
Abstract—Hereditary spastic paraplegia (HSP) is a heterogeneous group of neurodegenerative disorders predominantly characterized by damage to the pyramidal tract. The major, single symptom of HSP is progressive weakness and spasticity of lower extremities, which ultimately leads to difficulties in walking. HSP has various types of inheritance and clinical characteristics. The development of molecular genetics and the emergence of high-throughput sequencing techniques, such as New Generation Sequencing (NGS) in particular, revealed a pronounced genetic heterogeneity of HSP. Currently, about 80 genetic loci for HSP are designated as spastic paraplegia genes (SPGs) (with numbering according to the mapping history), and the vast majority of causative genes within loci have been identified. The most frequent autosomal dominant forms of HSP are SPG4 and SPG3; SPG11, SPG15, SPG7 are common autosomal recessive HSPs. The mechanisms of HSP molecular pathogenesis are variable and encompassing defects of neuronal membrane transport and the disturbance of myelination processes, lipid metabolism, mitochondrial dysfunctions, etc. The differential diagnosis for HSPs is difficult due to the existence of many geno- and phenocopies within the other neuropathologies that can be partially distinguished by neuroimaging methods. The only method for the precise diagnostics of HSP and the number of genomic copies is DNA diagnostics—the search for mutations in individual genes and/or the analysis of many genes simultaneously by NGS methods (gene panel, whole-exome sequencing, and whole-genome sequencing). Such DNA diagnostics opens up the opportunity to perform prenatal or preimplantation analysis of the fetus for families carrying HSP mutations to predict the risk of HSP inheritance. Currently, HSP treatment is directed against individual symptoms (antispastic drugs, etc.), and the discovery of the pathogenic action of different genes requires the development of new therapeutic approaches.

Similar content being viewed by others
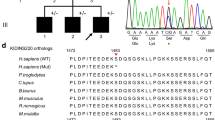
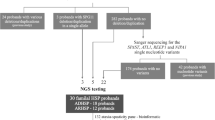

REFERENCES
Abdollahpour, H., Alawi, M., Kortum, F., et al., An AP4B1 frameshift mutation in siblings with intellectual disability and spastic tetraplegia further delineates the AP-4 deficiency syndrome, Eur. J. Hum. Genet., 2015, vol. 23, no. 2, pp. 256–259. https://doi.org/10.1038/ejhg.2014.73
Abou Jamra, R., Philippe, O., Raas-Rothschild, A., et al., Adaptor protein complex 4 deficiency causes severe autosomal-recessive intellectual disability, progressive spastic paraplegia, shy character, and short stature, Am. J. Hum. Genet., 2011, vol. 88, no. 6, pp. 788–795. https://doi.org/10.1016/j.ajhg.2011.04.019
Akhmetgaleeva, A.F., Molecular-genetic study of spastic paraplegia in the Republic of Bashkortostan, Extended Abstract of Cand. Sci. (Biol.) Dissertation, Ufa: Inst. Biochem. Genet., Ufa Sci. Center, Russ. Acad. Sci., 2017.
