Oleaceae) are due to multiple, overlapping inversions. Molec Biol Evol. 2007;24:1161–80.
Jansen RK, Wojciechowski MF, Sanniyasi E, Lee SB, Daniell H. Complete plastid genome sequence of the chickpea (Cicer arietinum) and the phylogenetic distribution of rps12 and clpP intron losses among legumes (Leguminosae). Molec Phylogen Evol. 2008;48:1204–17. https://doi.org/10.1016/j.ympev.
Johansson JT. There large inversions in the chloroplast genomes and one loss of the chloroplast gene rps16 suggest an early evolutionary split in the genus Adonis (Ranunculaceae). Pl Syst Evol. 1999;218:133–43.
Palmer JD. Plastid chromosomes: structure and evolution. In: Vasil IK, Bogorad L, editors. Cell culture and somatic cell genetics in plants, vol. 7A. The molecular biology of plastids. San Diego: Academic Press. p. 1991:5–53.
Pombert JF, Lemieux C, Turmel M. The complete chloroplast DNA sequence of the green alga Oltmannsiellopsis viridis reveals a distinctive quadripartite architecture in the chloroplast genome of early diverging ulvophytes. BMC Biol. 2006;4:3.
Raubeson LA, Jansen RK. Chloroplast genomes of plants. In: Henry R, editor. Diversity and evolution of plants—genotypic and phenotypic variation in higher plants. Oxfordshire: CABI Publishing; 2005. p. 45–68.
Huang H, Shi C, Liu Y, et al. Thirteen Camellia chloroplast genome sequences determined by high-throughput sequencing: genome structure and phylogenetic relationships. BMC Evol Biol. 2014;14:151.
Bock R, Knoop V. Genomics of chloroplasts and mitochondria (advances in photosynthesis and respiration). Adv Photosynth Respir. 2012;35(3):377–377.
Perry AS, Wolfe KH. Nucleotide substitution rates in legume chloroplast DNA depend on the presence of the inverted repeat. J Mol Evol. 2002;55:501–8.
Shaw J, Lickey EB, Schilling EE, Small RL. Comparison of whole chloroplast genome sequences to choose noncoding regions for phylogenetic studies in angiosperms: the tortoise and the hare III. Am J Bot. 2007;94(3):275–88.
Shaw J, Small RL. Chloroplast dna phylogeny and phylogeography of the north american plums (prunus subgenus prunus section prunocerasus, rosaceae). Am J Botany. 2005;92:2011–30.
Curtis SE, Clegg MT. Molecular evolution of chloroplastDNA sequences. Mol Biol Evol. 1984;1:291–301.
Gaut BS. Molecular clocks and nucleotide substitution rates in higher plants. Evol Biol. 1998;30:93–120.
Wolfe KH, Li WH, Sharp PM. Rates of nucleotide substitution vary greatly among plant mitochondrial, chloroplast, and nuclear DNAs. Proc Natl Acad Sci USA. 1987;84:9054–8.
Weng ML, Blazier JC, Govindu M, et al. Reconstruction of the ancestral plastid genome in Geraniaceae reveals a correlation between genome rearrangements, repeats, and nucleotide substitution rates. Mol Biol Evol. 2014;31(3):645–59.
Sazanov LA, Burrows PA, Nixon PJ. The plastid ndh genes code for an NADH-specific dehydrogenase: Isolation of a complex I analogue from pea thylakoid membranes. Proc Natl Acad Sci. 1998;95(3):1319–24.
Fan X, Zhang J, Li W, Peng L. The NdhV subunit is required to stabilize the chloroplast NADH dehydrogenase-like complex in Arabidopsis. Plant J. 2015:82(2):221–31.
Liqiang W, Hui Z. Complete plastome sequence of Iodes cirrhosa Turcz the first in the Icacinaceae, comparative genomic analyses and possible split of Idoes species in response to climate changes. Peerj. 2019;7:e6663.
Bisson GP, et al. Upregulation of the phthiocerol dimycocerosate biosynthetic pathway by rifampin-resistant, rpoB mutant Mycobacterium tuberculosis. J Bacteriol. 2012;194:6441–52.
Wolfe, K. H. Protein-coding genes in chloroplast DNA:compilation of nucleotide sequences, data base entries, and rates of molecular evolution. In L. Bogorad and I. K. Vasil [eds.], Cell culture and somatic cell genetics of plants, vol. 1991:7B, 467–482.
