
Abstract—
Humic substances (HS) are heterogeneous and polydisperse compounds formed in soils, sediments and waters during the decay and transformation of organic residues (the process called humification). The role of extracellular free-radical condensation reactions (secondary synthesis) in the formation of HS is a subject of debate. Here we have studied the formation of HS from a mixture of monomers under the dynamic conditions and at low substrate concentrations in the presence of laccase from the fungus Coprinus comatus F-2940. Laccase was immobilized on kaolinite modified with aluminum hydroxide. We have investigated some properties of the enzyme, reactivity of phenolic acids and amino acids in the presence of laccase. The optimum of 2.6-dimethoxyphenol (2.6-DMP) oxidation by free laccase was at pH 6.0. Upon immobilization, it shifted to the acidic region (pH 4.4), the thermal stability decreased, however the kinetic parameters of 2.6-DMP oxidation remained unchanged. In terms of reactivity (% of substrate conversion by free laccase) the individual phenolic acids formed a series: caffeic (72) > ferulic (53) > gallic = syringic (43) > protocatechuic (5.5) > vanillic = p-hydroxybenzoic (0). In the mixture of phenolic acids, gallic acid was most efficiently oxidized (50%), while the other acids were oxidized in comparable amounts (13–17%). The conversion of phenolic acids increased in the presence of lysine. When a mixture of gallic, protocatechuic, syringic, ferulic acids (0.01 mM each) and lysine (0.02 mM) was passed through a flow-through microcolumn, immobilized laccase effectively oxidized the phenolic acids, the reaction products bound to the mineral phase, staining it dark. According to high performance liquid chromatography, the molecular weights of compounds extracted from the mineral phase did not exceed 900 Da, thus fulvic acid-like substances were formed. Results of the study suggest an important role of free-radical heterophase reactions in the formation of the molecular composition of the liquid phase and organo-mineral complexes.
Similar content being viewed by others


INTRODUCTION
Soils are the main reservoir of organic carbon in terrestrial ecosystems. The amount of carbon stored in humus (1500 Gt C) is three times larger than that in the aboveground biomass (550 Gt C) [19]. Humus determines the fertility of soils and their functions in the biosphere [9], the processes of humus formation and decomposition have the profound influence on modern climate. However, the nature and composition of substances, comprising humus, as well as the mechanisms of their long-term stabilization in soils remain the subject of debate [1, 10, 29, 37, 47].
According to traditional views, humus is represented mostly by humic substances (HS), heterogeneous and polydisperse dark-colored compounds formed de novo during the degradation and transformation of organic residues (humification) [5, 8]. It is hypothesized that extracellular condensation reactions play an important role in the formation of HS in soils [5, 8, 20, 65]. This point of view was reflected in Stevenson’s definition of HS as “a series of relatively high molecular weight compounds, yellow to black in color and formed by secondary synthesis reactions” [65]. According to solubility characteristics, HS are divided into humic acids (HA, soluble under the alkaline conditions, precipitate at pH<2), fulvic acids (FA, soluble at all pH values) and humin (insoluble residue). Humic and fulvic acids are enriched by polar functional groups and are most reactive components of humus [8, 65].
Humic substances have long time been described as macromolecular aromatic compounds (5–100 kDa), resistant to biodegradation [8, 9]. This view is now being reconsidered. The notion of HS as supramolecular associations of relatively small molecules (about 2–6 kDa) held together by weak dispersive forces is practically generally accepted [59, 66]. Secondary synthesis has been questioned, as well as the existence of HS as mass products of this process [47]. A soil continuum model has been proposed which describes soil organic matter as a continuum of biomolecules at different stages of decomposition [47]. As a result of the renewed debate, HS have been redefined by International Humic Substances Society (IHSS) as a “complex and heterogeneous mixtures of polydispersed materials formed in soils, sediments, and natural waters by biochemical and chemical reactions during the decay and transformation of plant and microbial remains (a process called humification)” (www.humic-substances.org, access April, 2022).
A number of reviews have been published recently defending HS as specific compounds of soils and other natural environments [28, 37, 56] and supporting secondary synthesis reactions as playing an important role in synthesis of HS [29]. Oxidation of phenols to phenoxy radicals and quinones followed by spontaneous free-radical coupling with nitrogenous and other precursors [5, 20, 65] is a classic example of such reactions, underlying the condensation theory of humification [5, 65]. The oxidation of phenols is a catalytic process, which is carried out in the presence of extracellular enzymes (phenol oxidases and peroxidases of microorganisms) [5, 21, 36] or abiotic catalysts—for example, the ions of Fe3+ and Mn4+ in soil minerals [41]. Indeed, the formation of dark-colored compounds from monomeric phenolic and nitrogenous precursors has been shown in vitro in the presence of peroxidase [5, 14], laccase [52, 75], tyrosinase [55] or soil minerals [41, 43]. The formation of HA-like substances from phenolic fungal metabolites and nitrogenous compounds have been shown in vivo in the liquid cultures of microscopic fungi such as Epicoccum nigrum, Stachybotrys atra, S. chartarum, Aspergillus sydowi (Haider and Martin, 1967; Martin and Haider, 1969; Haider and Martin, 1970; cited from [61]). The enzymatic catalysis of condensation reactions is more effective than abiotic one; the molecular weights of the products are higher [12, 21, 58, 55].
