
Abstract
Reversible protein phosphorylation represents a key mechanism by which signals are transduced in eukaryotic cells. Dysregulated phosphorylation is also a hallmark of carcinogenesis and represents key drug targets in the precision medicine space. Thus, methods that preserve phosphoprotein integrity in the context of clinical tissue analyses are crucially important in cancer research. Here we investigated the impact of UV laser microdissection (UV LMD) and IR laser capture microdissection (IR LCM) on phosphoprotein abundance of key cancer signaling protein targets assessed by reverse-phase protein microarray (RPPA). Tumor epithelial cells from consecutive thin sections obtained from four high-grade serous ovarian cancers were harvested using either UV LMD or IR LCM methods. Phosphoprotein abundances for ten phosphoproteins that represent important drug targets were assessed by RPPA and revealed no significant differences in phosphoprotein integrity from those obtained using higher-energy UV versus the lower-energy IR laser methods.
Similar content being viewed by others


Background
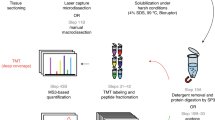
Laser microdissection (LMD) and laser capture microdissection (LCM) enable target populations of cells to be selectively harvested from heterogeneous admixtures of cells in the tissue microenvironment (TME), either from histological fresh-frozen or formalin-fixed paraffin-embedded (FFPE) tissue thin sections [1,2,3]. The ability to harvest and enrich homogenous cell populations and/or remove undesired tissue regions using LMD is broadly enabling to numerous analytical workflows for biomedical research. One such application is for cancer research as the cancer TME comprises a highly varied admixture of non-tumor cells, including fibroblasts, infiltrating lymphocytes, macrophages, along with fibrotic and/or necrotic regions. Thus, it is becoming increasingly evident that biomolecular analyses of each of these cell populations is important to make new strides in our understanding of this complex cellular ecosystem. This workflow is likely to lead to new advances in multi-omic analyses of cancer tissues, including proteomics, particularly since analysis of proteins extracted from whole tissue specimens limits detection of disease-related proteins due to the heterogeneous mixture of tumor with non-tumor cells [4,5,6,7,8,9,10]. Studies employing LMD as part of pre-analytical preparation of samples have identified cancer-associated molecular changes and prognostic biomarkers for many cancers including ovarian cancer [11,12,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28], cervical cancer [29,30,31], vulvar cancer [32, 33], and uterine cancer [34,35,36,37]. Many cancer signaling pathways regulating cell growth, proliferation, differentiation, and metastasis are mediated by the concerted actions of kinases and phosphatases and are frequently disrupted or dysregulated in cancer (reviewed in [38,39,40]). It is therefore critically important that pre-analytical preparation of specimens for phosphoproteomic analysis does not disrupt this labile post-translational modification, including collection of tissue samples by LMD. In practice, the two general laser microscopy platforms differ mechanistically in the way cells are procured, namely via low energy infrared (IR) contact laser-capture microdissection (LCM) [1, 2] or by high energy ultraviolet (UV) cutting LMD [3, 41,42,43,44].
The IR LCM and UV LMD platforms each rely on light microscopy to visualize and identify target cell populations for harvest from tissue sections. The IR LCM platform uses a thermolabile polymer film containing ethylene vinyl acetate impregnated with a dye that absorbs light at near-IR wavelengths which is brought into physical contact with the tissue section on a glass slide [1, 2]. The film is situated in a plastic support cap that optically focuses the laser in the same plane as the tissue section. An IR laser heats the film where it contacts the selected target regions; cells in contact with the heated polymer attach and are sheared from the remaining tissue. An extraction buffer is used to free the embedded cells from the polymer for subsequent molecular analysis. In contrast, the UV platform employs polyethylene napthalate (PEN), polyethylene tetraphthalate (PET) or polyphenylene sulfide (PPS) [3, 41, 42] membrane slides onto which thin tissue sections are cut. Target cells are harvested through the action of a UV laser that cuts the selected membrane and tissue elements that are collected by gravity into a collection vessel located below the slide or are catapulted by a laser pulse into a cap above the slide.
