
Abstract
Tris[2-(3′-cyanopropoxy)phenyl]phosphine oxide has been obtained via the alkylation of tris(2hydroxyphenyl)phosphine oxide with 4-bromobutyronitrile in the presence of K2CO3. The product structure has been elucidated by means of X-ray diffraction analysis. The terminal cyano groups in the obtained phosphine oxide have been converted into tetrazole rings via the click-reaction with NH4N3 to give a new hybrid tripodal propeller ligand, tris{2-[3′-(tetrazol-5′′-yl)propoxy]phenyl}phosphine oxide. Palladium(II) complexes of the prepared ligand and its short-linker analog, tris[2-(tetrazol-5′-ylmethoxy)phenyl]phosphine oxide, have been synthesized. Cytotoxicity of the ligands and their Pd(II) complexes has been studied.
Similar content being viewed by others

The compounds bearing tetrazole fragments are interesting objects for coordination chemistry as ligands binding metal ions [1–3] and building blocks for the creation of novel materials: coordination polymers [4, 5] metal-organic frameworks (MOFs) [6], energetic materials [7], supercapacitors [8], and spin crossovers [9, 10].
Several tripodal ligands with tetrazole pendant groups have been described in the literature [5, 6, 8, 11–13]. Their complexes with d- and f-block elements have been suggested as light-emitting materials [1, 2] and contrasting agents [14].
At the same time, tetrazoles are of special interest for of medicinal chemistry, since they exhibit a wide range of biological activity, even being atypical for natural objects [15]. Tetrazoles are carboxylic acid bioisosters metabolically stable toward many biological transformations [16]. Tetrazoles possess antiangiogenic [17], antibacterial [18], anticancer [19, 20], fungicide [21], antimalarial [22], antituberculous [23], and antiviral [24] activity.
The use of triphenylphosphine oxide fragment as the core of a tripodal ligand can favor the complex formation with metal ions [25–29]. The combination of the P=O fragment and tetrazole functional groups in hybrid ligands affords the compounds promising as ligands and biologically active molecules.
This study aimed to synthesize hybrid ligand 1 containing the phosphine oxide core and tetrazole side groups and to investigate cytotoxicity of the ligand and its palladium complex toward tumor cells. It was also interesting to compare ligand 1 to its analog 2 with a shorter linker [Bykhovskaya, O.V. et al., Russ. J. Gen., Chem., 2019, vol. 89, no. 12] (Scheme 1).

1.
The target ligand 1 was prepared in two stages as follows. In the first stage, the reaction of phosphine oxide 3 [30] with 4-bromobutyronitrile in the presence of K2CO3 in anhydrous DMF gave tris[2-(3′-cyanopropoxy)phenyl]phosphine oxide 4 in 96.2% yield (Scheme 2). That compound was isolated as white crystalline powder, readily soluble in organic solvents (CH2Cl2, CHCl3, DMF, DMSO, etc.).

2.
Composition and structure of the obtained trinitrile 4 were confirmed by the data of elemental analysis as well as IR and NMR (1Н, 13С, and 31Р) spectroscopy. IR spectrum of compound 4 contained the ν(С≡N) bands at 2241 cm–1 and the ν(P=O) bands at 1176 cm–1. The 1H NMR spectrum of the solution in CDCl3 revealed the signals of the linker methylene protons as triplets or quintets at 1.6–4.1 ppm. The signals of the phenyl protons of the triphenylphosphine oxide core revealed chemical shifts and multiplicities typical of the related compounds [31] and appeared as doublets and triplets at 6.8–7.7 ppm. The 13C NMR spectrum contained the singlet signals of the linker methylene groups at 13– 66 ppm and the CN group signals at 119.3 ppm. The signals of the carbon atoms of the triphenylphosphine oxide core appeared as doublets at 111–161 ppm with typical spin-spin coupling constants of JPС 1.7 to 111.2 Hz. The 31P NMR spectrum contained a singlet at 28.1 ppm, typical of triarylphosphine oxides.
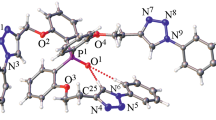
Molecular structure of tris[2-(3′-cyanopropoxy)phenyl]phosphine oxide 4 was confirmed by the X-ray diffraction analysis method (Fig. 1). Selected bond lengths are collected in Table 1. The molecule of compound 4 was asymmetric, with two of three ortho-substituents located to the same side as the P=O group with respect to the carbon atoms of the POC3 moiety and the third substituent was directed oppositely. The O1P1C22O4 angle corresponding to the substituent located oppositely to the phosphine oxide group equaled –172.2(1)°, two other substituents forming the OPCO angles of 38.6(1) to 64.8(1)°. In the earlier described cases of tris(tetrazoles) and tris(alkanols) [31], the molecule conformation has been additionally stabilized by two C–H···O(P) bonds. Molecule of compound 4 also contained two O–H···O(P) intramolecular bonds formed by the substituents which were to the same side as the P=O group (Fig. 1). The bond lengths and angles were typical of the earlier described phosphoryltris(2,1-phenylene)oxyalkenes [27, 29, 31–33].

