
Abstract
Background
Primitive neuroectodermal tumor commonly occurs in bones and is equivalent to Ewing’s sarcoma. Very few cases have been reported in the literature and they had a very different presentation and very aggressive behavior.
Case presentation
We present a case of a young 23-year-old male who presented with complaints of pain in left lumbar region of abdomen since 8 months and hematuria off and on. CT scan of the abdomen was done which revealed a large heterogeneously enhancing mass lesion in the left lumbar region arising from the superior and mid-pole of left kidney showing multiple non-enhancing necrotic areas. A diagnosis of left renal cell carcinoma was kept. Through the transperitoneal approach, the left kidney was approached and a left radical nephrectomy was done. The histopathology report revealed the tumor cells to be positive for CD99 and focally positive for Vimentin and negative for cytokeratin thereby making a diagnosis of primary neuroectodermal tumor (PNET).
Conclusions
Renal PNET is a rare renal malignancy that should be kept in the differential diagnosis of a renal SOL especially when it is a presenting feature in adolescent and young adult. It has a very aggressive course and multimodal therapy has to be considered in its treatment
Similar content being viewed by others


1 Background
Primitive neuroectodermal tumor of kidney is very rare. The peripheral primitive neuroectodermal tumor (PNET) was first described by Stout in 1918 and was included in the family of small round cell tumors. It commonly occurs in bones and is equivalent to Ewing’s sarcoma microscopically, immunohistochemically, and genetically [1]. Very few cases have been reported in the literature, and they had a very different presentation and very aggressive behavior [2]. They are seen in relatively young population. The diagnosis of renal PNET is mainly based on histopathology and immunohistochemistry, supported by cytogenetic analysis; thus, preoperative diagnosis is challenging [3].
Kakkar et al. in 2014 have shown an interesting case of a 23-year-old male who came with neck pain and progressive quadriparesis and was diagnosed as a case of poorly differentiated malignant tumor with a differential of lymphoma versus metastatic renal cell carcinoma. Later on the patient's condition deteriorated fast and he had a rapid downhill course [4].
2 Case presentation
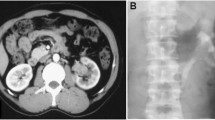
We present a case of a young 23-year-old male who presented to the surgery OPD with complaints of pain in left lumbar region of abdomen since 8 months. This pain was dull in nature, non-radiating and mostly confined to the left lumbar region. He also gave history of hematuria off and on since 1 month. This hematuria was associated with clots sometimes. He also had fever off and on. There was no history of other symptoms like malaise and weight loss. There was no history of any other major complaints or surgery in the past. His general examination did not reveal anything significant. His per abdominal examination revealed a lump in the left lumbar region which was approximately 8 cm below the costal margin and it was firm in consistency. The lump moved a little on respiration craniocaudally and it was non-tender. His genital examination was normal. Ultrasonography revealed two well-defined heterogeneously echotextured lesions predominantly hypoechoic in mid- and upper pole of left kidney with cystic changes within showing peripheral vascularity on color Doppler imaging causing anterior displacement of spleen. His blood biochemistry was normal and so was X-ray of the chest. CT scan of the abdomen was done which revealed a large heterogeneously enhancing mass lesion in the left lumbar region arising from the superior and mid-pole of left kidney showing multiple non-enhancing necrotic areas within with extensions with associated minimal fat stranding and lymphadenopathy. The right kidney was normal in function (Fig. 1).

CT scan of left renal PNET tumour
With this presumptive diagnosis of renal cell carcinoma, an open left radical nephrectomy was planned. With a left subcostal incision, through the transperitoneal approach the left kidney was approached and a left radical nephrectomy was done. Intra-operative and post-operative course was normal. The sample was formalin fixed and sent for histopathology. The histopathology report revealed the tumor cells to be positive for CD99 and focally positive for Vimentin and negative for cytokeratin. The cut surgical margins were free of tumor. A diagnosis of primary neuroectodermal tumor was made. These are shown in Figs. 2 and 3.