In the case of IR LCM, the dye within the film absorbs IR light to reduce damage to cellular components. Additionally, the IR laser typically used is low in energy and instantaneously applied in a pulsed fashion to further minimize IR-induced cell damage [1]. However, because the harvested tissue is effectively “melted” onto the LCM membrane, efficient cellular lysis and recovery of biomolecules requires harsh detergents that can be problematic for some downstream chemical manipulations and/or analytical applications, such as mass spectrometry. UV LMD harvests represent substantial versatility because tissue recovery does not involve melting of a membrane to the tissue. Some concern remains, however, regarding whether UV LMD provides a comparably suitable acquisition technique for analyzing labile macromolecules such as phosphoproteins in histologically prepared samples because of the potential for higher energy UV exposure to cells and molecules in direct field proximity to the laser. Analysis of clinical samples by IR LCM coupled to reverse-phase protein array (RPPA) has generated highly accurate and reliable phosphoprotein data from FFPE and frozen tissues [6, 8, 27, 45,46,47,48]. Conversely, staining and routine sample processing following LMD (aside from microdissection itself) have been shown to negatively impact a variety of molecular analyses, including phosphoprotein levels in frozen tissues [49], as well as RNA integrity [50, 51] and the availability and/or resolution of proteins and phosphoproteins [45, 52,53,54,55,56]. UV LMD has recently been proposed as faster and more precise than IR LCM as a preparatory tool for certain types of molecular analyses [57].
This study aimed to assess whether there is a measurable difference between UV- and IR-mediated laser microdissection on phosphoprotein integrity by analyzing key cancer phosphoprotein abundances by RPPA from four high grade serous epithelial ovarian carcinoma (HGSOC) specimens. Our results demonstrate that there is no significant difference between UV- and IR-mediated laser microscopy on phosphoprotein integrity.
Materials and methods
Tissue specimens
Snap-frozen tissue specimens were obtained from four ovarian high grade serous ovarian cancer (HGSOC) patients within 30 min of surgical resection under an IRB exempt protocol (Western IRB approved). Two consecutive thin tissue sections (10 µm) embedded in optimal cutting temperature (OCT) medium (Fisher Scientific) were cut by microtome from each patient specimen. One section from each specimen was placed on PEN membrane slides (Leica Microsystems) for UV LMD and the other was placed on uncharged glass slides (Premium Glass Microscope Slides, Daigger) for IR LCM. Tissue sections were imaged before and after UV LMD or IR LCM using an Aperio AT2 digital whole slide scanner (Leica).
Histologic staining
Slides were thawed and stained immediately prior to laser microscopy harvest. One glass and one PEN membrane slide prepared from consecutive tissue slices were stained for IR LCM and UV LMD, respectively for downstream RPPA analysis. Slides were fixed in 70% ethanol, rehydrated in deionized water, stained using Mayer’s Hematoxylin, and rinsed using deionized water and Scott’s Tap Water (Thermo Fisher Scientific). After staining and color development, slides were dehydrated in graded ethanol washes with two final rinses in xylene. Protease inhibitors (Roche) were added to all solutions except for the 100% ethanol and xylene washes.
Laser microdissection
IR LCM and UV LMD were performed independently on one consecutive tissue section each from each patient. UV LMD was performed on a LMD7 (Leica Microsystems) and IR LCM was performed on a PixCell II system (Arcturus). Two mm2 of tumor epithelium (yielding a final diluted protein concentration of approximately 0.25 µg/µl) from each slide was microdissected within 30 min for RPPA. A separate Hematoxylin and Eosin (H&E)-stained slide was used as a reference to map tissue zones for tumor epithelium collection by both LMD techniques (Fig. 1).

Micrographs of HGSOC tissue sections before and after tumor epithelial cell harvest by laser microdissection. A representative H&E-stained section for each patient (n = 4) is shown at 0.3× (1st column) and 5× (2nd column) magnification. Tumor epithelium from tissue thin sections (10 µm) was enriched via ultraviolet laser microdissection (UV LMD) or infrared laser capture microdissection (IR LCM). Representative areas following microdissection of tumor epithelium are shown at 0.3× magnification (3rd column); the images shown are post-UV LMD
For UV LMD, the 2 mm2 of tumor epithelium was collected into a dry tube to which the extraction/lysis buffer consisting of a 1:1 mixture of Tissue Protein Extraction Reagent (T-PER; Pierce), Novex 2× Tris–Glycine SDS Sample Buffer (Invitrogen), and 2.5% v/v 2-mercaptoethanol was added. Collected tissue was briefly centrifuged and frozen at − 80 °C. For IR LCM, protein extraction was performed as previously described [46]. The LCM caps were visually examined for tissue debris or non-specific tissue adhesion, which was removed by blotting the cap with a CapSure cleanup pad (Arcturus). In brief, LCM caps and LMD tubes were similarly incubated with extraction buffer, cell lysates were then collected and boiled for 10 min before storage at − 80 °C.