Condensation processes are facilitated by the presence of an interfaces. The amounts of HA-like substances formed in fungal cultures increased in the presence of clay minerals (Martin et al., 1972; cited from [61]). More high molecular weight products were synthesized from monomeric phenolic and nitrogenous precursors in the presence of immobilized laccase, than in a homogeneous system [75, 76]. Formation of HA-like dark-colored compounds was observed during solid-state cultivation of the white rot fungus on wheat straw [73]. It was also shown that low-molecular weight products of HA degradation can repolymerize on the fungal mycelium during submerged cultivation of ligninolytic fungus [77].
A significant drawback of laboratory experiments on secondary synthesis is that they are carried out in closed systems and at high precursor concentrations, usually 1–10 mg/mL [5, 64]. This corresponds to 5–50 mM at molecular mass of precursors (phenolic acids, amino acids) of about 200 g/mol while the concentration of phenolic acids in soil solutions usually do not exceed tens of micromoles [7, 57]. Free amino acids in soil solutions account for less than 1% of dissolved organic nitrogen, concentrations are usually 0.1–50 µM [46, 74]. The question arises about the possibility of biocatalytic secondary synthesis reactions at low substrate concentrations in open dynamic soil systems. To what extent does polymerization occur under such conditions?
Here we have studied formation of humic compounds in heterophase system at low precursor concentrations and under flow-through conditions. Laccase (EC 1.10.3.2., benzenediol : oxygen oxidoreductase) was used as a biocatalyst. Laccase is common in soils [23, 63]. The enzyme belongs to the family of copper-containing oxidases, contains 4 copper atoms in the active center and catalyzes the oxidation of a broad range of phenolic substrates and aromatic amines by molecular oxygen, which is reduced to water [33, 45]. Phenoxy radicals and quinones formed at catalytic stage of substrate oxidation can undergo spontaneous condensation reactions to form oligomers and polymers [45, 72]. Laccase is produced by ascomycetes, basidiomycetes, and deuteromycetes [16, 72], laccase-like enzymes have been also found in bacteria [45]. The enzyme shows activity in litter and soil humus horizons [17, 30], which suggests its involvement in heterophase condensation reactions during humification.
Laccase from soil basidiomycete Coprinus comatus F 2940 was used in the study. The enzyme was immobilized on kaolinite modified with aluminum hydroxide. We have studied some properties of free and immobilized laccase, its substrate specificity towards phenolic acids and the ability of certain amino acids to form complexes with phenolic acids in the presence of laccase. Experiments on heterophase synthesis of HS were carried out in a flow-through microcolumn [3, 4].
MATERIALS AND METHODS
Mineral. Kaolinite (Prosyanovskoe deposit, Ukraine) modified with amorphous aluminum hydroxide at the amount of 2.5 mmol Al/g clay have been used as the mineral phase. Some properties of the mineral were described earlier [3].
Phenolic acids and amino acids. Gallic (GAL), protocatechic (PCAT), p-hydroxybenzoic (HDB), vanillic (VAN), ferulic (FER) and syringic (SYR) acids (Sigma-Aldrich, USA) were used as phenolic precursors of HS [4]. Tyrosine, L-DOFA, glycine, lysine, arginine and tryptophan were used as nitrogenous precursors of HS. All the experiments with phenolic and amino acids were carried out in 5 mM Na-acetate buffer (pH 4.5)—buffer A, using ultra-pure distilled water with conductivity of 18 μS.
Laccase. The strain of Coprinus comatus VKM F-2940 was taken from the All-Russian Collection of Microorganisms (VKM, http://vkm.ru/). For enzyme production the inoculum was grown for 7 days on medium of the following composition (g/L): peptone—2; yeast extract—2; MgSO4—0.2; KH2PO4—0.3; and glucose, 2%. The inoculum was introduced into 750-mL flasks containing 200 mL of Kirk’s high nitrogen medium in 20 mM tartrate buffer (pH 4.5) (g/L): CaCl2—0.01; MgSO4—0.1; KH2PO4—0.3; glucose—1%; Tween 80—0.05%; α-asparagine—0.9; NH4NO3—0.5 and laccase inducers—Cu2+ ions (0.1 mM) and Mn2+ ions (0.5 mM). On day 10, the culture liquid was separated from mycelium for enzyme purification.
Laccase was purified to an electrophoretically homogeneous state. The purification stages were as follows: 1—desalting. The enzyme was precipitated from the culture liquid by (NH4)2SO4 at 90% saturation followed by centrifugation at 15000 g for 30 min. The precipitate was dissolved in 20 mM Na-acetate buffer, pH 5.0 (buffer B) and dialyzed against this buffer (10 kDa membrane); 2—ion-exchange chromatography on DEAE-Toyopearl. Protein preparation was applied to the column equilibrated with buffer B, washed with three volumes of this buffer and eluted at the rate of 1 mL/min with 0–1 M gradient of NaCl; 3—gel filtration on HiLoad 26/60 Superdex 200 column (GE Healthcare, USA). Protein preparation was applied to a column equilibrated with buffer B containing 0.1 M NaCl and eluted at a rate of 1 mL/min. The fractions containing laccase activity were dialyzed against buffer A; 4—ion-exchange chromatography on an UNOQ6 column (Bio-Rad, Hercules, CA, USA). The dialyzed preparation was applied to the column equilibrated with buffer B, washed with three volumes of this buffer and eluted by a linear gradient of 0–1 M NaCl in buffer B. The purified laccase preparation was dialyzed against buffer B and used for further work.