Almontashiri, N.A., Chen, H.H., Mailloux, R.J., et al., SPG7 variant escapes phosphorylation-regulated processing by AFG3L2, elevates mitochondrial ROS, and is associated with multiple clinical phenotypes, Cell Rep., 2014, vol. 7, no. 3, pp. 834–847. https://doi.org/10.1016/j.celrep.2014.03.051
Arnoldi, A., Crimella, C., Tenderini, E., et al., Clinical phenotype variability in patients with hereditary spastic paraplegia type 5 associated with CYP7B1 mutations, Clin. Genet., 2012, vol. 81, no. 2, pp. 150–157. https://doi.org/10.1111/j.1399-0004.2011.01624.x
Balicza, P., Grosz, Z., Gonzalez, M.A., et al., Genetic background of the hereditary spastic paraplegia phenotypes in Hungary—an analysis of 58 probands, J. Neurol. Sci., 2016, vol. 364, pp. 116–121. https://doi.org/10.1016/j.jns.2016.03.018
Bauer, P., Winner, B., Schule, R., et al., Identification of a heterozygous genomic deletion in the spatacsin gene in SPG11 patients using high-resolution comparative genomic hybridization, Neurogenetics, 2009, vol. 10, no. 1, pp. 43–48. https://doi.org/10.1007/s10048-008-0144-2
Blackstone, C., Cellular pathways of hereditary spastic paraplegia, Annu. Rev. Neurosci., 2012, vol. 35, pp. 25–47. https://doi.org/10.1146/annurev-neuro-062111-150400
Boukhris, A., Feki, I., Elleuch, N., et al., A new locus (SPG46) maps to 9p21.2-q21.12 in a Tunisian family with a complicated autosomal recessive hereditary spastic paraplegia with mental impairment and thin corpus callosum, Neurogenetics, 2010, vol. 11, no. 4, pp. 441–448. https://doi.org/10.1007/s10048-010-0249-2
Bross, P., Naundrup, S., Hansen, J., et al., The Hsp60-(p.V98I) mutation associated with hereditary spastic paraplegia SPG13 compromises chaperonin function both in vitro and in vivo, J. Biol. Chem., 2008, vol. 283, no. 23, pp. 15694–15700. https://doi.org/10.1074/jbc.M800548200
Buermans, H.P. and den Dunnen, J.T., Next generation sequencing technology: advances and applications, Biochim. Biophys. Acta, Mol. Basis Dis., 2014, vol. 1842, no. 10, pp. 1932–1941. https://doi.org/10.1016/j.bbadis.2014.06.015
Chrestian, N., Dupre, N., Gan-Or, Z., et al., Clinical and genetic study of hereditary spastic paraplegia in Canada, Neurol. Genet., 2017, vol. 3, no. 1, art. ID e122. https://doi.org/10.1212/NXG.0000000000000122
Crimella, C., Baschirotto, C., Arnoldi, A., et al., Mutations in the motor and stalk domains of KIF5A in spastic paraplegia type 10 and in axonal Charcot–Marie–Tooth type 2, Clin. Genet., 2012, vol. 82, no. 2, pp. 157–164. https://doi.org/10.1111/j.1399-0004.2011.01717.x
de Souza, P.V., de Rezende Pinto, W.B., de Rezende Batistella, G.N., et al., Hereditary spastic paraplegia: clinical and genetic hallmarks, Cerebellum, 2017, vol. 16, no. 2, pp. 525–551. https://doi.org/10.1007/s12311-016-0803-z
Denora, P.S., Santorelli, F.M., and Bertini, E., Hereditary spastic paraplegias: one disease for many genes, and still counting, Handb. Clin. Neurol., 2013, vol. 113, pp. 1899–1912. https://doi.org/10.1016/B978-0-444-59565-2.00060-5
Ebbing, B., Mann, K., Starosta, A., et al., Effect of spastic paraplegia mutations in KIF5A kinesin on transport activity, Hum. Mol. Genet., 2008, vol. 17, no. 9, pp. 1245–1252. https://doi.org/10.1093/hmg/ddn014
Elert-Dobkowska, E., Stepniak, I., Krysa, W., et al., Molecular spectrum of the SPAST, ATL1 and REEP1 gene mutations associated with the most common hereditary spastic paraplegias in a group of Polish patients, J. Neurol. Sci., 015, vol. 359, nos. 1–2, pp. 35–39. https://doi.org/10.1016/j.jns.2015.10.030
Erichsen, A.K., Koht, J., Stray-Pedersen, A., et al., Prevalence of hereditary ataxia and spastic paraplegia in southeast Norway: a population-based study, Brain, 2009, vol. 132, no. 6, pp. 1577–1588. https://doi.org/10.1093/brain/awp056
Ferreirinha, F., Quattrini, A., Pirozzi, M., et al., Axonal degeneration in paraplegin-deficient mice is associated with abnormal mitochondria and impairment of axonal transport, J. Clin. Invest., 2004, vol. 113, no. 2, pp. 231–242. https://doi.org/10.1172/JCI20138
Fink, J.K., Hereditary spastic paraplegia: clinico-pathologic features and emerging molecular mechanisms, Acta Neuropathol., 2013, vol. 126, no. 3, pp. 307–328. https://doi.org/10.1007/s00401-013-1115-8
Gan-Or, Z., Bouslam, N., Birouk, N., et al., Mutations in CAPN1 cause autosomal-recessive hereditary spastic paraplegia, Am. J. Hum. Genet., 2016, vol. 98, no. 5, pp. 1038–1046. https://doi.org/10.1016/j.ajhg.2016.04.002
Garone, C., Tadesse, S., and Hirano, M., Clinical and genetic spectrum of mitochondrial neurogastrointestinal encephalomyopathy, Brain, 2011, vol. 134, no. 11, pp. 3326–3332. https://doi.org/10.1093/brain/awr245
Goizet, C., Boukhris, A., Maltete, D., et al., SPG15 is the second most common cause of hereditary spastic paraplegia with thin corpus callosum, Neurology, 2009, vol. 73, no. 14, pp. 1111–1119. https://doi.org/10.1212/WNL.0b013e3181bacf59
Gruenenfelder, F.I., Thomson, G., Penderis, J., et al., Axonglial interaction in the CNS: what we have learned from mouse models of Pelizaeus–Merzbacher disease, J. Anat., 2011, vol. 219, no. 1, pp. 33–43. https://doi.org/10.1111/j.1469-7580.2011.01363.x
Harding, A.E., Classification of the hereditary ataxias and paraplegias, Lancet, 1983, vol. 321, no. 8334, pp. 1151–1155.