Reverse phase protein microarray
After thawing, all lysates for RPPA analysis were heated at 100 °C for 2 min in a dry heat block, cooled to ambient temperature, centrifuged, and used for printing microarrays. Using a 2470 Aushon Arrayer (Aushon BioSystems), samples were immobilized onto nitrocellulose-coated glass slides (Grace Biolabs) in technical triplicates as previously described [46]. Selected arrays were stained with Sypro Ruby Protein Blot Stain (Molecular Probes), according to manufacturer’s instructions to estimate the total amount of protein in each sample [46].
Before immunostaining, remaining slides were treated with Reblot Plus Mild Antibody stripping solution (Chemicon) for 15 min at room temperature, washed twice with PBS, and incubated in I-block solution (Tropix) for at least 4 h. Immunostaining was performed on an automated system (Dako) and each array was probed with one antibody targeting a protein of interest. Samples were probed with a total of ten antibodies targeting the phosphorylated forms of Akt S473 (Cell Signaling catalog #9271; 1:100), c-Abl T735 (Cell Signaling catalog #2864; 1:50), EGFR Y1068 (Cell Signaling catalog #2234; 1:50), HER2 Y1248 (Imgenex catalog #90189-1; 1:500), HER3 Y1289 (Cell Signaling catalog #4791; 1:200), ERK1/2 T202/Y204 (Cell Signaling catalog #9101; 1:1000), p70S6K T389 (Cell Signaling catalog #9205; 1:100), PDGFR Y751 (Cell Signaling catalog #3161; 1:50), Rb S780 (Cell Signaling catalog #3590; 1:2000), and RET Y905 (Cell Signaling catalog #3221; 1:100). Antibody specificity and linear dynamic range were previously tested [58]. Samples were then incubated with a secondary biotinylated goat anti-rabbit (Vector Laboratories; 1:7500) and with the commercially available tyramide-based Catalyzed Signal Amplification System (CSA, Dako) coupled with a fluorescent streptavidin-conjugated IRDye680 dye (LI-COR Biosciences). One slide was probed with secondary antibody only and used as a negative control for normalization purposes.
Stained arrays were scanned with a laser PowerScanner (TECAN) using the appropriate wavelength channel. Image analysis was performed using a commercially available software (MicroVigene v5.1.0.0, VigeneTech, Inc.). The software automatically performs spot finding, subtraction of local background and of nonspecific signal collected through the negative control slide(s). Samples were then normalized to the amount of protein and averaged across replicates.
Results
Tumor epithelial cells were harvested using UV LMD or IR LCM from consecutive snap-frozen HGSOC patient tumor tissue thin sections (n = 4) (Fig. 1). The levels of ten key phosphoproteins involved in PI3K/AKT/mTOR signal transduction and related pathways which are frequently activated in ovarian cancer [59, 60] were analyzed using a standardized analytical panel of antibodies [58] (Additional file 1: Table S1). Comparative analyses revealed that the abundance level of all phosphoproteins remained consistent between the UV- and IR-mediated harvests (Fig. 2). A non-parametric Mann–Whitney-based comparison between matched UV LMD and IR LCM indicated that rank orders were not statistically different for the measured phosphoproteins, with the exception of pRET Y905 (p = 0.0286, Table 1). Pearson and Spearman correlations among all phosphoprotein abundances measured per patient confirmed high concordance between samples microdissected by both techniques (Fig. 3, Table 2). The Pearson r values were 0.9996 (p = 8.647 × 10−13), 0.9393 (p = 2.838 × 10−07), 0.8459 (p = 6.213 × 10−05), and 0.9847 (p = 3.423 × 10−09) for Patients 1–4, respectively. Similarly, Spearman’s rho (ρ) values showed high correlation and were 0.9890, 0.9941, 0.9934, and 0.9216 for Patients 1–4, respectively.