The molecular weight of laccase (SDS-PAAG) was 71.3 kDa; the protein concentration in the laccase preparation was 0.03 mg/mL (Bradford technique).
The enzyme activity was determined spectrophotometrically (Shimadzu 1800 spectrophotometer, Japan) by the rate of oxidation of ABTS (2.2-azino-bis-(3-ethylbenzthiazoline-6-sulfonate) in buffer A at 420 nm. The coefficient of extinction of ABTS at 420 nm is 36 000/M/cm [38]. The amount of enzyme catalyzing the oxidation of 1 micromole of ABTS per one minute was taken as one unit (U) of laccase activity.
Laccase immobilization. The mineral was equilibrated with buffer A by shaking the suspension for 5 min at 1400 rpm (Biosan thermoshaker TS-100C, Latvia). The mixture was then centrifuged for 1 min at 18 000 g (Elmi CM-50 centrifuge, Latvia). The supernatant was replaced with 0.5 mL of enzyme preparation in buffer A and shaken for 1 h at 25°C. Then, the mixture was centrifuged and the activity in the supernatant was measured using 1 mM ABTS in buffer A as a substrate. The mineral was washed with 1 mL of buffer A and the activity was assessed in the washings. The efficiency of immobilization (%) was determined as:
where Ainitial—activity added to the mineral, Asupern—activity in the supernatant, Awashings—activity in the washings.
We have used 0.08 µg of laccase and 2 mg of the mineral when studied the properties of the immobilized enzyme and 0.71 µg of laccase and 20 mg of the mineral in the dynamic experiment. The immobilization efficiency was 90–100%.
The activity of the immobilized laccase was measured by adding 1.5 mL of 1.0 mM ABTS in buffer A to the mineral. The mixture was shaken on a thermoshaker (2 min), centrifuged (1 min), the 100 µL aliquot of the supernatant was taken and diluted in 900 µL of buffer A. The procedure was repeated three times. The activity was estimated as an increase in optical density at 420 nm and calculated as U of activity per unit weight of the mineral taken for the experiment.
Properties of laccase. The рН-optimum of free and immobilized enzyme was determined with 2 mM 2.6 dimethoxyphenol (2.6-DMP) in 5 mM Na-acetate buffer in the pH range of 4.0–5.8. Thermostability was determined by incubation of the enzyme at a given temperature for 30 min (1400 rpm, 25°С) and measuring the residual activity with 1 mM ABTS. Kinetic constants were determined with 2.6-DMP in buffer A at рН 4.5.
Oxidation of individual phenolic acids by free laccase. Reactions were performed in 2 mL Eppendorf tubes. Substrate specificity of laccase was assessed using 1 mL reaction mixtures containing 2 mM of each acid and 0.05 U of laccase in buffer A. The incubation time was 1, 3 and 5 hours. Reactions were performed at 25°C and 1400 rpm (on thermoshaker) and terminated with 10 mM NaN3. Conversion of phenolic acids by laccase was evaluated by loss of the initial substrate in the incubation mixtures. Concentration of phenolic acids was measured by reverse phase high pressure liquid chromatography (RP-HPLC) as described below.
Oxidation of a mixture of phenolic acids by free laccase of C. comatus in the presence and absence of amino acids. Reactions were performed in 2 mL Eppendorf tubes. The control mixture of phenolic acids contained gallic, protocatechuic, vanillic, ferulic and syringic acids (2 mM each in 1 mL of buffer A). The mixtures with amino acids contained one of the following acids: tyrosine, glycine, L-DOPA, lysine, arginine, or tryptophan (2 mM each in 1 mL of buffer A). The reactions were initiated by addition of laccase (0.08 U). The mixtures were incubated for 1 h at 25°C and 1400 rpm, reaction was terminated by 10 mM NaN3. Concentrations of phenolic acids were determined by RP-HPLC. The ability of amino acids to interact with phenolic acids in the presence of laccase was assessed by comparing the loss of phenolic acids in the systems with amino acids and control without amino acids.
Transformation of phenolic acids in the presence of immobilized laccase under the dynamic conditions. Flow-through experiment. Dynamic experiment was performed in Teflon flow-through microcolumn with inner diameter of 7 mm, height 28 mm, total volume 1 ml. Schematic diagram of the column is given in [1]. The double-layer paper filters (Apexlab, Russia) were used at the column inlet and outlet to retain the mineral sample in the column. The column was connected to a BioLogic LP chromatographic system (BioRad, USA) with a fraction collector (Model 2110, BioRad, USA). A mixture of phenolic acids (gallic, protocatechuic, syringic, ferulic) or a mixture or phenolic acids with lysine were used in the experiment. For the preparation of the stock solution of phenolic acids (0.1 mM), they were weighted into Eppendorf tubes, dissolved in 200 µL of ethanol, transferred to a volumetric flask and diluted to a volume of 100 mL by buffer A. The stock solution of lysine (0.2 mM) was prepared in a 100 mL volumetric flask. Aliquots of the stock solutions were used to prepare working solutions—200 mL in buffer A. A working solution of the phenolic acid contained 0.01 mM of each acid in buffer A; a working solution of the phenolic acids—lysine mixture contained 0.01 mM of each acid and 0.02 mM of lysine in buffer A.
