
Abstract
Ewing sarcoma/primitive neuroectodermal tumor (ES/PNET) is a childhood malignancy, typically occurring in the bone and rarely in any other part of the body. We herein present a case of ES/PNET of the renal pelvis. A 37-year-old male patient presented with a chief complaint of pain in the left flank and gross hematuria. The tumor had caused moderate hydronephrosis, and ureteroscopic biopsy findings were highly suspicious of sarcoma. Subsequently, radical nephroureterectomy was performed. On the basis of the pathological and cytogenetic findings, a final diagnosis of primary ES/PNET of left renal pelvis was made. Adjuvant chemotherapy with adriamycin and ifosfamide was initiated as ES/PNET often exhibits aggressive biological behavior. The patient was disease-free at his last regular follow-up visit 18 months after the surgery. To our knowledge, this is the first reported case of primary ES/PNET of the renal pelvis.
Similar content being viewed by others


Background
Ewing sarcoma/primitive neuroectodermal tumor (ES/PNET) is the second most common form of bone malignancy (after osteosarcoma) occurring in childhood [1]. However, ES/PNET of extra-skeletal origin is an extremely rare entity. With the exception of ES/PNET arising from the renal parenchyma, ES/PNET of the renal pelvis has never been reported in the literature. To help establish the biological nature of primary ES/PNET of the renal pelvis, we present the clinical, diagnostic, and therapeutic aspects of this rare neoplasm along with a review of the literature.
Case presentation
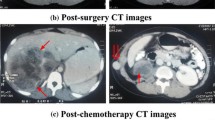
A 37-year-old male patient was admitted with a chief complaint of intermittent pain in the left plank and gross hematuria. His past medical history and findings of a physical examination were unremarkable. Results of his blood chemistry and routine blood tests were within the normal range. Urine cytology showed no signs of malignancy. Ultrasonography demonstrated a 4-cm hypoechoic mass occupying the dilated left renal pelvis, the presence of which was subsequently confirmed by enhanced computed tomography (CT) (Figure 1) and left retrograde pyelography. Cytology of the washout fluid obtained from the retrograde catheterization was also negative for malignancy. Further clinical investigations showed no evidence of metastasis. A preoperative ureteroscopic biopsy performed, with findings highly suspicious of sarcoma. The patient subsequently underwent open, left radical nephroureterectomy with excision of the bladder cuff. No intraoperative or postoperative complications developed.
Macroscopic examination showed that a 4.0 × 2.3 × 1.5 cm soft tissue mass was situated in the renal pelvis, with lobulated contours, a gray-white cut surface, and an intact renal parenchyma. Microscopic examination revealed a small round cell tumor with focal necrosis, infiltrating the smooth muscle layer of the renal pelvis and without renal parenchyma involvement. On immunohistochemistry, the tumor cells stained positive for CD99 (Figure 2), CD56, S-100, and vimentin; focally positive for epithelial membrane antigen and Ki-67; and negative for Bcl-2, cytokeratin 7, cytokeratin 20, desmin, WT-1, muscle specific actin, and Syn. On the basis of these findings, a pathological diagnosis of primary ES/PNET of the left renal pelvis was made. The diagnosis was further confirmed by fluorescence in situ hybridization (FISH) to detect the presence of EWS/FLI-1 fusion products. Adjuvant chemotherapy with adriamycin and ifosfamide was administered as ES/PNET often exhibit highly aggressive biological behavior. The patient was in excellent physical and mental condition without recurrence or metastasis as confirmed by careful follow-up at 18 months postoperatively.

Contrast-enhanced CT demonstrating a 4-cm soft-tissue mass occupying the dilated left renal pelvis. (A) Enhanced CT. (B) Left retrograde pyelography.