General view of a molecule of tris[2-(3′-cyanopropoxy)phenyl]phosphine oxide 4 (CCDC 2085004) with the atoms displayed as thermal ellipsoids (p 50%). The C–H···O intramolecular interactions are shown in dotted lines.
In the second stage, trinitrile 4 was introduced in the reaction with NH4N3 prepared in situ from NH4Cl and NaN3, affording ligand 1 in 74.2% yield (Scheme 2). The product structure was elucidated by means of IR, Raman, and NMR (1H, 13C, and 31P) spectroscopy. Tris[2-(tetrazol-5′-ylmethoxy)phenyl]phosphine oxide 2 has been prepared by us earlier [Bykhovskaya, O.V. et al., Russ. J. Gen., Chem., 2019, vol. 89, no. 12] using the same method from the corresponding cyano derivative [25].
IR and Raman spectra of ligand 1 were compared to those earlier described for ligand 2 with a known structure (Table 2). IR spectrum of compound 1 did not contain the bands of the starting trinitrile 4 at 2241 cm–1 [ν(С≡N)] and 1176 cm–1 [ν(P=O)]. Instead, the ν(P=O) at 1086 cm–1 appeared in the spectrum as in the case of ligand 2, corresponding to a phosphoryl group involved in a bifurcate hydrogen bond. The spectra of ligands 1 and 2 were similar in the range of the ν(NH) vibrations. It should be noted that the spectrum of a solution of ligand 1 in DMSO-d6 contained the ν(P=O) band assigned to the phosphoryl group involved in hydrogen bond (1157 cm–1) and, in contrast to ligand 2, the band of the free P=O group (1185 cm–1). That feature could be due to longer linker providing higher mobility of the pendant group.
Assignment of the ring vibrational frequencies was based on the calculation of the frequency and shape of normal vibrations in 5-substituted tetrazoles [34]. The absorption bands at 1600–1400 cm–1 mainly related to the ν(C=N) and ν(N=N) vibrations overlapped with strong bands assigned to the phenoxy groups in the spectra of compounds 1 and 2 and thus could not be reliably detected. The band at 1480 cm–1 in the spectrum of ligand 1 (Table 2) was assigned mainly to the ν(C–N) vibrations, the band at 1242 cm–1 corresponded to the N=N bonds vibrations mixed with ring deformations, whereas the bands at 1124 and 1110 cm–1 were assigned to the δ(NH) deformational vibrations. The band at 1040 cm–1 was due to the out-of-plane ρ(NH) vibrations mixed with the ring deformations, and the band at 990 cm–1 was assigned to the deformation of the cycle. Dissolution of ligand 1 in DMSO-d6 led to the disruption of the intermolecular hydrogen bonds and an intramolecular Р=О∙∙∙HN hydrogen bond, as evidenced by the change in the δ(NH) and ν(P=O) bands (Table 2).
1H NMR spectrum of ligand 1 contained the signals of the linker methylene groups as three broadened singlets at 1.7–3.9 ppm. The aryl protons appeared as multiplets at 6.8–7.3 and 7.4–7.7 ppm. The 1H NMR spectrum did not contain the signals of the NH group, this feature is typical of the tetrazoles spectra. The 13C NMR spectrum revealed three singlet signals of the linker methylene groups at 19– 67 ppm, a singlet of the tetrazole carbon atom at 155.67 ppm, and several doublets of the aryl carbon atoms at 113–160 ppm. The 31P NMR spectrum contained a singlet at 28 ppm, typical of triarylphosphine oxides.
Palladium complexes with different heterocyclic compounds have been recognized for the antitumor activity [35]. In view of that, we synthesized the 1 : 1 complexes of the new ligand 1 (n = 3) and the previously prepared ligand 2 (n = 1) with PdCl2: complexes 5 and 6, respectively (Scheme 3).

3.
The complexes were isolated and characterized by means of elemental analysis as well as IR and NMR spectroscopy. According to the elemental analysis data, ligands 1 and 2 formed the 1 : 1 complexes with PdCl2. In contrast to the lanthanum complex with deprotonated ligand 2 described by us earlier [Bykhovskaya, O.V. et al., Russ. J. Gen., Chem., 2019, vol. 89, no. 12], the amino groups capable of hydrogen bonding were retained in complex 6, which could affect the coordination properties of the ligand. The ν(NH) bands of the palladium complex were changed in comparison with the free ligand spectrum: the maximum at 2700 cm–1 became weaker, and the second maximum appeared at ~3100 cm–1, which could be assigned to weaker bound NH groups and the ν(ОH) vibrations of bound water. The ν(Р=О) band, as in the ligand spectrum, appeared at about 1090 cm–1, evidencing preservation of the intramolecular hydrogen bons of the phosphoryl oxygen atom with two tetrazole rings, the ring vibrational bends being changed (Table 2). The Raman spectrum of complex 6 contained a band at 300 cm–1, which could be assigned to the νs(Pd–Cl) symmetric vibrations in the trans complex [36], hence palladium evidently coordinated two tetrazole rings in the trans configuration. In view of the 1 : 1 complex composition, it could be suggested that those tetrazole rings belonged to different molecules, and the dimers with one of the tetrazole rings acting as bridge were formed. Moreover, a water molecule could be bound to proton as well as nitrogen atom, linking the complexes.