Hensiek, A., Kirker, S., and Reid, E., Diagnosis, investigation and management of hereditary spastic paraplegias in the era of next-generation sequencing, J. Neurol., 2015, vol. 262, no. 7, pp. 1601–1612. https://doi.org/10.1007/s00415-014-7598-y
HGMD. https://www.portal.biobase-international.com/ hgmd/pro/start.php.
Hirst, J., Edgar, J.R., Esteves, T., et al., Loss of AP-5 results in accumulation of aberrant endolysosomes: defining a new type of lysosomal storage disease, Hum. Mol. Genet., 2015, vol. 24, no. 17, pp. 4984–4996. https://doi.org/10.1093/hmg/ddv220
Illarioshkin, S.N., Rudenskaya, G.E., Ivanova-Smolenskaya, I.A., et al., Nasledstvennye ataksii i paraplegii (Inherited Ataxia and Paraplegia), Moscow: MEDpress-inform, 2006, pp. 286–390.
Ishiura, H., Takahashi, Y., Hayashi, T., et al., Molecular epidemiology and clinical spectrum of hereditary spastic paraplegia in the Japanese population based on comprehensive mutational analyses, J. Hum. Genet., 2014, vol. 59, no. 3, pp. 163–172. https://doi.org/10.1038/jhg.2013.139
Kara, E., Tucci, A., Manzoni, C., et al., Genetic and phenotypic characterization of complex hereditary spastic paraplegia, Brain, 2016, vol. 139, no. 7, pp. 1904–1918. https://doi.org/10.1093/brain/aww111
Klebe, S., Lossos, A., Azzedine, H., et al., KIF1A missense mutations in SPG30, an autosomal recessive spastic paraplegia: distinct phenotypes according to the nature of the mutations, Eur. J. Hum. Genet., 2012, vol. 20, no. 6, pp. 645–649. https://doi.org/10.1038/ejhg.2011.261
Klebe, S., Stevanin, G., and Depienne, C., Clinical and genetic heterogeneity in hereditary spastic paraplegias: from SPG1 to SPG72 and still counting, Rev. Neurol., 2015, vol. 171, nos. 6–7, pp. 505–530. https://doi.org/10.1016/j.neurol.2015.02.017
Liu, Y.T., Laura, M., Hersheson, J., et al., Extended phenotypic spectrum of KIF5A mutations: from spastic paraplegia to axonal neuropathy, Neurology, 2014, vol. 83, no. 7, pp. 612–619. https://doi.org/10.1212/WNL.0000000000000691
Lo Giudice, T., Lombardi, F., Santorelli, F.M., et al., Hereditary spastic paraplegia: clinical-genetic characteristics and evolving molecular mechanisms, Exp. Neurol., 2014, vol. 261, pp. 518–539. https://doi.org/10.1016/j.expneurol.2014.06.011
Lopez, E., Casasnovas, C., Gimenez, J., et al., Characterization of Alu and recombination-associated motifs mediating a large homozygous SPG7 gene rearrangement causing hereditary spastic paraplegia, Neurogenetics, 2015, vol. 16, no. 2, pp. 97–105. https://doi.org/10.1007/s10048-014-0429-6
Lynch, D.S., Koutsis, G., Tucci, A., et al., Hereditary spastic paraplegia in Greece: characterization of a previously unexplored population using next-generation sequencing, Eur. J. Hum. Genet., 2016, vol. 24, no. 6, pp. 857–863. https://doi.org/10.1038/ejhg.2015.200
Magzhanov, R.V., Saifullina, E.V., Idrisova, R.F., et al., Epidemiology of hereditary spastic paraplegias in Bashkortostan Republic, Med. Genet., 2013, vol. 12, no. 7 (133), pp. 12–16.