Naciri Y, Linder P. Species delimitation and relationships: the dance of the seven veils. Taxon. 2015;64:3–16.
Nicola MV, Johnson LA, Pozner R. Unraveling patterns and processes of diversification in the South Andean-Patagonian Nassauvia subgenus Strongyloma (Asteraceae, Nassauvieae). Mol Phylogenet Evol. 2019;136:164–82.
Lin HY, Hao YJ, Li JH, Fu CX, Pamela SS, Douglas ES, Zhao YP. Phylogenomic conflict resulting from ancient introgression following 24 species diversification in Stewartia s.l. (Theaceae). Mol Phylogenet Evol. 2019;135:1–11.
Townsend JP, Leuenberger C. Taxon sampling and the optimal rates of evolution for phylogenetic inference. Syst Biol. 2011;60:358–65.
Townsend JP, Lopez-Giraldez F. Optimal selection of gene and ingroup taxon sampling for resolving phylogenetic relationships. Syst Biol. 2010;59:446–57.
Fan X, Zhang J, Li W, et al. The NdhV subunit is required to stabilize the chloroplast NADH dehydrogenase-like complex in Arabidopsis. Plant J. 2015;82(2):221–31.
Funding
This research was funded by National Natural Scientific Foundation of China (31660055 and 31660074) and by a start-up fund from Hainan University (kyqd1633).
Author information
Authors and Affiliations
Contributions
HW, XC, and JL designed this study; XC, HW and JH designed experiments, sequenced chloroplast genomes; ZZ analyzed the data; HW, XC, and JL drafted the manuscript; All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Fig. S1.
Bayesian inference (BI) and Maximum likelihood (ML) phylogram of Styracaceae based on LSC regions, with ambiguous sites excluded from analysis. The support values on the branches are bootstrap value/Bayesian posterior probability; “*”means 100%/1.0 support values. The genera of Styracaceae are indicated by different colors, which correspond to branch colors.
Additional file 2: Fig S2.
Bayesian inference (BI) and Maximum likelihood (ML) phylogram of Styracaceae based on SSC regions, with ambiguous sites excluded from analysis. The support values on the branches are bootstrap value/Bayesian posterior probability; “*”means 100%/1.0 support values. The genera of Styracaceae are indicated by different colors, which correspond to branch colors.
Additional file 3: Fig. S3.
Bayesian inference (BI) and Maximum like- lihood (ML) phylogram of Styracaceae based on IR regions, with ambiguous sites excluded from analysis. The support values on the branches are bootstrap value/Bayesian posterior probability; “*”means 100%/1.0 support values. The genera of Styracaceae are indicated by different colors, which correspond to branch colors.
Additional file 4: Fig. S4.
Bayesian inference (BI) and Maximum like-lihood (ML) phylogram of Styracaceae based on complete plastome sequences, with ambiguous sites excluded from analysis. The support values on the branches are bootstrap value/Bayesian posterior probability; “*”means 100%/1.0 support values. The genera of Styracaceae are indicated by different colors, which correspond to branch colors.
Additional file 5: Fig. S5.
Bayesian inference (BI) and Maximum likelihood (ML) phylogram of Styracaceae based on plastome LSC+SSC regions, with ambiguous sites excluded from analysis. The support values on the branches are bootstrap value/Bayesian posterior probability; “*”means 100%/1.0 support values. The genera of Styracaceae are indicated by different colors, which correspond to branch colors.
Additional file 6: Fig. S6.
Bayesian inference (BI) and Maximum likelihood (ML) phylogram of Styracaceae based on plastome noncoding regions, with ambiguous sites excluded from analysis. The support values on the branches are bootstrap value/Bayesian posterior probability; “*”means 100%/1.0 support values. The genera of Styracaceae are indicated by different colors, which correspond to branch colors.
Additional file 7: Table S1.
The results of partitionfinder models in the study.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Cite this article
Cai, XL., Landis, J.B., Wang, HX. et al. Plastome structure and phylogenetic relationships of Styracaceae (Ericales). BMC Ecol Evo 21, 103 (2021). https://doi.org/10.1186/s12862-021-01827-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12862-021-01827-4