Phosphoprotein abundances in laser microdissected ovarian cancer tumor epithelial cells assessed by reverse-phase protein array. ERK1/2 pT202/pY204 in the UV LMD enriched Patient 1 sample is reported at complete signal saturation (150,000 RFU). The asterisk (*) indicates a significant difference (p < 0.05) in rank orders determined by a non-parametric Mann–Whitney U Test between matched ultraviolet laser microdissection (UV LMD) and infrared laser capture microdissection (IR LCM) collections

Correlation of ten phosphoproteins from high grade serous ovarian cancer tumor epithelium after laser microdissection. Fluorescence measurements (RFU) of phosphoprotein abundances from four patients enriched via ultraviolet laser microdissection (UV LMD) (x-axis) or infrared laser capture microdissection (IR LCM) (y-axis). ERK1/2 pT202/pY204 in the UV LMD enriched Patient 1 sample was measured at complete signal saturation (150,000 RFU) and was therefore excluded from this figure as to not impact the correlation values
Discussion
In this study we conducted a first-of-its kind comparison between two popular cellular isolation techniques that are being extensively used in a number of important precision oncology programs such as the U.S. Department of Defense APOLLO program [61,62,63] and the I-SPY2 TRIAL series. Several prior precision studies have compared reproducibility, sampling heterogeneity, and laser effects for the LCM process, as well as accuracy studies comparing LCM generated HER2 and activated HER2 to FISH and IHC [8, 47, 48, 64, 65]. These previous studies have already demonstrated the precision and accuracy of the underpinning microdissection-RPPA workflow. Here we demonstrate that UV or IR-mediated laser microdissection does not significantly impact the quality of phosphoprotein analysis by RPPA. We found that the abundance of each phosphoprotein measured for the four HGSOC patient specimens tested was highly concordant in samples collected using either UV LMD or IR LCM techniques. Phosphoprotein levels in tissue sections microdissected with UV LMD were slightly higher in 31/40 (78%) of the comparisons. It is emphasized that this trend was not significant and that the results from each technique were highly correlated (with all p < 6.213 × 10−05). Thus, phosphoprotein abundances may be marginally improved for the analytes measured when microdissected using UV LMD, albeit these results did not achieve significance.
The choice of UV LMD and IR LCM will ultimately depend on the experimental aims and the heterogeneity of the tissue microenvironment itself. UV LMD confers ease of use and flexibility for sample collection, including higher sample processivity and is well suited for capturing large areas of relatively homogeneous cellular regions [57]. The non-contact UV LMD method allows for tissue from multiple sections or slides to be collected into the same tube, minimizing potential for sample loss. The current polymer caps used in IR LCM have a finite surface area, thus larger tissue sections or multiple tissue sections require additional caps for collection. This platform allows for direct capture of defined cellular regions, single cells and cell layers, and intermingled complex cellular microenvironments that could be present in any given tissue sample. Using this approach, another important hallmark is the ability to directly visualize and conduct pathology review of the cells captured without the need of complex difference imaging of regions collected before and after harvest, which is currently required for secondary pathology review of tissue regions harvested by UV LMD. The buffer necessary for extracting cells from the polymer caps however contain reagents that are incompatible with some downstream analytical approaches, including mass spectrometry (MS). Additional sample preparation steps such as filter-aided sample preparation (FASP) are required to remove the MS-incompatible reagents [66] from samples enriched using the current IR LCM caps. By comparison, the non-contact UV LMD method allows for a variety of buffer types to be added into the collection tube after LMD enrichment allowing for its incorporation into the standardized workflows of multiple analytical techniques.
Conclusions
Our analysis demonstrates no significant negative impact on phosphoprotein recovery using high energy UV LMD versus IR LCM enrichment as measured by RPPA analysis. Further analysis on an expanded number of phosphoproteins as well as evaluating UV LMD vs IR LCM in the context of analysis of smaller isolated cellular regions will be the focus of future studies.
Availability of data and materials
Not applicable.
Abbreviations
- LMD:
-
Laser microdissection
- LCM:
-
Laser capture microdissection
- TME:
-
Tissue microenvironment
- FFPE:
-
Formalin-fixed paraffin-embedded
- IR LCM:
-
Infrared contact laser capture microdissection
- UV LMD:
-
Ultraviolet cutting laser microdissection
- PEN:
-
Polyethylene napthalate
- PET:
-
Polyethylene tetraphthalate
- PPS:
-
Polyphenylene sulfide
- RPPA:
-
Reverse-phase protein array
- HGSOC:
-
High grade serous ovarian carcinoma
- OCT:
-
Optimal cutting temperature
- MS:
-
Mass spectrometry
- FASP:
-
Filter-aided sample preparation
- Akt:
-
Protein kinase B
- cAbl:
-
Tyrosine kinase ABL
- EGFR:
-
Epidermal growth factor receptor
- HER2:
-
Human epidermal growth factor receptor 2
- HER3:
-
Human epidermal growth factor receptor 3
- ERK1/2:
-
Extracellular signal-regulated kinase 1/2
- P70S6K:
-
Ribosomal protein S6 kinase
- PDGFR:
-
Platelet derived growth factor receptor
- Rb:
-
Retinoblastoma
- RET:
-
Rearranged during transcription
- PI3K:
-
Phosphoinositide 3-kinase
- mTOR:
-
Mammalian target of rapamycin
References
Emmert-Buck MR, Bonner RF, Smith PD, Chuaqui RF, Zhuang Z, Goldstein SR, et al. Laser capture microdissection. Science. 1996;274(5289):998–1001.