The ν(Р=О) band in the spectrum of complex 5, as in the case of complex 6, was not shifted in comparison with the free ligand spectrum, hence it could be suggested that the node with the bifurcate hydrogen bond was retained. Position of the ring bands was slightly changed (Table 2). The νs(Pd–Cl) band at 299 cm–1 in the Raman spectrum was stronger than that in the spectrum of complex 6 and was also typical of trans geometry of the coordination plane. Overall, comparison of the vibrational spectra revealed structural similarity of the obtained palladium complexes.
Hence, the analysis of the vibrational spectra suggested the trans structure of complexes 5 and 6 with coordination of tetrazole rings of two ligands and preservation of the bifurcate intramolecular hydrogen bond with the phosphoryl group.
Complexes 5 and 6 were slowly dissolved in DMSO, seemingly without decomposition (as per the IR spectroscopy data).
1Н NMR spectrum of the solution of complex 5 contained a broad singlet of the central methylene group of the linker at 1.74 ppm and a triplet of the CH2N group protons at 2.63 ppm, whereas the signal of the CН2О group protons was masked with the water signal (Table 3). The aryl protons of complex 5 were observed as triplets at 6.98 (Н5), 7.53 ppm (Н4) and a doublet of doublets at 7.08 (Н3), 7.47 ppm (Н6).
The 1Н NMR spectrum of complex 6 contained a broad singlet of the CН2О group at 5.38 ppm and the signals of the aromatic protons as triplets at 6.96 (Н5), 7.58 ppm (Н4), triplet of doublets at 7.34 ppm (Н3), and doublet of doublets at 7.26 ppm (Н6). Comparison of the signals of protons for the ligands 1, 2 and complexes 5, 6 (Table 3) revealed that the complex formation with PdCl2 did not significantly change the 1Н NMR spectra as well as the 31P NMR spectra: the signal of the phosphorus atom was shifted downfield by 0.1–2.5 ppm. Those data evidenced no coordination with the P=O group during the complex formation.
Ligands 1, 2 and their complexes with Pd(II) 5, 6 were tested for cytotoxicity against human cells cultures: А549 (lung carcinoma), НСТ116 (colon carcinoma), HeLa (cervix of the uterus adenocarcinoma), and MCF7 (breast adenocarcinoma). The obtained data are collected in Table 4.
The tested compounds revealed weaker activity in comparison with the reference (Camptothecin). The palladium complex 5 was inactive towards the used cell cultures, and other compounds were inactive toward the НCT116 cells (colon carcinoma). Ligands 1 and 2 revealed almost equal cytotoxicity against the А549, HeLa, and MCF7 cells. At the same time, complex 6 revealed twice higher cytotoxicity against the А549, HeLa, and MCF7 in comparison with the starting ligand 2. Overall, the Pd(II) complex of ligand 2 with the short linker was the most active of the tested compounds.
In summary, we prepared tris[2-(3′-cyanopropoxy)phenyl]phosphine oxide, and its molecular structure was elucidated by means of X-ray diffraction analysis. A new tripodal ligand, tris{2-[3′-(tetrazol-5′′-yl)propoxy]phenyl}phosphine oxide, and its complex with PdCl2 were synthesized. Anticancer activity of the obtained compounds towards human cells was determined. The complex with tris[2-(tetrazol-5′-ylmethoxy)phenyl]phosphine oxide was found to be the most promising for further investigation.
EXPERIMENTAL
Organic solvents (reagent grade) were dried and purified via conventional methods [37]. Deuterated solvents: CDCl3, DMSO-d6 (Acros), and 4-bromobutyronitrile (Aldrich, 97%) were used as received. The salts: PdCl2 (reagent grade, Reakhim), sodium azide (Acros), and NH4Cl (reagent grade, Reakhim) were used as received. K2CO3 (reagent grade, Reakhim) was calcined prior to use. Tris(2-hydroxyphenyl)phosphine oxide 3 was synthesized as described elsewhere [30]. Tris[2-(tetrazol-5′-ylmethoxy)phenyl]phosphine oxide 2 has been synthesized by us earlier.