Solid nests and organoid pattern

Small-to-medium cells with eosinophilic cytoplasm, uniform and central nuclei, salts and pepper chromatin
Considering our rural setup in central part of subcontinent and keeping in mind financial background of patient, we did not go for additional confirmatory tests as FL-1/EWS. The patient was later referred to medical oncology department. On further enquiry, it has been come to our knowledge that following regimen was started consisting of cyclophosphamide, doxorubicin, vincristine, ifosamide, and etoposide. Five cycles of this regimen have already been given and patient is currently doing well.
3 Discussion
PNET was initially identified as member of small round tumors by Arthur Purdy Stout in 1918. It typically occurs in bone or soft tissue and rarely reported a renal localization [3]. The origin of the tumor is debatable but studies show that it is derived from cells that have migrated from the neural tube with variable capability of ectodermal or neuronal differentiation and these tumors can arise anywhere from brain to the sacrum. The male-to-female ratio is about 3:1. The presenting symptoms and clinical signs are non-specific and similar to those of the classical renal tumors [4]. Ct images of renal PNET are also without characteristic signs, commonly revealing solitary, large, ill-defined irregular or lobulated heterogenous mass. Hence, the diagnosis of PNET is histological. Treatment strategy of renal PNET is multimodal including surgery, chemotherapy, and radiotherapy. Owing to the difficulty in preoperative diagnosis, a neo-adjuvant chemotherapy before surgery is not applicable. In case of local invasion of perinephric tissue or positive surgical margins, post-operative radiotherapy is recommended [5].
PNET demonstrates characteristic immunoreactivity for CD99, typically in a diffuse and membranous pattern, and similar expression is observed in a minority of cases of desmoplastic small round cell tumors (DSRCT). CD99 is a cell surface glycoprotein involved in leukocyte migration, T cell adhesion, and transmembrane protein transport and also in T-cell death by a caspase-independent pathway.
Since this is a highly aggressive tumor, chances of metastatic spread are high. Hence, the overall prognosis of this tumor is poor with a 5-year survival of 45% to 55% [6].
Approximately, 90% of Ewing sarcoma (ES)/PNET have a specific t (11;22) which results in a chimeric EWS-FLI-1 fusion protein. Immunohistochemical for the carboxy-terminus of FLI-1 is sensitive and highly specific for the diagnosis of the ES/PNET [4].
4 Conclusion
In general, renal PNET is a rare renal malignancy that should be kept in the differential diagnosis of a renal space occupying lesion (SOL) especially when it is a presenting feature in adolescent and young adult. These are rare tumors with high chance of metastasis. This diagnosis is reached after histopathology. The treatment is multimodal in nature and patient should be offered chemotherapy/radiotherapy. Frequent examinations and metastatic workup should be done periodically. Despite all modalities, overall survival is dismal in these patients.
Availability of data and materials
Available.
Abbreviations
- PNET:
-
primary neuroectodermal tumor
- SOL:
-
renal space occupying lesion
- DSRCT:
-
desmoplastic small round cell tumors
References
Rashid A, Madan R, Singh L, Sharma DN (2012) Primitive neuroectodermal tumor of the kidney. Clin Cancer Investig J 1(3):148
Erkılıç S, Özsaraç C, Koçer NE et al (2006) Primary primitive neuroectodermal tumor of the kidney: a case report. Int Urol Nephrol 38:199–202. https://doi.org/10.1007/s11255-006-6675-2/
Naimi A, Soltan M, Saberi E, Riahinezhad M (2018) Renal primitive neuroectodermal tumor in a child with brain metastasis: a case report. Diagn Pathol 3(143):2476–3024
Kakkar S, Gupta D, Kaur G, Rana V (2014) Primary primitive neuroectodermal tumor of kidney: a rare case report with diagnostic challenge. Indian J Pathol Microbiol 57:298–300
Ellinger J, Bastian PJ, Hauser S, Biermann K, Müller SC (2006) Primitive neuroectodermal tumor: rare, highly aggressive differential diagnosis in urologic malignancies. Urology 68(2):257–262. https://doi.org/10.1016/j.urology.2006.02.037
Angel JR, Alfred A, Sakhuja A, Sells RE, Zechlinski JJ (2010) Ewing’s sarcoma of the kidney. Int J Clin Oncol 15(3):314–318
Acknowledgements
Authors wish to Thanks Dr Pooja Batra & Dr Pradeep Bokariya for their support.
Funding
Self-financed.
Author information
Authors and Affiliations
Contributions
PR: Concepts, design, Literature search, Data acquisition, Data analysis, Statistical analysis, Manuscript preparation and Manuscript editing. BR: Concepts, design, Definition of intellectual content, Literature search, Data analysis, Statistical analysis, Manuscript preparation, Manuscript editing, Manuscript review and Guarantor. DP: Definition of intellectual content, Literature search, Data acquisition, Manuscript preparation, Manuscript editing and Manuscript review. GD: Data acquisition, Data analysis and Statistical analysis. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Yes (Written consent from subject concerned).
Competing interests
None.
Data analysis and statistical analysis
All authors have read and approved the manuscript.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Pandey, R., Batra, R., Dhaigude, P. et al. Primitive neuroectodermal tumor of the kidney: a rare case. Afr J Urol 27, 45 (2021). https://doi.org/10.1186/s12301-021-00138-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12301-021-00138-w