Bonner RF, Emmert-Buck M, Cole K, Pohida T, Chuaqui R, Goldstein S, et al. Laser capture microdissection: molecular analysis of tissue. Science. 1997;278(5342):1481–3.
Bohm M, Wieland I, Schutze K, Rubben H. Microbeam MOMeNT: non-contact laser microdissection of membrane-mounted native tissue. Am J Pathol. 1997;151(1):63–7.
Imbeaud S, Auffray C. ‘The 39 steps’ in gene expression profiling: critical issues and proposed best practices for microarray experiments. Drug Discov Today. 2005;10(17):1175–82.
Jacobsen M, Repsilber D, Gutschmidt A, Neher A, Feldmann K, Mollenkopf HJ, et al. Deconfounding microarray analysis—independent measurements of cell type proportions used in a regression model to resolve tissue heterogeneity bias. Methods Inf Med. 2006;45(5):557–63.
Wulfkuhle JD, Speer R, Pierobon M, Laird J, Espina V, Deng J, et al. Multiplexed cell signaling analysis of human breast cancer applications for personalized therapy. J Proteome Res. 2008;7(4):1508–17.
El-Serag HB, Nurgalieva ZZ, Mistretta TA, Finegold MJ, Souza R, Hilsenbeck S, et al. Gene expression in Barrett’s esophagus: laser capture vs whole tissue. Scand J Gastroenterol. 2009;44(7):787.
Silvestri A, Colombatti A, Calvert VS, Deng J, Mammano E, Belluco C, et al. Protein pathway biomarker analysis of human cancer reveals requirement for upfront cellular-enrichment processing. Lab Invest. 2010;90(5):787–96.
Liotta LA, Petricoin EF 3rd. Omics and cancer biomarkers: link to the biological truth or bear the consequences. Cancer Epidemiol Biomarkers Prev. 2012;21(8):1229–35.
Bateman NW, Conrads TP. Recent advances and opportunities in proteomic analyses of tumour heterogeneity. J Pathol. 2018;244(5):628–37.
Ni X, Zhang W, Huang KC, Wang Y, Ng SK, Mok SC, et al. Characterisation of human kallikrein 6/protease M expression in ovarian cancer. Br J Cancer. 2004;91:725–31.
Silasi DA, Alvero AB, Illuzzi J, Kelly M, Chen R, Fu HH, et al. MyD88 predicts chemoresistance to paclitaxel in epithelial ovarian cancer. Yale J Biol Med. 2006;79(3–4):153–63.
Socha MJ, Said N, Dai Y, Kwong J, Ramalingam P, Trieu V, et al. Aberrant promoter methylation of SPARC in ovarian cancer. Neoplasia. 2009;11(2):126–35.
Norquist BM, Garcia RL, Allison KH, Jokinen CH, Kernochan LE, Pizzi CC, et al. The molecular pathogenesis of hereditary ovarian carcinoma: alterations in the tubal epithelium of women with BRCA1 and BRCA2 mutations. Cancer. 2010;116(22):5261–71.
Wiegand KC, Shah SP, Al-Agha OM, Zhao Y, Tse K, Zeng T, et al. ARID1A mutations in endometriosis-associated ovarian carcinomas. N Engl J Med. 2010;363(16):1532–43.
Wong KK, Tsang YT, Deavers MT, Mok SC, Zu Z, Sun C, et al. BRAF mutation is rare in advanced-stage low-grade ovarian serous carcinomas. Am J Pathol. 2010;177(4):1611–7.
Steg AD, Bevis KS, Katre AA, Ziebarth A, Alvarez RD, Zhang K, et al. Stem cell pathways contribute to clinical chemoresistance in ovarian cancer. Clin Cancer Res. 2012;18(3):869–81.
Vang S, Wu HT, Fischer A, Miller DH, MacLaughlan S, Douglass E, et al. Identification of ovarian cancer metastatic miRNAs. PLoS ONE. 2013;8(3):e58226.