20 mg of a mineral containing 0.03 U of active enzyme (24 µL of the preparation corresponding to 0.72 µg of protein) or the same amount of inactivated laccase (enzyme was inactivated before immobilization by heating for 30 min at 80°C) was placed into the column. Then, 3 mL of buffer A was pumped through the column in the direction from its bottom to the top in order to equilibrate the mineral with the eluent. Then the remaining buffer solution was pumped out and 50 mL of phenolic acids solution (or phenolic acids with lysine) was pumped through at a rate of 0.1 mL/min. The solution at the column outlet was collected as 2 mL fractions. Concentration of phenolic acids was determined by RP-HPLC. Prior to analysis, some fractions were combined.
The loss of phenolic acids in the solution which passed through the column was calculated as follows:
where Cinitial is the initial concentration of each phenolic acid (0.01 mM = 10 nmol/mL), Сeluate is the concentration of phenolic acid in the eluate (nmol/mL), and V is the volume of the fraction (mL).
Desorption of phenolic acids. After the experiment, the remaining solution was pumped out of the column and phenolic acids were desorbed first by 16 mL of 5 mM Na-acetate buffer (pH 4.5, buffer A) and then by 24 mL of 50 mM Na-acetate buffer (pH 4.5) at a rate of 0.5 mL/min. Solution passed through the column was collected at the column outlet as 2 mL fractions and phenolic acids were analyzed by RP-HPLC.
Extraction of phenolic acids. The mineral suspension after the desorption experiment was quantitatively removed from the column and centrifuged (2 min, 18000 g), the supernatant was discarded. Compounds, bound to the mineral were extracted first by 1 mL of 0.1 М HCl and then by 1 mL of 0.1 М NaOH under the N2 atmosphere (each extraction—30 min at 1400 rpm, thermoshaker TS-100 C, Biosan, Latvia). Supernatants were separated from the mineral by centrifugation (18000 g) and 20 mg of NaF was added to the extracts in order to bind aluminum ions into soluble complexes. Concentration of phenolic acids in the extracts was analyzed by RP-HPLC, molecular weight distributions were obtained by high pressure liquid chromatography (HPLC) as described below. The pH of the extracts was adjusted to that of the eluents by addition of microquantities of 15% HCl. pH was controlled with a HI 1330 microelectrode (Hanna Instruments, USA).
Analysis of phenolic acids. Phenolic acids were analyzed by RP-HPLC using an Agilent 1100 Quat Pump chromatographic system equipped with a diode array detector, a column thermostat, and an injector (Rheodyne, Cotati, United States). A SunergiHydro-RP column (150 × 4.6 mm, 4 μm; Phenomenex, United States) was used for separation of phenolic acids. The starting solution (solution A) contained 90% of H2O, 5 vol % of acetonitrile and 5 vol % (0.1 mass %) of 3‑fluoroacetic acid. The gradient was created using solution B containing 95% of acetonitrile and 5 vol % of 0.1 wt % 3-fluoroacetic acid as follows: 0–20 min, 5–15% B; 20–30 min, 15–40% B; 30–40 min, 40% В. The elution rate was 0.5 mL/min and the sample volume 20 μL. The temperature of the column—30°С. Phenolic acids were quantified by evaluation of chromatogram peak areas as described earlier.
Molecular weight distributions of the reaction products. Alkaline extracts were analyzed by HPLC on an Agilent 1100 chromatographic system (see above) using TSK-2000 SW column; 0.1 M phosphate buffer (pH 7.0) with addition of 0.1% SDS and 0.05% NaN3 was used as an eluent. The flow rate was 0.5 mL/min. The column was calibrated using globular proteins—cytochrome (12.5 kDa), ribonuclease A (17.8 kDa), chymotrypsinogen A (25 kDa), ovalbumin (45 kDa), bovine serum albumin (67 kDa) (Sigma, USA), and polystyrene sulfonic acids (PSAs) with masses of 6.8, 10, 17, 32, 77 kDa (Sigma-Aldrich, USA). The void volume of the column (Vo) was determined by blue dextran (2000 kDa), the total volume (Vt) was determined by the reverse peak of the solvent. The molecular weights (MW) of the chromatographic peaks (kDa) were found according to the equations:
RESULTS AND DISCUSSION
Immobilization of laccase of Coprinus comatus and properties of the enzyme. Enzymes in soils are mainly bound to the solid phase [23], with reactions occurring in heterogeneous systems. Clay minerals, as well as poorly crystalline oxyhydroxides (the latter may exist as discrete phases or as precipitates on the mineral surfaces) are common inorganic sorbents for enzymes [42]. Kaolinite modified with aluminum hydroxide can therefore serve as an appropriate model of complex mineral phases. Modification of kaolinite leads to an increase in its surface area [3]. The mineral surface acquires a positive charge due to aluminum hydroxide (PZC 8.0–9.2 [40]). This facilitates the sorption of laccase (pI 3–3.5; www.brenda-enzymes.org), phenolic acids [4], and their oxidation products [76].