Montenegro, G., Rebelo, A.P., Connell, J., et al., Mutations in the ER-shaping protein reticulon 2 cause the axon-degenerative disorder hereditary spastic paraplegia type 12, J. Clin. Invest., 2012, vol. 122, no. 2, pp. 538–544. https://doi.org/10.1172/JCI60560
Murphy, S., Gorman, G., Beetz, C., et al., Dementia in SPG4 hereditary spastic paraplegia: clinical, genetic, and neuropathologic evidence, Neurology, 2009, vol. 73, no. 5, pp. 378–384. https://doi.org/10.1212/WNL.0b013e3181b04c6c
Musacchio, T., Zaum, A.K., Uceyler, N., et al., ALS and MMN mimics in patients with BSCL2 mutations: the expanding clinical spectrum of SPG17 hereditary spastic paraplegia, J. Neurol., 2017, vol. 264, no. 1, pp. 11–20. https://doi.org/10.1007/s00415-016-8301-2
Noetzli, L., Sanz, P.G., Brodsky, G.L., et al., A novel mutation in PLP1 causes severe hereditary spastic paraplegia type 2, Gene, 2014, vol. 533, no. 1, pp. 447–450. https://doi.org/10.1016/j.gene.2013.09.076
Novarino, G., Fenstermaker, A.G., Zaki, M.S., et al., Exome sequencing links corticospinal motor neuron disease to common neurodegenerative disorders, Science, 2014, vol. 343, no. 6170, pp. 506–511. https://www.ncbi.nlm.nih.gov/omim.https://doi.org/10.1126/science.1247363
OMIM. https://www.ncbi.nlm.nih.gov/omim.
Orlacchio, A., Montieri, P., Babalini, C., et al., Late-onset hereditary spastic paraplegia with thin corpus callosum caused by a new SPG3A mutation, J. Neurol., 2011, vol. 258, no. 7, pp. 1361–1363. https://doi.org/10.1007/s00415-011-5934-z
Reid, E., Science in motion: common molecular pathological themes emerge in the hereditary spastic paraplegias, J. Med. Genet., 2003, vol. 40, no. 2, pp. 81–86.
Ribai, P., Depienne, C., Fedirko, E., et al., Mental deficiency in three families with SPG4 spastic paraplegia, Eur. J. Hum. Genet., 2008, vol. 16, no. 1, pp. 97–104. https://doi.org/10.1038/sj.ejhg.5201922
Richard, S., Lavie, J., Banneau, G., et al., Hereditary spastic paraplegia due to a novel mutation of the REEP1 gene: case report and literature review, Medicine (Baltimore), 2017, vol. 96, no. 3, art. ID e5911. https://doi.org/10.1097/MD.0000000000005911
Rismanchi, N., Soderblom, C., Stadler, J., et al., Atlastin GTPases are required for Golgi apparatus and ER morphogenesis, Hum. Mol. Genet., 2008, vol. 17, no. 11, pp. 1591–1604. https://doi.org/10.1093/hmg/ddn046
Rudenskaya, G.E., Sermyagina, I.G., Illarioshkin, S.N., et al., Inherited spastic paraplegia type 4 (SPG4): clinical and molecular genetic characteristics, Zh. Nevropatol. Psikhiatr. im. S.S. Korsakova, 2010, vol. 110, no. 6, pp. 12–19.
Rydning, S.L., Wedding, I.M., Koht, J., et al., A founder mutation p.H701P identified as a major cause of SPG7 in Norway, Eur. J. Neurol., 2016, vol. 23, no. 4, pp. 763–771. https://doi.org/10.1111/ene.12937
Ryzhkova, O.P., Kardymon, O.L., Prokhorchuk, E.B., et al., Guidelines for the interpretation of massive parallel sequencing variants, Med. Genet., 2017, vol. 16, no. 7, pp. 4–17.