IR spectra of solid compounds 1, 5, and 6 were obtained using a Bruker Tensor 37 IR spectrometer (resolution 2 cm–1, range 4000–400 cm–1, KBr pellets or Nujol suspension). IR spectra of the solutions in DMSO-d6 (c 0.04 M, CaF2 cuvettes with pathlength of 0.06 mm) were recorded over the 4000–1100 cm–1 range. Raman spectra of the powders were recorded over the 3500– 100 cm–1 range using a Jobin-Ivon LabRAM 300 spectrometer equipped with a microscope and laser CCD detector. A 632.8 nm He–Ne laser (power not exceeding 2 mW) was used as the excitation source. 1Н, 13С{1Н}, and 31P{1Н} NMR spectra of the solutions in CDCl3 and DMSO-d6 were recorded using a Bruker Avance 400 instrument operating at 400.13, 100.61, and 161.98 MHz (1Н, 13С, and 31P, respectively). The signals of residual protons and carbon atoms were used as internal references in the 1Н and 13С NMR spectra, the accuracy of the chemical shifts determination being better than ±0.01 and ±0.03 ppm, respectively. The chemical shifts in the 31P NMR spectra were determined relative to 85% H3PO4 as external reference. The signals were assigned by comparison with the spectra of similar compounds [31]. Melting points were determined in a melting point apparatus using short Anschutz thermometers. The С, H, and N content was determined using a Carlo Erba 1106 instrument, the content of P was determined as described elsewhere [38].
Tris[2-(3′-cyanopropoxy)phenyl]phosphine oxide (4). A mixture of 0.74 g (0.005 mol) of 4-bromobutyronitrile, 2.1 g (0.015 mol) of freshly calcined K2CO3, and 0.49 g (0.0015 mol) of tris(2-hydroxyphenyl)phosphine oxide 3 in 20 mL of anhydrous DMF was heated at 95°C with stirring during 4 h. The solvent was removed, and 15 mL of CH2Cl2 was added to the dry residue; the obtained mixture was washed with water (4×15 mL). The organic solution was dried with Na2SO4, and the solvent was removed. Light yellow oil crystallizing during keeping was obtained. The product was recrystallized from a CH2Cl2–Et2O mixture to obtain white crystalline substance. Yield 0.76 g (96.2%), mp 159°С. The crystals suitable for X-ray diffraction analysis were prepared via recrystallization from ethanol. IR spectrum (KBr), ν, cm–1: 2935 m, 2875 w, 2241 m (C≡N), 1591 s, 1575 m, 1481 m, 1465 m, 1441 vs, 1383 m, 1282 vs, 1253 m, 1176 m (P=O), 1157 m, 1136 m, 1086 m, 1047 s, 953 m, 933 m, 826 m, 772 s, 754 m, 739 m, 705 m, 615 w, 558 s, 530 m, 509 m, 414 m. 1Н NMR spectrum (CDCl3), δ, ppm: 1.77 quintet (6Н, CH2CH2CH2, 3JHH 6.0 Hz), 2.03 t (6Н, CH2CN, 3JHH 7.0 Hz), 4.02 t (6Н, CH2O, 3JHH 5.4 Hz), 6.98 d. d (3Н, Н3, 3JHH 8.4, 4JPH 5.6 Hz), 7.10 t. d (3Н, Н5, 3JHH 7.5, 4JPH 2.1 Hz), 7.48 d. d. d (3Н, Н6, 3JPH 14.8, 3JHH 7.6, 4JHH 1.2 Hz), 7.57 t. d (3Н, Н4, 3JHH 7.8, 4JHH 1.4 Hz). 13С NMR spectrum (CDCl3), δC, ppm: 13.79 (CH2CH2CH2), 24.82 (CH2CN), 65.60 (СН2О), 111.96 d (С3, 3JPС 6.5 Hz), 119.27 (CN), 119.48 d (С1, 1JPС 111.2 Hz), 121.27 d (С5, 3JPС 12.8 Hz), 134.24 d (С6, 2JPС 9.2 Hz), 134.44 d (С4, 4JPС 1.7 Hz), 160.32 d (С2, 2JPС 1.9 Hz). 31P{1Н} NMR spectrum (CDCl3): δP 28.1 ppm. Found, %: C 67.80; H 5.90; N 7.75; P 5.74. C30H30N3O4P. Calculated, %: C 68.30; H 5.73; N 7.97; P 5.87.