The efficiency of immobilization of C. comatus laccase on modified kaolinite was 90–100%. It has been shown previously, that sorption of Panus tigrinus laccase on this sorbent was one order of magnitude higher than on pure kaolinite [76]; thus, laccase is bound predominantly to aluminum hydroxide. High affinity to aluminum hydroxide (sorption isotherms of H-type) was also shown for laccase of Trametes villosa [13].
The optimum of 2.6-DMP oxidation by free laccase was at pH 6.0 (Table 1), which agrees with the literature data [18, 34]. The pH-optimum of the immobilized enzyme shifted to the acidic region (pH 4.4). Such changes in the pH optimum of an enzyme upon immobilization is a well-known phenomenon [49], associated with possible conformational changes of the enzyme during sorption and changes in the degree of ionization of its constituent amino acids [32]. The thermostability of free laccase of C. comatus was higher than that of immobilized one (Table 1) although immobilization often decreases the sensitivity of an enzyme to high temperatures [31, 50]. Activities of both free and immobilized laccase was completely lost within 30 min at 80°C.
The catalytic activity of an enzyme may decrease or increase upon immobilization due to conformational changes of the protein during sorption, affecting its active center [31, 72]. However, kinetic parameters of 2.6-DMP oxidation by laccase of Coprinus comatus remained practically unchanged. Similar results were obtained for laccase from T. villosa immobilized on aluminum hydroxide [13]. In the present work, this may be due to a shift in the pH-optimum upon immobilization. Kinetic constants were measured at pH 4.5, which is close to the optimum of the immobilized laccase (pH 4.4), whereas the activity of free laccase comprised 70% of maximum at this pH value (data not shown).
Thus, laccase of C. comatus retain high catalytic activity upon immobilization. The pH optimum shifts to the values characteristic of acidic soils. This should favor the participation of the enzyme in biocatalytic processes in such soils.
Substrate specificity of laccase from C. comatus. Phenolic acids under the study are typical products of lignin degradation and are considered as precursors of humic acids [65].
In terms of oxidation rate, the individual phenolic acids formed a series: CAF > FER > GAL = SYR > PCAT > VAN = HDB (Fig. 1a). Hydroxycinnamic acids such as caffeic and ferulic acids were most efficiently oxidized by laccase (72 and 53% in 1 h), followed by hydroxybenzoic acids—gallic and syringic (43%). Vanillic acid showed very weak reactivity (6% after 5 h of incubation), p-hydroxybenzoic acid was not oxidized at all.

Transformation of phenolic acids by free laccase of C. comatus in 5 mM Na–acetate buffer (рН 4.5): (а) oxidation of individual phenolic acids (2 mM each) by laccase (0.05 U of activity) during (1) 1, (2) 3, and (3) 5 h of reaction; (b) oxidation of phenolic acids by laccase in the absence and presence of amino acids (2 mM each, 0.08 U of laccase). Reactions were performed on a thermoshaker (25 С, 1100 rpm) in 1 mL incubation mixtures. Phenolic acids: (1) gallic, (2) protocatechuic, (3) vanillic, (4) syringic, and (5) ferulic. Amino acids: (6) L-DOPA and (7) tryptophan.
The one-electron transfer from the substrate to type 1 copper site (T1 site) of laccase is the rate-limiting step in the oxidation of substrates by laccases [27]. The reactivity of laccases is determined by the difference in redox potentials (E°') of the substrate and T1 copper site of the enzyme [54] as well as by steric factors [70]. The redox potential of the T1 site of laccases from various producers is in the range of 0.4–0.8 V. The highest redox potentials are that of the laccases of white rot fungi (0.78 V), the intermediate one is that of soil basidiomycetes and ascomycetes (0.47–0.71 V), and the lowest one is that of bacteria (0.42 V) [27, 54]. The redox potential of phenolic compounds is in the range of 0.5–1.2 V [33]. Small electron-donating substituents (OH, NH2–, Cl–, OCH3–, CH3), especially in ortho-position to OH group, increase an electron density at phenoxy group and facilitate oxidation process [67, 70]; steric factors of such small substituents play minor role [70]. The electron-withdrawing substituents (СООН, СОН, СОR, NO2) reduce the electron density at the phenoxy group thus making it more difficult to be oxidized [26, 70]. The presence of two electron-donating OH-groups in gallic acid and two –OCH3 groups in syringic acid in the ortho-position to the phenoxy group can explain the high oxidation rate of gallic and syringic acids as compared to protocatechuic and vanillic acids (Fig. 1a). The absence of the electron-withdrawing –COOH group directly bound to benzene ring and the positive effect of the Cα=Cβ bond on electron delocalization [26] contribute to the efficient oxidation of caffeic and ferulic acids. Laccase of C. comatus showed unusually weak reactivity with vanillic acid, which is a typical substrate for laccases of the white rot fungi [24]. This is most likely due to the low redox potential of laccases from C. comatus [34].
The transformation of each of the phenolic acids by laccase in their mixture (Fig. 1b) was significantly different from the reactions of laccase with individual acids (Fig. 1a). In the mixture, phenolic acids formed the following order (% of acids conversion in 1 h): GAL (49.9) > FER (16.9) > SYR (14.7) > PCAT (13.5) > VAN (13.0) (Fig. 1b). According to their redox potentials (V), phenolic acids form a series: CAF (0.53) < SYR (0.75) < FER (0.82) < VAN (0.88) [39]; GAL (0.26) < CAF (0.31) < FER (0.53) (vs. Ag/AgCl, [26]); CAF (0.45) < SYR (0.49) < VAN (0.73) [62]. The effective oxidation of gallic acid in the mixture can be explained by its lower redox potential compared with the other acids. Vanillic acid was also oxidized in the mixture which can be explained by possible action of the other acids as redox mediators [24].