Smith AL, Sun M, Bhargava R, Stewart NA, Flint MS, Bigbee WL, et al. Proteomic analysis of matched formalin-fixed, paraffin-embedded specimens in patients with advanced serous ovarian carcinoma. Proteomes. 2013;1(3):240–53.
Wegdam W, Argmann CA, Kramer G, Vissers JP, Buist MR, Kenter GG, et al. Label-free LC-MSe in tissue and serum reveals protein networks underlying differences between benign and malignant serous ovarian tumors. PLoS ONE. 2014;9(9):e108046.
Worley MJ, Liu S, Hua Y, Kwok JSL, Samuel A, Hou L, et al. Molecular changes in endometriosis-associated ovarian clear cell carcinoma. Eur J Cancer. 2015;51(13):1831–42.
Moran-Jones K, Gloss BS, Murali R, Chang DK, Colvin EK, Jones MD, et al. Connective tissue growth factor as a novel therapeutic target in high grade serous ovarian cancer. Oncotarget. 2015;6(42):44551–62.
Wang Q, Tang Y, Yu H, Yin Q, Li M, Shi L, et al. CCL18 from tumor-cells promotes epithelial ovarian cancer metastasis via mTOR signaling pathway. Mol Carcinog. 2016;55(11):1688–99.
Phippen NT, Bateman NW, Wang G, Conrads KA, Ao W, Teng P, et al. NUAK1 (ARK5) is associated with poor prognosis in ovarian cancer. Front Oncol. 2016;6:213.
Yang J, Zhou Y, Ng SK, Huang KC, Ni X, Choi PW, et al. Characterization of MicroRNA-200 pathway in ovarian cancer and serous intraepithelial carcinoma of fallopian tube. BMC Cancer. 2017;17(1):422.
Vafaee F, Colvin EK, Mok SC, Howell VM, Samimi G. Functional prediction of long non-coding RNAs in ovarian cancer-associated fibroblasts indicate a potential role in metastasis. Sci Rep. 2017;7(1):10374.
Sereni MI, Baldelli E, Gambara G, Ravaggi A, Hodge KA, Alberts DS, et al. Kinase-driven metabolic signalling as a predictor of response to carboplatin-paclitaxel adjuvant treatment in advanced ovarian cancers. Br J Cancer. 2017;117(4):494–502.
Sereni MI, Baldelli E, Gambara G, Deng J, Zanotti L, Bandiera E, et al. Functional characterization of epithelial ovarian cancer histotypes by drug target based protein signaling activation mapping: implications for personalized cancer therapy. Proteomics. 2015;15(2–3):365–73.
Arnouk H, Merkley MA, Podolsky RH, Stöppler H, Santos C, Álvarez M, et al. Characterization of molecular markers indicative of cervical cancer progression. Proteomics Clin Appl. 2009;3(5):516–27.
Herfs M, Yamamoto Y, Laury A, Wang X, Nucci MR, McLaughlin-Drubin ME, et al. A discrete population of squamocolumnar junction cells implicated in the pathogenesis of cervical cancer. Proc Natl Acad Sci USA. 2012;109(26):10516–21.
Royse KE, Zhi D, Conner MG, Clodfelder-Miller B, Srinivasasainagendra V, Vaughan LK, et al. Differential gene expression landscape of co-existing cervical pre-cancer lesions using RNA-seq. Front Oncol. 2014;4:339.
Pinto AP, Miron A, Yassin Y, Monte N, Woo TY, Mehra KK, et al. Differentiated vulvar intraepithelial neoplasia contains Tp53 mutations and is genetically linked to vulvar squamous cell carcinoma. Mod Pathol. 2010;23(3):404–12.
Wang Z, Trope CG, Suo Z, Troen G, Yang G, Nesland JM, et al. The clinicopathological and prognostic impact of 14-3-3 sigma expression on vulvar squamous cell carcinomas. BMC Cancer. 2008;8:308.
Dubil EA, Tian C, Wang G, Tarney CM, Bateman NW, Levine DA, et al. Racial disparities in molecular subtypes of endometrial cancer. Gynecol Oncol. 2018;149(1):106–16.
Bateman NW, Dubil EA, Wang G, Hood BL, Oliver JM, Litzi TA, et al. Race-specific molecular alterations correlate with differential outcomes for black and white endometrioid endometrial cancer patients. Cancer. 2017;123(20):4004–12.
Lacey JV, Mutter GL, Ronnett BM, Ioffe OB, Duggan MA, Rush BB, et al. PTEN expression in endometrial biopsies as a marker of progression to endometrial carcinoma. Cancer Res. 2008;68(14):6014–20.