Tris{2-[3′-(tetrazol-5′′-yl)propoxy]phenyl}phosphine oxide (1). 0.59 g (0.009 mol) of NaN3 and 0.48 g (0.009 mol) of NH4Cl were added at vigorous stirring to a solution of 0.53 g (0.001 mol) of tris(3-cyanopropoxyphenyl)phosphine oxide in 15 mL of anhydrous DMF at room temperature. The obtained suspension was kept at 130°С during 80 h and cooled to room temperature; 25 mL of water was then added. The obtained solution was extracted with СНСl3 (5×10 mL). The organic fractions were united, washed with water (4×10 mL), and dried with Na2SO4. After removal of the solvent, the residue was a low-viscous oil which was recrystallized from diethyl ether. Yield 0.49 g (74.2%), mp 155–156°С. IR spectrum, ν, cm–1: 3435 br. w (H2O), 2700 br. m (NH)), 1900 br. vs, 1592 vs, 1578 s, 1574 sh, 1567 sh, 1561 w. sh, 1477 vs, 1444 vs, 1388 m, 1280 vs, 1249 m, 1236 m, 1211 s, 1160 m, 1142 s, 1116 m, 1098 sh, 1089 s, 1050 s, 1040 s, 1027 sh, 1009 m, 925 w, 853 w, 802 sh, 764 s, 750 m, 730 s, 720 s, 698 m, 587 w, 577 w, 563 m, 552 m, 532 m, 514 m, 487 m, 450 w, 429 w. 1Н NMR spectrum (DMSO-d6), δ, ppm: 1.76 br. s (6Н, CH2CH2CH2), 2.65 br. s (6Н, CH2-tetrazole), 3.87 br. s (6Н, СН2О), 6.85–7.25 m (6Н, Н3, Н5), 7.42–7.60 m (6Н, Н4, Н6). 13С NMR spectrum (DMSO-d6), δC, ppm: 19.28 (CH2CH2CH2), 26.52 (CH2-tetrazole), 66.62 (СН2О), 113.03 d (С3, 4JPС 6.0 Hz), 119.28 d ( С1, 1JPС 115.8 Hz), 120.95 d (С5, 3JPС 12.5 Hz), 134.31 d (С6, 2JPС 8.4 Hz), 134.68 (С4), 155.67 (С=N), 160.49 d (С2, 2JPС 1.7 Hz). 31Р{1Н} NMR spectrum (DMSO-d6): δP 28.0 ppm. Found, %: C 54.22; H 5.28; N 25.02; P 4.71. C30H33N12O4P∙0.5H2O. Calculated, %: C 54.13; H 5.15; N 25.25; P 4.65.
Dichloro(tris{2-[3′-(tetrazol-5′′-yl)propoxy]phenyl}phosphine oxide)palladium(II) (5). 21.3 mg of PdCl2 (0.12 mmol) was dissolved in 2 mL of EtOH on heating with addition of several drops of 6 M HCl. The obtained solution was added dropwise with stirring to a solution of ligand 1 in 5 mL of EtOH; the formed light yellow precipitate was filtered off, washed with diethyl ether, and dried. Yield 0.10 g (95.2%), mp >280°С (decomp.). IR spectrum (KBr), ν, cm–1: 3446 br, 3100–2200 (NH), 1900 br. w, 1590 vs, 1575 s, 1558 sh, 1543 sh, 1473 s, 1443 vs, 1386 w, 1282 vs, 1245 s, 1166 m, 1141 m, 1111 m, 1087 m (P=O), 1063 sh, 1042 br. m, 942 br. m, 829 w, 758 m, 706 w, 557 m, 519 br. m. Raman spectrum, ν, cm–1: 3071, 2937, 2885, 1588, 1481, 1436, 1404, 1281, 1247, 1164, 1143, 1040, 943, 825, 801, 670, 604, 349, 299. 1Н NMR spectrum (DMSO-d6), δ, ppm: 1.74 br. s (6H, CH2CH2CH2), 2.63 t (6H, CH2-tetrazole, 3JHH 5.6 Hz), CH2О signal was overlapped with the water signal, 6.98 t (3H, H5, 3JHH 7.0 Hz), 7.08 d. d (3H, H3, 3JHH 8.4, 4JPH 5.4 Hz), 7.47 d. d (3H, H6, 3JPH 15.2, 3JHH 7.6 Hz), 7.53 t (3H, H4, 3JHH 7.8 Hz). 31Р{1Н} NMR spectrum (CDCl3): δP 28.1 ppm. Found, %: С 40.99; H 4.16; N 18.67; Cl 7.84; P 3.13. C30H33Cl2N12O4PPd·2.5H2O. Calculated, %: С 40.99; H 4.36; N 19.12; Cl 8.07; P 3.52.