Interaction of amino acids with phenolic acids in the presence of laccase. Condensation of amino acids with phenolic compounds is considered as one of the mechanisms of nitrogen incorporation into HA structure [5, 65]. In order to study the reactivity of amino acids in oxidative coupling reactions, the following compounds were taken: aliphatic neutral amino acid glycine; aliphatic basic amino acids lysine and arginine; aromatic amino acids tyrosine, tryptophan and dihydroxyphenylalanine (L-DOPA). All these compounds are present in soil solutions [25, 68, 69]. Lysine, arginine and tryptophan are known for their high reactivity due to presence of free amino group (‒NH2), a guanidine group (–C(NH2)2) and an indole ring respectively. These groups interact with carbonyl group (C=O) of aldehydes, ketones and lipids to form dark-colored condensates (Maillard reaction) [6], or interact with quinone group to form N‑containing heterocomplexes (HA and FA-like compounds). L-DOPA is a precursor of melanins, pigments which are similar in properties to HAs.
The ability of amino acids to interact with phenolic acids was assessed indirectly by comparing the conversion of phenolic acids by laccase in mixtures with amino acids and in mixtures without amino acids (Fig. 1b). Laccase of C. comatus oxidized L-DOPA and tryptophan. The amounts of phenolic acids oxidized by laccase changed significantly in the presence of lysine and L-DOPA. The conversion of gallic acid decreased significantly (1.4 times) in the presence of L-DOPA. The conversion of each of the phenolic acids increased approximately 1.5-fold in the presence of lysine. This indicates that lysine interacts with all phenolic acids in the mixture. The high reactivity of lysine with phenolic substrates has been shown previously [35]. Glycine, often used as a model amino acid in humification reactions [43, 65], did not interact with phenolic acids in this study.
Dynamic flow-through experiment. Gallic, protocatechuic, ferulic, syringic acid and lysine were used to study secondary synthesis reactions under the dynamic conditions. The above mentioned phenolic acids were efficiently oxidized by laccase in a homogeneous system and conversion increased in the presence of lysine (Fig. 1b).
In the control experiment with inactivated laccase, phenolic acids formed the following order by their loss in the eluate: GAL > PCAT \( \gg \) FER > SYR (Table 2, Fig. 2). The loss was due to sorption of phenolic acids by the mineral and possibly by the protein immobilized on its surface [60]. Effective sorption of gallic and protocatechuic acids by modified kaolinite was shown by us earlier [3, 4]. Ferulic and syringic acids were very weakly adsorbed in the presence of ortho-diphenolic acids due to competition for binding sites [3, 4].

Transformation of phenolic acids (0.01 mM each) by immobilized laccase of C. comatus in the absence (open symbols) and presence (closed symbols) of lysine (0.02 mM) in the flow-through microcolumn. Circles—active laccase (0.03 U, 0.7 μg of protein), triangles—control with inactivated enzyme (0.7 μg of protein). GAL—gallic acid, PCAT—protocatechuic acid, SYR—syringic acid, FER—ferulic acid. The total loss of compound in the eluate (nmol) was calculated as Ʃ(Сinitial – Сeluate) · Vfraction, where Сinitial is the initial concentration of phenolic acid (0.01 mM = 10 nmol/mL), Сeluate is the concentration of phenolic acid on the column outlet (nmol/mL), and V is the volume of the fraction (mL).
In the experiment with active laccase the amounts of syringic and ferulic acids in the column eluate reduced by 7.0 and 3.2 times, respectively, compared to control with inactivated enzyme (Table 2). The loss of phenolic acids in the eluate was due to: 1) sorption of the initial monomers on the mineral phase; 2) oxidation/condensation of phenolic acids in the liquid phase; and 3) sorption of the oxidation products by the mineral. Laccase efficiently oxidized syringic, ferulic and gallic acids acids under dynamic conditions despite of low substrate concentrations. The effect of laccase was least pronounced for protocatechuic acid, which agrees with the substrate specificity of the free enzyme (Fig. 1a).
For all phenolic acids except gallic acid, the effect of the biocatalyst was observed from the first portion of the eluate (Table 2). The loss of gallic acid at the beginning of the experiment was mainly due to its sorption, as evidenced by quite similar loss values in the variants with active and inactivated laccase. Sorption reduced the concentration of gallic acid in the solution and, consequently, the rate of its oxidation by laccase. When active sites on the mineral were filled up, the effect of sorption on the oxidation of gallic acid was reduced. At the same time, the removal of gallic acid from the reaction mixture due to sorption promoted efficient oxidation of syringic and ferulic acids. Note, that in the homogeneous system, the presence of gallic acid hindered oxidation of syringic and ferulic acids (Fig. 1b). Thus, the high affinity of a substrate to the mineral phase [3] has a significant influence on heterophasic enzymatic catalysis, determining the composition of the reaction products.
