
Abstract
Background
Primitive neuroectodermal tumors (PNET) are highly aggressive tumors belonging to the Ewing’s sarcoma family. Primitive neuroectodermal tumor is a group of bone and soft tissue tumor arising from neuroectodermal tissues. They are characterized by the expression of MIC2 and neural markers like neuron-specific enolase, synaptophysin, S-100, vimentin, Leu-7, and the presence of the EWS-FLI1 translocation. It is a rare malignancy with only about 150 cases being published worldwide. The tumor usually presents with non-specific symptoms such as abdomen pain and hematuria. Inferior vena cava (IVC) involvement by renal PNET is extremely rare with only a handful cases reported in the literature and is a clinically challenging scenario. There is a dearth of data on the management of this disease; further, the management of this disease in current scenarios is not well reported. We report our experience with three cases over 11 years which spans all three spectrums of the disease: localized, locally advance and metastasis.
Case presentation
Case 1: 37-year-old gentleman presenting with hematuria found to have right renal tumor with IVC invasion which was found to be unresectable intraoperatively. Patient was started on chemotherapy but deteriorated and died 3 months later. Case 2: 38-year-old gentleman presenting with abdominal pain found to have locally advanced left renal tumor. Patient developed local recurrence and metastasis 15 months after resection but had stable disease for nearly 4 years before developing acute myeloid leukemia and withdrawing consent for treatment. Case 3: 13-year-old girl who presented with right abdominal mass and subsequently found to have localized disease. After aggressive chemotherapy, patient is now recurrence-free after nearly 11 years.
Conclusions
Our study aims to show the spectrum of renal primitive neuroectodermal tumors with outcomes in all three clinical scenarios, with the aim of reinforcing that high index of suspicion for this disease is required in young patients presenting with aggressive disease, and immunohistochemistry should be used when in doubt, as outcomes are greatly improved with adjuvant chemotherapy.
Similar content being viewed by others


1 Background
Primitive neuroectodermal tumors (PNET) are small round cell tumors belonging to the Ewing’s Sarcoma family which arise from the central nervous system and in peripheral tissues. Renal presentation of PNET is a rare phenomenon, with about 150 cases reported in the literature, with even fewer being reported with vascular involvement. PNETs usually occur in young adults most often between 20 and 30 years. The male/female ratio is about 3:1 [1]. The average age for those affected is about 26 years, with 38% of all patients with the tumor between the ages of 4 and 69 [2]. Most patients present with non-specific symptoms, such as abdominal pain or hematuria. These tumors are aggressive in nature, with early metastasis and poor prognosis. The mainstay of treatment is surgery followed by chemotherapy and radiotherapy, or chemotherapy in the neoadjuvant setting, which may improve outcome. Even with a multi-modality treatment, the prognosis is poor with 5 year survival of 45–50% and a recurrence rate of 80% in those treated with conservative methods [3]. There is a paucity of data on the topic and also a lack of outcomes in the present era of intensive chemotherapy regimens. We report our experience with the disease which spans the spectrum from localized to metastatic disease.
2 Case presentation
2.1 Case 1
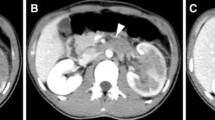
37-year-old gentleman presented with history of total, painless, gross hematuria with clots since the last 2 months. There was no history of pedal edema or abdominal distenstion and no significant medical/surgical/family history. The patient had no history of smoking/tobacco intake and alcohol consumption was occasional. Patient was evaluated with ultrasonogram (USG) abdomen at a local hospital which showed a large right renal mass. Patient subsequently underwent contrast-enhanced computed tomography (CECT) abdomen which showed a right renal mass of 9.3 × 7.4 cm with infiltration into renal vein and infra-hepatic inferior vena cava (IVC) proximally and the right external iliac vein distally. A magnetic resonance imaging (MRI) abdomen was performed which showed infiltration into bilateral external iliac veins, which was confirmed with color Doppler. Due to significant vascular involvement, cardiothoracic team was involved in the surgery. The patient underwent an open radical nephrectomy with IVC thrombectomy and IVC repair with paracaval node excision. Intraoperatively, there was a large tumor thrombus involving the right renal vein, suprarenal IVC extending up to the retro hepatic IVC for about 7 cm till the hepatic vein-IVC junction, along with a dense adherent bland thrombus extending up to the femoral veins, as a result of which tumor thrombectomy could not be performed. The IVC was ligated as there were collateral vessels and the patient started on anticoagulation postoperatively. The tumor was grossly 9.6 × 8.7x8.6 cm in size, arising from the upper pole. Microscopically, hematoxylin & eosin (H&E) staining revealed atypical small round cells with hyperchromatic nuclei and irregular nuclear membranes. There were multiple areas of apoptotic debris and necrosis with frequent mitosis and extensive lymphovascular emboli. Further confirmation was sought with immunohistochemical analysis which however positivity for CD99, FLI1 and vimentin with Ki-67 of 22%. Postoperatively, the patient was initiated on anticoagulation to reduce embolization risk. The patient was started on chemotherapy but had clinical deterioration after the first cycle and discontinued treatment. The patient died 3 months later with and overall survival of 5 months after diagnosis (Fig. 1).