Dichloro(tris{2-[3′-(tetrazol-5′′-yl)methoxy]phenyl}phosphine oxide)palladium(II) (6). 29.8 mg of PdCl2 (0.168 mmol) was dissolved in 2 mL of EtOH on heating with addition of several drops of 6 M. HCl. The obtained solution was added dropwise with stirring to a solution of ligand 2 in 5 mL of MeOH; the formed light yellow precipitate was filtered off, washed with diethyl ether, and dried. Yield 0.12 g (93.0%), mp >240°C (decomp.). IR spectrum (KBr), ν, cm–1: 3446 br, 3100–2200 (NH), 1900 br. w, 1591 vs, 1576 s, 1556 sh, 1476 vs, 1442 vs, 1282 vs, 1219 m, 1167 m, 1142 m, 1120 sh, 1090 m (P=O), 1064 sh, 1041 sh, 1028 s, 857 w, 803 w, 758 m, 698 m, 557 m, 500 sh. Raman spectrum, ν, cm–1: 3068, 2942, 1588, 1575, 1474, 1451, 1284, 1257, 1162, 1221, 1144, 1041, 851, 799, 662, 587, 357, 300. 1H NMR spectrum (DMSO-d6), δ, ppm: 5.38 br. s (6H, CH2), 6.96 t (3H, H5, 3JHH 7.0 Hz), 7.26 d. d (3H, H6, 3JPH 14.8, 3JHH 7.2 Hz), 7.34 t. d (3H, H3, 3JHH 8.4, 4JPH 6.0 Hz), 7.58 t (3H, H4, 3JHH 7.6 Hz). 31P{1H} NMR spectrum (CDCl3): δP 29.5 ppm. Found, %: C 37.51; H 3.00; N 21.52; Cl 8.79; P 3.77. C24H21Cl2N12O4PPd·H2O. Calculated, %: C 37.54; H 3.02; N 21.89; Cl 9.23; P 4.03.
X-ray diffraction analysis of tris[2-(3′-cyanopropoxy)phenyl]phosphine oxide 4. Single crystals of compound 4 were obtained via crystallization from ethanol. Intensity of 309409 reflections was measured using a Bruker Apex II CCDc diffractometer with graphite monochromator (MoKα radiation, λ 0.71073 Å, 2θ-scanning) at 120.0(2) K. Crystallographic data for compound 4: C30H30N3O4P, M 527.54, monoclinic crystals, space group P21/n, a 11.0546(4), b 14.3483(5), c 17.0770(6) Å, β 96.596(1)°, V 2690.7(2) Å3, Z 4, dcalc 1.302 g/cm3, μ 0.143 mm–1, 12087 independent reflections (Rint 0.0394), 8661 observed reflections, final convergence factors R1 [I > 2σ(I)] 0.047, wR(F2) 0.130 and GOF 1.01. The structure was solved using SHELXT software [39] and refined via full-matrix least squares method over F2. The non-hydrogen atoms were refined under anisotropic approximation, the positions of the hydrogen atoms were calculated, and the hydrogen atoms were included in the refinement using the rider model with Uiso(H) = 1.2Ueq(C). The calculations were performed using SHELXL2014 [40] and OLEX2 [41] software package. CCDC 2085004 contains additional crystallographic information relevant to this paper. These data are available free of charge via http://www.ccdc.cam.ac.uk/structures/.
Cytotoxicity (in vitro МТТ-test). The А549 and НСТ116 human cells were cultivated in DMEM medium, and MCF7 and HeLa human cells were cultivated in ЕМЕМ medium with addition of 10% fetal calf serum, 2 mM of glutamine, and 1% gentamicin as antibiotic at 37°С under humid СО2 atmosphere (5%). The cells of adhesion cultures А549, НСТ116, MCF7, and HeLa were seeded at concentration of 1×104 cells/200 μL in a 96-well plate and cultivated at 37°С under humid atmosphere with 5% of СО2. Upon 24 h of incubation, 100 to 1.56 рМ of the studied compounds was added to the cell cultures, and they were cultivated for further 72 h under the same conditions. The experiments were run in triplicate at each concentration. The compounds were dissolved in DMSO, final concentration of which in the well did not exceed 1% and was not toxic for the cells. Pure solvent (final concentration 1%) was added to the reference cells. Upon the incubation, 20 μL of МТТ (5 mg/mL) was added to each well, and the plate was incubated during 2 h. The medium was then removed from the wells, and 100 μL of DMSO was added to each well for dissolution of the formed formazan crystals. Absorbance at 536 nm was measured using a Cytation3 plate scanner (BioTeklnstruments, Inc). The value of the concentration inducing 50% inhibition of the cells growth (IC50, μM) was determined from the dose-dependent curves using OriginPro 9.0 software.