The contribution of laccase to conversion of phenolic acids under flow-through conditions was estimated from the difference between the loss of phenolic acids in the system with the active enzyme and the control with inactivated laccase. By the degree of conversion by laccase phenolic acids formed a series: GAL > FER > SYR > PCAT (Table 2), consistent with the patterns established in the homogeneous system. However, at low substrate concentrations the difference in oxidation rate of gallic acid and the other phenolic acids was not as significant as in homogeneous system (Fig. 1b), due to the reasons discussed above. The conversion of all phenolic acids except protocatechuic acid increased in the presence of lysine. Thus, we have shown that free-radical reactions between phenolic and nitrogenous compounds can occur at low substrate concentrations characteristic of natural soil environment.
Desorption of phenolic acids and molecular composition of organic-mineral complexes. The composition of the condensation products can be judged indirectly by the amount and composition of monomers desorbed from the mineral in variants with active and inactivated enzyme. Therefore, desorption of phenolic acids with acetate buffer of increasing concentration and then extraction with 0.1 M HCl were carried out. It is supposed that 5 mM buffer desorbs phenolic acids weakly bound by electrostatic and hydrogen bonds, while 50 mM buffer desorbs phenolic acids which form inner sphere complexes with Al on the surface of modified kaolinite [4]. Phenolic acids bound with aluminum hydroxide and retained after the desorption with Na-acetate buffer can be transferred into solution by 0.1 M HCl, which dissolves the aluminum hydroxide.
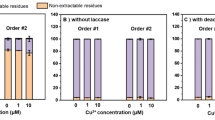
Ferulic and syringic acids were almost completely desorbed by 5 mM Na-acetate buffer (Fig. 3, Table 2), which is in agreement with the data obtained earlier [4]. The amounts of phenolic acids desorbed from the mineral by 5 and 50 mM Na-acetate buffer were significantly lower in the variants with active laccase (Figs. 3a, 3b, Table 2) compared with the control with inactivated enzyme (Figs. 3c, 3d). The same trend was observed in the acidic extracts. The total amounts of gallic, syringic and ferulic acids desorbed and extracted from the mineral surface in the control with inactivated enzyme comprised 27–56% of their loss in the solution; the value for protocatechuic acid was about 80% (Fig. 4). In case of active laccase, the recovery was only 5–8% for gallic, syringic and ferulic acids and 30–40% for protocatechuic acid, which corresponds to substrate specificity of the enzyme. The observed difference can be considered as an indirect evidence of condensation reactions with products distributed between the solid and liquid phases. It can be assumed that oxidation products of gallic acid prevailed on the mineral phase, as evidenced by its high loss in the solution, relatively small recovery from the mineral phase (Fig. 3, Table 2), as well as the dark coloring of the mineral surface.

Desorption of phenolic acids from the mineral by 5 and 50 mM Na-acetate buffer in the flow-through experiment: (a) the mixture of phenolic acids without lysine, laccase 0.08 U; (b) the miхture of phenolic acids with lysine, laccase 0.08 U; (c) the mixture of phenolic acids without lysine, inactivated laccase; (d) the mixture of phenolic acids with lysine, inactivated laccase.

The amount of phenolic monomers recovered from the mineral by Na-acetate buffer and 0.1 М HCl in the dynamic experiment with active and inactivated laccase (% of total loss in the solution passed through the column).
Molecular weight distributions of the reaction products bound to the mineral surface. The elution profiles of alkaline extracts consisted of a main peak near the total volume of the column and a number of “sorption” peaks beyond the total volume of the column (Figs. 5a, 5b) belonging to ferulic and syringic acids (spectroscopy data not shown). The molecular weight of the main peak was about 270 Da according to calibration by globular proteins and 900 Da according to calibration by PSA. The spectrum of the main peak in the variant with active laccase differed from that in the variant with inactivated enzyme by a significant increase in absorbance in visible region (see inset in Fig. 5). This may indicate the formation of dark-colored oligomers. It is well known that phenolic compounds such as vanillic and syringic [48], gallic [11], ferulic [22], caffeic [15] acids form dimers and trimers (including quinone dimers) via C–C and C–O–C bonds during their oxidation by laccase and peroxidase. Dimerization with formation of conjugated double bonds [53] is also well known. In the present work, the formation of dimers/oligomers and their binding to the solid phase is also evidenced by the significant difference between the amounts of monomers recovered from the mineral surface in the variants with active and inactivated enzyme (Fig. 4, Table 2).

Molecular weight distributions of compounds, desorbed from the mineral by 0.1 М NaOH after the extraction by 0.1 M HCl: (a) without lysine and (b) with lysine. Chromatographic system Agilent 1100, TSK-2000 SW column, 0.1 М phosphate buffer (pH 7.0) with addition of 0.1% SDS and 0.05% NaN3.
Previously [75] we have shown the formation of polymeric HA-like substances (10–75 kDa, gel-filtration) in the presence of laccase of the white rot fungus Panus tigrinus immobilized on modified kaolinite. The precursor mixture contained caffeic acid, gallic acid, vanillic acid, ferulic acid, p-hydroxybenzoic acid (0.15 mg or 0.8 mM each), the amino acids tryptophan (0.10 mg or 0.5 mM), phenylalanine (0.1 mg or 0.6 mM) and arginine (0.05 mg or 0.3 mM) and 0.08 U of laccase. Efficient polymerization in that work could be due to several reasons: (1) higher concentration of substrates; (2) higher redox potential of the enzyme—P. tigrinus laccase oxidizes phenolic acids more efficiently than C. comatus laccase (data not shown); (3) composition of precursor mixture including caffeic acid, which is oxidized more efficiently than gallic acid (Fig. 1a); and (4) longer interaction time (24–72 h).