Sanchez-Ferrero, E., Coto, E., Beetz, C., et al., SPG7 mutational screening in spastic paraplegia patients supports a dominant effect for some mutations and a pathogenic role for p.A510V, Clin. Genet., 2013, vol. 83, no. 3, pp. 257–262. https://doi.org/10.1111/j.1399-0004.2012.01896.x
Schüle, R. and Schöls, L., Genetics of hereditary spastic paraplegias, Semin. Neurol., 2011, vol. 31, no. 5, pp. 484–493. https://doi.org/10.1055/s-0031-1299787
Schüle, R., Schlipf, N., Synofzik, M., et al., Frequency and phenotype of SPG11 and SPG15 in complicated hereditary spastic paraplegia, J. Neurol., Neurosurg., Psychiatry, 2009, vol. 80, no. 12, pp. 1402–1404. https://doi.org/10.1136/jnnp.2008.167528
Shaw, M., Yap, T.Y., Henden, L., et al., Identical by descent L1CAM mutation in two apparently unrelated families with intellectual disability without L1 syndrome, Eur. J. Med. Genet., 2015, vol. 58, nos. 6–7, pp. 364–368. https://doi.org/10.1016/j.ejmg.2015.04.004
Silver, J.R. Familial spastic paraplegia with amyotrophy of the hands, J. Neurol., Neurosurg., Psychiatry, 1996, vol. 29, pp. 135–144.
Solowska, J.M. and Baas, P.W., Hereditary spastic paraplegia SPG4: what is known and not known about the disease, Brain, 2015, vol. 138, no. 9, pp. 2471–2484. https://doi.org/10.1093/brain/awv178
Stevanin, G., Azzedine, H., Denora, P., et al., Mutations in SPG11 are frequent in autosomal recessive spastic paraplegia with thin corpus callosum, cognitive decline and lower motor neuron degeneration, Brain, 2008, vol. 131, no. 3, pp. 772–784. https://doi.org/10.1093/brain/awm293
Synofzik, M. and Schüle, R., Overcoming the divide between ataxias and spastic paraplegias: shared phenotypes, genes, and pathways, Mov. Disord., 2017, vol. 32, no. 3, pp. 332–345. https://doi.org/10.1002/mds.26944
Tesson, C., Koht, J., and Stevanin, G., Delving into the complexity of hereditary spastic paraplegias: how unexpected phenotypes and inheritance modes are revolutionizing their nosology, Hum. Genet., 2015, vol. 134, no. 6, pp. 511–538. https://doi.org/10.1007/s00439-015-1536-7
Windpassinger, C., Auer-Grumbach, M., Irobi, J., et al., Heterozygous missense mutations in BSCL2 are associated with distal hereditary motor neuropathy and Silver syndrome, Nat. Genet., 2004, vol. 36, no. 3, pp. 271–276. https://doi.org/10.1038/ng1313
Zhao, G.H. and Liu, X.M., Clinical features and genotype-phenotype correlation analysis in patients with ATL1 mutations: a literature reanalysis, Transl. Neurodegener., 2017, vol. 6, p. 9. https://doi.org/10.1186/s40035-017-0079-3
Zhu, P.P., Patterson, A., Lavoie, B., et al., Cellular localization, oligomerization, and membrane association of the hereditary spastic paraplegia 3A (SPG3A) protein atlastin, J. Biol. Chem., 2003, vol. 278, no. 49, pp. 49063–49071. https://doi.org/10.1074/jbc.M306702200
Zinchenko, R.A., El’chinova, G.I., Baryshnikova, N.V., Polyakov, A.V., and Ginter, E.K., Prevalences of hereditary diseases in different populations of Russia, Russ. J. Genet., 2007, vol. 43, no. 9, pp. 1038–1045.
Author information
Authors and Affiliations
Corresponding author
Additional information
Translated by M. Batrukova
Rights and permissions
About this article

Cite this article
Kadnikova, V.A., Ryzhkova, O.P., Rudenskaya, G.E. et al. Molecular Genetic Diversity and DNA Diagnostics of Hereditary Spastic Paraplegia. Biol Bull Rev 9, 145–156 (2019). https://doi.org/10.1134/S2079086419020063
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S2079086419020063