REFERENCES
Gaponik, P.N., Voitekhovich, S.V., and Ivashkevich, O.A., Russ. Chem. Rev., 2006, no. 75, p. 507. https://doi.org/10.1070/RC2006v075n06ABEH003601
Popova, E.A., Trifonov, R.E., and Ostrovskii, V.A., Arkivoc, 2012, vol. 45. https://doi.org/10.3998/ark.5550190.0013.102
Massi, M., Stagni, S., and Ogden, M.I., Coord. Chem. Rev., 2018, vol. 375, p. 164. https://doi.org/10.1016/j.ccr.2017.11.017
Aromi, G., Barrios, L.A., Roubeau, O., Gamez, P., Coord. Chem. Rev., 2011, vol. 255, p. 485. https://doi.org/10.1016/j.ccr.2010.10.038
Feng, Y., Cai, S.-L., Gao, Y., and Zheng, S.-R., J. Solid State Chem., 2018, vol. 265, p. 64. https://doi.org/10.1016/j.jssc.2018.05.031
He, H., Song, Y., Sun, F., Zhao, N., and Zhu, G., Cryst. Growth Des., 2015, vol. 15, p. 2033. https://doi.org/10.1021/acs.cgd.5b00229
Haiges, R. and Christe, K.O., Inorg Chem., 2013, vol. 52, p. 7249. https://doi.org/10.1021/ic400919n
Wang, J., Deng, S.-Q., Zhao, T.-T., Zheng, S.-R., Cai, S.-L., Fan, J., and Zhang, W.-G., Dalton Trans., 2020, vol. 4, p. 12150. https://doi.org/10.1039/d0dt01666g
Zeni, W., Seifried, M., Knoll, C., Welch, J.M., Giester, G., Stöger, B., Artner, W., Reissner, M., Müller, D., and Weinberger, P., Dalton Trans., 2020, vol. 49, p. 17183. https://doi.org/10.1039/d0dt03315d
Kusz, J., Gütlich, P., and Spiering, H., Top. Curr. Chem., 2004, vol. 234, p. 129. https://doi.org/10.1007/b95415
Wei, W., Wang, G., Zhang, Y., Jiang, F., Wu, M., and Hong, M., Chem. Eur. J., 2011, vol. 17, p. 2189. https://doi.org/10.1002/chem.201002246
Białońska, A., Bronisz, R., Rudolf, M.F., and Weselski, M., Inorg. Chem., 2012, vol. 51, p. 237. https://doi.org/10.1021/ic201535x
Boland, Y., Hertsens, P., Marchand-Brynaert, J., and Garcia, Y., Synthesis, 2006, vol. 9, p. 1504. https://doi.org/10.1055/s-2006-926439
Giraud, M., Andreiadis, E.S., Fisyuk, A.S., Demadrille, R., Pécaut, J., Imbert, D., and Mazzanti, M., Inorg. Chem., 2008, vol. 47, p. 3952. https://doi.org/10.1021/ic8005663
Ostrovskii, V.A., Trifonov, R.E., and Popova, E.A., Russ. Chem. Bull., 2012, vol. 61, p. 768. https://doi.org/10.1007/s11172-012-0108-4
Zou, Y., Liu, L., Liu, J., and Liu, G., Future Med. Chem., 2020, vol. 12, p. 91. https://doi.org/10.4155/fmc-2019-0288
Li, Y., Pasunooti, K.K., Li, R.J., Liu, W., Head, S.A., Shi, W.Q., and Liu, J.O., J. Med. Chem., 2018, vol. 61, p. 11158. https://doi.org/10.1021/acs.jmedchem.8b01252
Gao, F., Xiao, J., and Huang, G., Eur. J. Med. Chem., 2019, vol. 184, p. 111744. https://doi.org/10.1016/j.ejmech.2019.111744
Zhang, J.Y., Wang, S., Ba, Y.Y., and Xu, Z., Eur. J. Med. Chem., 2019, vol. 178, p. 341. https://doi.org/10.1016/j.ejmech.2019.05.071
Popova, E.A., Protas, A.V., and Trifonov, R.E., Anti-Cancer Agents Med. Chem., 2018, vol. 17, p. 1856. https://doi.org/10.2174/1871520617666170327143148
Wang, S.Q., Wang, Y.F., and Xu, Z., Eur. J. Med. Chem., 2019, vol. 170, p. 225. https://doi.org/10.1016/j.ejmech.2019.03.023
Gao, C., Chang, L., Xu, Z., Yan, X.-F., Ding, C., Zhao, F., Wu, X., and Feng, L.-S., Eur. J. Med. Chem., 2019, vol. 163, p. 404. https://doi.org/10.1016/j.ejmech.2018.12.001
Roh, J., Karabanovich, G., Vlčková, H., Carazo, A., Němeček, J., Sychra, P., Valášková, L., Pavliš, O., Stolaříková, J., Klimešová, V., Vávrová, K., Pávek, P., and Hrabálek, A., Bioorg. Med. Chem., 2017, vol. 25, p. 5468. https://doi.org/10.1016/j.bmc.2017.08.010
Zhan, P., Li, Z., Liu, X., and Clercq, D.E., Mini Rev. Med. Chem., 2009, vol. 9, p. 1014. https://doi.org/10.2174/138955709788681618
Kudryavtsev, I.Yu., Baulina, T.V., Pasechnik, M.P., Matveev, S.V., Petrovskii, P.V., and Nifant’ev, E.E., Russ. Chem. Bull., 2013, vol. 62, no. 4, p. 1086. https://doi.org/10.1007/s11172-013-0146-6
Kudryavtsev, I.Yu., Baulina, T.V., Pasechnik, M.P., Matveev, S.V., and Matveeva, A.G., Phosphorus, Sulfur, Silicon, Relat. Elem., 2014, vol. 189, nos. 7–8, p. 946. https://doi.org/10.1080/10426507.2014.904865