Immunohistochemistry showed intense positive membranous CD99 staining.
Conclusion
During the past two decades, nine cases of primary sarcoma of the renal pelvis have been described in the English literature: six of leiomyosarcoma, two of rhabdomyosarcoma, and one of cystic embryonal sarcoma (Table 1) [2–10]. To our knowledge, this is the first case of primary ES/PNET of the renal pelvis to be reported in the literature. Although being a rare entity, sarcoma should be included in the preoperative differential diagnosis of renal pelvis tumors. However, preoperative diagnosis of sarcoma is often difficult because of its rare incidence, non-specific clinical presentation, and radiological features (similar to those of urothelial carcinoma). Tsai et al. [8] and Chow et al. [9] reported the use of fine-needle aspiration biopsy with good results for primary renal pelvis sarcoma. Furthermore, for tumors of the upper urinary tract, ureteroscopic biopsy may be useful in establishing a more accurate preoperative diagnosis and deciding on the surgical modality, as was performed in the case reported here.
Histologically, lymphoblastic lymphoma, neuroblastoma, rhabdomyosarcoma, poorly differentiated synovial sarcoma, and Wilms tumor are included in the differential diagnosis of ES/PNET because they are all small round cell tumors. High levels of universal membranous CD99 expression are seen in ES/PNET cells, but this is not specific for ES/PNET. Thus, a broad immunohistochemistry panel may aid in differentiating these unique entities, even though this is sometimes difficult. Recently, molecular techniques have been increasingly used to confirm the diagnosis of ES/PNET, as it is one of the few solid tumors for which underlying chromosomal translocations have been described [11]. The most common translocation is the t(11, 22) (q24; q12), detected in more than 85% of ES/PNET by FISH or reverse transcription-polymerase chain reaction. Accurate diagnosis is essential, as these tumors require different treatment strategies.
As ES/PNET rarely spreads to lymph nodes, the single most important factor considered when planning initial treatment is the extent of disease at presentation, with the lungs being the most common site of metastasis. Initial presentation is the main factor that influences prognosis. The 5-year survival rate associated with extraskeletal ES/PNET in adults is 60% and 33% for non-metastatic and metastatic disease [1, 12], respectively. Surgical resection may be the treatment of choice for local control in ES/PNET. However, ES/PNET often results in rapid recurrence or metastasis, even after complete resection. Therefore, patients with ES/PNET may require multidisciplinary management, combining systemic neoadjuvant and/or adjuvant chemotherapy with local control measures (surgery and/or radiation). Ifosfamide-based protocols appear to confer a survival advantage in ES/PNET patients [12, 13]. Novel molecular-targeted therapies for the treatment of ES/PNET have now transitioned from the laboratory to the clinical setting. In a recent phase I trial of a fully human IgG2 monoclonal antibody targeting the insulin-like growth-factor-1 receptor, figitumumab, a number of patients with refractory, advanced ES/PNET responded unexpectedly well [14]. Such patients should be informed about relevant clinical trials and encouraged to enter these trials. Due to the rarity of this condition, the impact of different treatment modalities (simple nephrectomy vs. radical nephrectomy vs. radical nephroureterectomy with or without chemotherapy and/or radiotherapy) on outcomes cannot be evaluated until more cases have been reported.
References
Balamuth NJ, Womer RB: Ewing’s sarcoma. Lancet Oncol. 2010, 11: 184-192. 10.1016/S1470-2045(09)70286-4.
Dhamne SA, Gadgil NM, Padmanabhan A: Leiomyosarcoma of the renal pelvis. Indian J Pathol Microbiol. 2009, 52: 549-551. 10.4103/0377-4929.56161.
Chung YG, Kang SC, Yoon SM, Han JY, Seong DH: Leiomyosarcoma arising from the blind end of a bifid renal pelvis. Yonsei Med J. 2007, 48: 557-560. 10.3349/ymj.2007.48.3.557.
Kartsanis G, Douros K, Zolota V, Perimenis P: Case report: leiomyosarcoma of the renal pelvis. Int Urol Nephrol. 2006, 38: 211-213. 10.1007/s11255-006-0019-0.
Minami H, Ueki O, Tanaka T, Nishida H, Hashimoto T, Kawaguchi K: Case of leiomyosarcoma of the renal pelvis. Int J Urol. 2004, 11: 122-124. 10.1111/j.1442-2042.2004.00747.x.
Moudouni SM, En-Nia I, Rioux-Leclerq N, Guille F, Lobel B: Leiomyosarcoma of the renal pelvis. Scand J Urol Nephrol. 2001, 35: 425-427. 10.1080/003655901753224530.
Ito J, Monobe Y, Sakamoto K, Tanaka H: Embryonal sarcoma of adult and pediatric kidneys: report of a case with localized submucosal invasion of the renal pelvis and long-term survival. Pathol Int. 2000, 50: 832-838. 10.1046/j.1440-1827.2000.01113.x.
Tsai WC, Lee SS, Cheng MF, Lee HS: Botryoid-type pleomorphic rhabdomyosarcoma of the renal pelvis in an adult. A rare case report and review of the literature. Urol Int. 2006, 77: 89-91. 10.1159/000092943.
Chow LT, Chan SK, Chow WH: Fine needle aspiration cytodiagnosis of leiomyosarcoma of the renal pelvis. A case report with immunohistochemical study. Acta Cytol. 1994, 38: 759-763.
Kren L, Goncharuk VN, Votava M, Hermanova M, Ross JS, Nazeer T, Dvorak K: Botryoid-type of embryonal rhabdomyosarcoma of renal pelvis in an adult. A case report and review of the literature. Cesk Patol. 2003, 39: 31-35.
Delattre O, Zucman J, Plougastel B, Desmaze C, Melot T, Peter M, Kovar H, Joubert I, de Jong P, Rouleau G, Aurias A, Thomas G: Gene fusion with an ETS DNA-binding domain caused by chromosome translocation in human tumours. Nature. 1992, 359: 162-165. 10.1038/359162a0.
Iwamoto Y: Diagnosis and treatment of Ewing’s sarcoma. Jpn J Clin Oncol. 2007, 37: 79-89. 10.1093/jjco/hyl142.
Grier HE, Krailo MD, Tarbell NJ, Link MP, Fryer CJ, Pritchard DJ, Gebhardt MC, Dickman PS, Perlman EJ, Meyers PA, Donaldson SS, Moore S, Rausen AR, Vietti TJ, Miser JS: Addition of ifosfamide and etoposide to standard chemotherapy for Ewing’s sarcoma and primitive neuroectodermal tumor of bone. N Engl J Med. 2003, 348: 694-701. 10.1056/NEJMoa020890.
Olmos D, Postel-Vinay S, Molife LR, Okuno SH, Schuetze SM, Paccagnella ML, Batzel GN, Yin D, Pritchard-Jones K, Judson I, Worden FP, Gualberto A, Scurr M, de Bono JS, Haluska P: Safety, pharmacokinetics, and preliminary activity of the anti-IGF-1R antibody figitumumab (CP-751,871) in patients with sarcoma and Ewing’s sarcoma: a phase 1 expansion cohort study. Lancet Oncol. 2010, 11: 129-135. 10.1016/S1470-2045(09)70354-7.
Acknowledgments
Due to a limit on the number of references, many primary papers could not be cited. We apologize for this.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
YL designed the research project. ZL, XW, YL, and LC prepared the manuscript and collected the documents. All authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article

Cite this article
Liu, Z., Wang, X., Lu, Y. et al. Primary Ewing sarcoma/primitive neuroectodermal tumor of the renal pelvis: a case report. World J Surg Onc 12, 293 (2014). https://doi.org/10.1186/1477-7819-12-293
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1477-7819-12-293