T2 HASTE MRI images showing A the right upper polar tumor and the tumor thrombus in the IVC B the thrombus in the IVC and the distal extent in the iliac vein
2.2 Case 2
38-year-old gentleman presented with history of vague abdominal discomfort since 1 month. On evaluation with CECT, kidney ureter bladder (KUB) was found to have left renal mass of 10 × 6.3 cm with invasion of perihilar fat. Patient underwent open radical nephrectomy. Histopathology showed pT3b PNET. Patient was started on adjuvant chemotherapy with doxorubicin, vincristine and cyclophosphamide. Patient was recurrence-free for 15 months, following which he developed local recurrence with pulmonary metastasis. Patient subsequently underwent palliative chemotherapy with ifosfamide and etoposide plus radiotherapy to the renal bed. Patient had stable disease for nearly 5 years when he developed pyrexia of unknown origin (PUO) and was found to have developed acute myeloid leukemia with acute kidney injury. Patient attenders withdrew consent for further management, and patient died 1 month later, with an overall survival of 6.8 years (Fig. 2).

A and B Hematoxylin and eosin stained tissue with 10 × and 40 × magnification, C CD99-tumor cells with membranous positivity, D FLI-1-Tumor cells with nuclear positivity
2.3 Case 3
13-year-old girl was brought by parents with history of abdominal mass, noticed 1 week back. On evaluation was found to have a right renal mass about 11 × 5.4 cm. Patient underwent radical nephrectomy 1 week later, with histopathology showing pT2 PNET. Patient completed six cycles of chemotherapy with ifosfamide, vincristine and etoposide. Patient is symptom-free and found to be free of recurrence 11 years from diagnosis.
3 Discussion
Primitive neuroectodermal tumors were first described by Stout in 1918 [4]. A rare malignancy of the neuroectoderm, they are fast-growing small round cell neoplasms, occurring very rarely in the kidney. PNETs account for less than 1% of sarcomas. PNETs are now classified as part of the Ewing sarcoma family.
The first description of renal PNET was given by Seemayer et al. in 1975 [5]. The tumor is highly malignant and grows rapidly, with early metastases to lung, bone, and lymph nodes. This aggressive behavior is responsible for the poor prognosis of the disease. Usual presentation is in the younger age group. All patients in our series were younger than 40 years, which is consistent with the finding.
Contrast-enhanced computed tomography (CECT) is used for staging. Magnetic resonance imaging can be used for imaging vascular invasion, if suspected. On CECT, PNETs are typically presented as a large heterogeneous mass with central low-density areas due to necrosis or hemorrhage. Areas of calcification may be noted. Tumor extension into the renal vein, IVC, right atrium have been described in few reports, although we believe this is the first reported case with such extensive distal IVC invasion. There are at least two reports of psoas muscle invasion. Hepatic metastases, when present, are manifested as low attenuation lesions. [2, 6, 7]
The definite diagnosis of renal ES/PNET is based on histopathology and immunohistochemistry of resected specimens. Ewing’s sarcoma/PNET are a part of the small round cell tumor group. These tumors are composed of sheets of small cells with high nuclear to cytoplasmic ratio. The cytoplasm is scant, eosinophilic, and usually contains glycogen. The nuclei are round, with finely dispersed chromatin, and one or more tiny nucleoli. This tumor frequently undergoes necrosis, and areas of hemorrhage are evident. Cells show membranous expression of CD99 or MIC2 on immunohistochemistry. Antibody against FLI1 present in the nucleus is specific for PNET. Neuron-specific enolase (NSE), synaptophysin, and S-100 protein may also be expressed by the tumor. IHC report was positive for CD99, FLI 1 and vimentin for all the cases, which is consistent with the findings in other studies [8].