Matveeva, A.G., Kudryavtsev, I.Yu., Pasechnik, M.P., Vologzhanina, A.V., Baulina, T.V., Vavina, A.V., Sukat, G.Ya., Matveev, S.V., Godovikov, I.A., Turanov, A.N., Karandashev, V.K., and Brel, V.K., Polyhedron, 2018, vol. 142, p. 71. https://doi.org/10.1016/j.poly.2017.12.025
Turanov, A.N., Matveeva, A.G., Kudryavtsev, I.Yu., Pasechnik, M.P., Matveev, S.V., Godovikova, M.I., Baulina, T.V., Karandashev, V.K., and Brel, V.K., Polyhedron, 2019, vol. 161, p. 276. https://doi.org/10.1016/j.poly.2019.01.036
Kudryavtsev, I.Y., Bykhovskaya, O.V., Matveeva, A.G., Baulina, T.V., Pasechnik, M.P., Matveev, S.V., Vologzhanina, A.V., Turanov, A.N., Karandashev, V.K., and Brel, V.K., Monat. Chem., 2020, vol. 151, p. 1705. https://doi.org/10.1007/s00706-020-02702-6
Kudryavtsev, I.Yu., Baulina, T.V., Khrustalev, V.N., Petrovskii, P.V., Pasechnik, M.P., and Nifant’ev, E.E., Doklady Chem., 2013, vol. 448, no. 2, p. 55. https://doi.org/10.1134/S0012500813020092
Baulina, T.V., Pasechnik, M.P., Kudryavtsev, I.Yu., Bykhovskaya, O.V., Sukat, G.Ya., Smol’yakov, A.F., Anikina, L.V., and Brel, V.K., J. Mol. Struct., 2020, vol. 1217, p. 128324. https://doi.org/10.1016/j.molstruc.2020.128324
Livant, P.D., Mao, J., and Webb, T.R., Acta Crystallogr., 1996, vol. 52, p. 2924. https://doi.org/10.1107/S0108270196008906
Baulina, T.V., Kudryavtsev, I.Y., Smolyakov, A.F., Pasechnik, M.P., and Brel, V.K., Heteroatom. Chem., 2018, p. e21454. https://doi.org/10.1002/hc.21454
Matveeva, N.A., Sushko, N.I., Makarevich, N.I., Gaponik, P.N., Ivashkevich, O.A., and Koren’, A.O., Zh. Prikl. Spektr., 1992, vol. 57, nos. 5–6, p. 442.
Al-Janabi, A.S.M., Alheety, M.A., Al-Samrai, O.A.Y., Shaaban, S., Kibar, B., and Cacan, E., Inorg. Chem. Commun., 2020, vol. 121, p. 108193. https://doi.org/10.1016/j.inoche.2020.108193
Nakamoto, K., Infrared and Raman Spectra of Inorganic and Coordination Compounds, Hoboken: J. Wiley & Sons, Inc., 2009, pt. B.
Armarego, W.L.F., Purification of Laboratory Chemicals, Amsterdam: Elsevier, 2017. https://doi.org/10.1016/B978-0-12-805457-4.50008-2
Gel’man, N.E., Terent’eva, E.A., Shanina, T.M., and Kiparenko, L.M., Metody kolichestvennogo organicheskogo elementnogo mikroanaliza (Quantitative Organic Elemental Microanalysis Methods), Moscow: Khimija, 1987, p. 296.
Sheldrick, G.M., Acta Crystalogr. (A), 2015, vol. 71, no. 1, p. 3. https://doi.org/10.1107/S2053273314026370
Sheldrick, G.M., Acta Crystallogr. (C), 2015, vol. 71, no. 1, p. 3. https://doi.org/10.1107/S2053229614024218
Dolomanov, O.V., Bourhis, L.J., Gildea, R.J., Howard, J.A.K., and Puschman, H., J. Appl. Cryst., 2009, vol. 42, no. 2, p. 339. https://doi.org/10.1107/S0021889808042726
Funding
This study was financially supported by the Russian Science Foundation (project no. 20-13-00329). Elemental analysis and recording of NMR, IR, and Raman spectra were financially supported by the Ministry of Science and Higher Education of the Russian Federation and performed using the equipment of the Center for Molecular Structure Studies, Nesmeyanov Institute of Organoelement Compounds, RAS.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
V.K. Brel is a member of the Editorial Board of Russian Journal of General Chemistry. No conflict of interest was declared by other authors.
Additional information
Translated from Zhurnal Obshchei Khimii, 2021, Vol. 91, No. 10, pp. 1617–1626 https://doi.org/10.31857/S0044460X2110019X.
Rights and permissions
Open Access. This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Baulina, T.V., Kudryavtsev, I.Y., Bykhovskaya, O.V. et al. Tripodal Phosphine Oxide Ligand with Tetrazole Functionality. Russ J Gen Chem 91, 2052–2060 (2021). https://doi.org/10.1134/S1070363221100194
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S1070363221100194