Li J, Xing X, Li D, Zhang B, Mutch DG, Hagemann IS, et al. Whole-genome DNA methylation profiling identifies epigenetic signatures of uterine carcinosarcoma. Neoplasia. 2017;19(2):100–11.
Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100(1):57–70.
Bild AH, Yao G, Chang JT, Wang Q, Potti A, Chasse D, et al. Oncogenic pathway signatures in human cancers as a guide to targeted therapies. Nature. 2006;439(7074):353–7.
Harsha H, Pandey A. Phosphoproteomics in cancer. Mol Oncol. 2010;4(6):482–95.
Schutze K, Becker I, Becker KF, Thalhammer S, Stark R, Heckl WM, et al. Cut out or poke in–the key to the world of single genes: laser micromanipulation as a valuable tool on the look-out for the origin of disease. Genet Anal. 1997;14(1):1–8.
Schermelleh L, Thalhammer S, Heckl W, Posl H, Cremer T, Schutze K, et al. Laser microdissection and laser pressure catapulting for the generation of chromosome-specific paint probes. Biotechniques. 1999;27(2):362–7.
Schutze K, Posl H, Lahr G. Laser micromanipulation systems as universal tools in cellular and molecular biology and in medicine. Cell Mol Biol. 1998;44(5):735–46.
Micke P, Ostman A, Lundeberg J, Ponten F. Laser-assisted cell microdissection using the PALM system. Methods Mol Biol. 2005;293:151–66.
Espina V, Mueller C, Liotta LA. Phosphoprotein stability in clinical tissue and its relevance for reverse phase protein microarray technology. Methods Mol Biol. 2011;785:23–43.
Baldelli E, Calvert V, Hodge A, VanMeter A, Petricoin EF, Pierobon M. Reverse phase protein microarrays. In: Espina V, editor. Molecular profiling: methods and protocols. New York: Springer; 2017. p. 149–69.
Wulfkuhle JD, Berg D, Wolff C, Langer R, Tran K, Illi J, et al. Molecular analysis of HER2 signaling in human breast cancer by functional protein pathway activation mapping. Clin Cancer Res. 2012;18(23):6426–35.
Baldelli E, Haura EB, Crino L, Cress DW, Ludovini V, Schabath MB, et al. Impact of upfront cellular enrichment by laser capture microdissection on protein and phosphoprotein drug target signaling activation measurements in human lung cancer: implications for personalized medicine. Proteomics Clin Appl. 2015;9(9–10):928–37.
Collaud S, Wiedl T, Cattaneo E, Soltermann A, Hillinger S, Weder W, et al. Laser-capture microdissection impairs activity-based protein profiles for serine hydrolase in human lung adenocarcinoma. J Biomol Tech. 2010;21(1):25–8.
Wang H, Owens JD, Shih JH, Li MC, Bonner RF, Mushinski JF. Histological staining methods preparatory to laser capture microdissection significantly affect the integrity of the cellular RNA. BMC Genomics. 2006;7:97.
Kaneko T, Okiji T, Kaneko R, Suda H, Nör JE. Gene expression analysis of immunostained endothelial cells isolated from formaldehyde-fixated paraffin embedded tumors using laser capture microdissection—a technical report. Microsc Res Tech. 2009;72(12):908–12.
Grellner W, Vieler S, Madea B. Transforming growth factors (TGF-alpha and TGF-beta1) in the determination of vitality and wound age: immunohistochemical study on human skin wounds. Forensic Sci Int. 2005;153(2–3):174–80.
Grellner W, Madea B. Demands on scientific studies: vitality of wounds and wound age estimation. Forensic Sci Int. 2007;165(2–3):150–4.
Espina V, Edmiston KH, Heiby M, Pierobon M, Sciro M, Merritt B, et al. A portrait of tissue phosphoprotein stability in the clinical tissue procurement process. Mol Cell Proteomics. 2008;7(10):1998–2018.
Mueller C, Edmiston KH, Carpenter C, Gaffney E, Ryan C, Ward R, et al. One-step preservation of phosphoproteins and tissue morphology at room temperature for diagnostic and research specimens. PLoS ONE. 2011;6(8):e23780.
Mouledous L, Hunt S, Harcourt R, Harry JL, Williams KL, Gutstein HB. Lack of compatibility of histological staining methods with proteomic analysis of laser-capture microdissected brain samples. J Biomol Tech. 2002;13(4):258–64.