The usual presentation is with symptoms of abdomen pain, hematuria or flank mass, all three of which were present in our case series. PNETs are known for their aggressive course. They invade local tissues early and metastasise to lung, bone and liver. As a result, the prognosis of the disease is poor, with a 5 year survival of 45–50%, which holds true for our experience. Stage at presentation is the most important prognostic factor. As noted in our series, localized tumor treated with radical resection and aggressive chemotherapy has a good outcome, with metastatic disease having a survival less than 6 months.
The usual treatment is radical excision followed by aggressive chemotherapy regimens. Vincristine(V), dactinomycin(D), adriamycin(A), cyclophosphamide(C), ifosfamide(I), and etoposide(E) have been found to be effective against the tumor. Our patients received VDAC in the initial course which was followed by IE. The current chemotherapeutic management of this disease uses this combination regimen. [9]. Radiotherapy has been reported for locally advanced disease or when there is involvement of Gerota’s fascia and was administered to the patient with local recurrence [7].
We acknowledge that our experience is limited by the number of cases and the time period over which they were distributed.
4 Conclusions
The most common renal pathology presenting with inferior vena cava invasion is clear cell renal carcinoma, however, renal PNET should be considered as a differential in young patients presenting with an aggressive renal mass. The histopathological picture is characteristic which can be further aided with immunohistochemistry. Although the outcome of the patients with advanced disease is better after biopsy and neoadjuvant chemotherapy, due to the rarity of this tumor, we cannot recommend change of standard practice of radical nephrectomy (RN). RN is curative in localized disease, with good prognosis, however, advanced disease requires multimodal therapy and has a poor survival.
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- PNET:
-
Primitive ectodermal tumor
- IVC:
-
Inferior vena cava
- USG:
-
Ultrasonography
- CECT:
-
Contrast-enhanced computed tomography
- KUB:
-
Kidney ureter bladder
- PUO:
-
Pyrexia of unknown origin
References
Abolhasani M, Salarinejad S, Moslemi MK (2016) Ewing sarcoma/primitive neuroectodermal tumor of the kidney: a report of three cases. Int J Surg Case Rep 28:330–334
Ellinger J, Bastian PJ, Hauser S et al (2006) Primitive neuroectodermal tumor: rare, highly aggressive differential diagnosis in urologic malignancies. Urology 68:257–262
Casella R, Moch H, Rochlitz C et al (2001) Metastatic primitive neuroectodermal tumor of the kidney in adults. Eur Urol 39:613–617
Stout AP (1918) A tumor of the ulner nerve. Proc NY Pathol Soc 18:2–12
Seemayer TA, Thelmo WL, Bolande RP et al (1975) Peripheral neuroectodermal tumors. Perspect Pediatr Pathol 2:151–172
Ng AW, Lee PS, Howard RG (2004) Primitive neuroectodermal kidney tumour. Australas Radiol 48:211–213
Doerfler O, Reittner P, Groell R et al (2001) Peripheral primitive neuroectodermal tumour of the kidney: CT findings. Pediatr Radiol 31:117–119
Desai SS, Jambhekar NA (2010) Pathology of Ewing’s sarcoma/PNET: current opinion and emerging concepts. Indian J Orthop 44(4):363–368
Zhang S, Li Y, Wang R, Song B (2019) Ewing’s sarcoma/primitive neuroectodermal tumor of the kidney: a case report and literature review. Transl Androl Urol 8(5):562–566
Acknowledgements
None.
Funding
None.
Author information
Authors and Affiliations
Contributions
AS is the assistant surgeon for case 1, principal author and contributed to data collection and compilation. NK contributed to data analysis and is the operating surgeon for case 1. MA contributed to data analysis and is the operating surgeon for case 1. DM contributed to data acquisition and is the operating surgeon for case 3. BK contributed to data analysis and is the operating surgeon for case 2. The manuscript has been read and approved by all the contributing authors and the criteria for contribution have been met.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Approval has been obtained from the Institutional Ethics Committee of Kovai Medical Center and Hospital, Coimbatore, Tamil Nadu, India, vide number EC/AP/928/02/2022, dated 25/02/2022. Consent to participate was obtained from the patients.
Consent for publication
Consent for publication was obtained from the patient/next of kin.
Competing interests
The authors declare that they have no competing interests.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Sharma, A., Kuppurajan, N., Anandan, M. et al. Primitive neuroectodermal tumor of the kidney: an experience of three cases. Afr J Urol 28, 37 (2022). https://doi.org/10.1186/s12301-022-00304-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12301-022-00304-8