Vandewoestyne M, Goossens K, Burvenich C, Van Soom A, Peelman L, Deforce D. Laser capture microdissection: should an ultraviolet or infrared laser be used? Anal Biochem. 2013;439(2):88–98.
Pierobon M, Ramos C, Wong S, Hodge KA, Aldrich J, Byron S, et al. Enrichment of PI3K-AKT-mTOR pathway activation in hepatic metastases from breast cancer. Clin Cancer Res. 2017;23(16):4919–28.
Mabuchi S, Kuroda H, Takahashi R, Sasano T. The PI3K/AKT/mTOR pathway as a therapeutic target in ovarian cancer. Gynecol Oncol. 2015;137(1):173–9.
Gasparri ML, Bardhi E, Ruscito I, Papadia A, Farooqi AA, Marchetti C, et al. PI3K/AKT/mTOR pathway in ovarian cancer treatment: are we on the right track? Geburtshilfe Frauenheilkd. 2017;77(10):1095–103.
Conrads TP, Petricoin EF. The Obama administration’s cancer Moonshot: a call for proteomics. Clin Cancer Res 2016;22(18):4556–58
Fiore LD, Rodriguez H, Shriver CD. Collaboration to accelerate proteogenomics cancer care: the Department of Veterans Affairs, Department of Defense, and the National Cancer Institute’s Applied Proteogenomics OrganizationaL Learning and Outcomes (APOLLO) network. Clin Pharmacol Ther 2017;101(5):619-621
Lee JSH, Darcy KM, Hu H, Casablanca Y, Conrads TP, Dalgard CL, Freymann JB, Hanlon SE, Huang GD, Kvecher L, Maxwell GL, Meng F, Moncur JT, Turner C, Wells JM, Wilkerson MD, Zhu K, Ramoni RB, Shriver CD. From discovery to practice and survivorship: building a national real-world data learning healthcare framework for military and veteran cancer patients. Clin Pharmacol Ther. 2019;106(1):52–7.
Mueller C, deCarvalho AC, Mikkelsen T, Lehman NL, Calvert V, Espina V, et al. Glioblastoma cell enrichment is critical for analysis of phosphorylated drug targets and proteomic-genomic correlations. Cancer Res. 2014;74(3):818–28.
Pierobon M, Silvestri A, Spira A, Reeder A, Pin E, Banks S, et al. Pilot phase I/II personalized therapy trial for metastatic colorectal cancer: evaluating the feasibility of protein pathway activation mapping for stratifying patients to therapy with imatinib and panitumumab. J Proteome Res. 2014;13(6):2846–55.
Ni MW, Wang L, Chen W, Mou HZ, Zhou J, Zheng ZG. Modified filter-aided sample preparation (FASP) method increases peptide and protein identifications for shotgun proteomics. Rapid Commun Mass Spectrom. 2017;31(2):171–8.
Acknowledgements
Not applicable.
Funding
This study was supported in part by the U.S. Department of Defense - Uniformed Services University of the Health Sciences (HU0001-16-2-0006 and HU0001-16-2-0014) and the Ovarian Cancer Research Program from the Congressionally Directed Medical Research Program (W81XWH-16-2-0038). Disclaimer: The views expressed herein are those of the authors and do not reflect the official policy of the Department of Army/Navy/Air Force, Department of Defense, or U.S. Government.
Author information
Authors and Affiliations
Contributions
Contributed to conception and design: ALH, EFP, TPC. Contributed to identification and acquisition of patient specimens: GG, DM, JO, GLM, TPC. Contributed to acquisition, analysis and/or interpretation of data: ALH, MP, EB, EFP, TPC. Drafted and/or revised the article: ALH, MP, NWB, EFP, TPC. Acquired funding for the research: GLM, EFP, TPC. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This study was conducted under an IRB exempt protocol (Western IRB approved).
Consent for publication
Not applicable.
Competing interests
TPC receives research funding from AbbVie. EFP receives research funding from Genentech, Pfizer, AbbVie, and is a co-inventor of the RPPA technology described herein and receives royalties on the related license agreements.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1: Table S1.
Normalized reverse phase protein array abundance values. The asterisk (*) indicates complete signal saturation for the ERK1/2 pT202/pY204 measured in the UV LMD enriched Patient 1 sample.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Hunt, A.L., Pierobon, M., Baldelli, E. et al. The impact of ultraviolet- and infrared-based laser microdissection technology on phosphoprotein detection in the laser microdissection-reverse phase protein array workflow. Clin Proteom 17, 9 (2020). https://doi.org/10.1186/s12014-020-09272-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12014-020-09272-z