The low concentrations of phenolic acids and their sorption to the mineral phase were the factors suppressing polymerization under the conditions of the flow-through experiment. The substrate concentrations of 0.01 mM were 1–2 orders of magnitude lower than the KM values (0.13–10 mM) of the oxidation of phenolic substrates by laccase [70]. Nevertheless, even under such unfavorable conditions, effective conversion of gallic, syringic and ferulic acids by laccase takes place (Figs. 2, 4).
CONCLUSIONS
Under the flow-through conditions and at precursor concentrations characteristic of soil solutions, low molecular weight FA-like products were formed during laccase-catalyzed heterophasic condensation reactions. The structure of phenolic acids plays an important role in their distribution between solid and liquid phases. Ortho-diphenols (gallic acid, protocatechuic acid) interact much more efficiently with modified kaolinite than methoxy-substituted compounds (syringic and ferulic acids). In the presence of laccase, the distribution of phenolic acids between the solid and liquid phases is determined both by the reactivity of phenolic acids, and by interactions with the mineral phase. At low substrate concentration and free active sites on the mineral, selective binding of one of the compounds in a mixture (e.g. gallic acid) can either interfere (in the case of gallic acid) or promote (in the case of ferulic and syringic acids) enzymatic catalysis in the heterogeneous system. Among phenolic acids under the study, gallic acid possessed the highest reactivity when present in a mixture, followed by syringic and ferulic acids. Lysine shows high reactivity in the formation of complexes with phenolic acids in the presence of laccase. The results of the study suggest an important role of heterophasic free-radical reactions in the formation of the molecular composition of the liquid phase and organo-mineral complexes in soil.
REFERENCES
M. S. Ermolin, N. N. Fedyunina, V. K. Karandashev, and P. S. Fedotov, “Study of the mobility of cerium oxide nanoparticles in soil using dynamic extraction in a microcolumn and a rotating coiled column,” J. Anal. Chem. 74, 825–833 (2019). https://doi.org/10.1134/S1061934819080070
A. G. Zavarzina, N. N. Danchenko, V. V. Demin, Z. S. Artemyeva, and B. M. Kogut, “Humic substances: hypotheses and reality (a review),” Eurasian Soil Sci. 54, 1826–1854 (2021).
A. G. Zavarzina, M. S. Ermolin, V. V. Demin, and P. S. Fedotov, “Interaction of the mixture of phenolic acids with modified kaolinite under batch and dynamic conditions,” Eurasian Soil Sci. 51, 938–946 (2018). https://doi.org/10.1134/S1061934819080070
A. G. Zavarzina, M. S. Ermolin, V. V. Demin, and P. S. Fedotov, “The effect of acetic acid and acetate ions on sorption–desorption of a mixture of phenolic acids by modified kaolinite,” Eurasian Soil Sci. 53, 1046–1055 (2020). https://doi.org/10.1134/S1064229320080177
M. M. Kononova, Soil Organic Matter (Academy of Sciences of the USSR, Moscow, 1963) [in Russian].
O. V. Kosmachevskaya, “Ubiquitous Maillard reaction,” Khim. Zhizn’, No. 2, (2012). https://hij.ru/read/92/.
M. S. Malinina and S. V. Ivanilova, “Phenol compounds in solutions of soils of different types in the central forest state biosphere reserve,” Eurasian Soil Sci. 41, 377–385 (2008). https://doi.org/10.1134/S1064229308040030
D. S. Orlov, Soil Humic Acids and the General Theory of Humification (Moscow State Univ., Moscow, 1990) [in Russian].
D. S. Orlov, O. N. Biryukova, and N. I. Sukhanova, Organic Matter of Soils of the Russian Federation (Nauka, Moscow, 1996) [in Russian].
V. M. Semenov, A. S. Tulina, N. A. Semenova, and L. A. Ivannikova, “Humification and nonhumification pathways of the organic matter stabilization in soil: a review,” Eurasian Soil Sci. 46, 355–368 (2013). https://doi.org/10.1134/S106422931304011X
B. J. Ahn, K. K. Gaikwad, and Y. S. Lee, “Characterization and properties of LDPE film with gallic-acid-based oxygen scavenging system useful as a functional packaging material,” J. Appl. Polym. Sci. 133 (43), 44138 (2016). https://doi.org/10.1002/app.44138
M. Y. Ahn, C. E. Martinez, D. D. Archibald, A. R. Zimmerman, J.-M. Bollag, and J. Dec, “Transformation of catechol in presence of a laccase and birnessite,” Soil Biol. Biochem. 38, 1015–1020 (2006). https://doi.org/10.1016/j.soilbio.2005.08.016
M. Y. Ahn, A. R. Zimmerman, C. E. Martinez, D. D. Archibald, J.-M. Bollag, and J. Dec, “Characteristics of Trametes villosa laccase adsorbed on aluminum hydroxide,” Enzyme Microb. Technol. 41, 141–148 (2007). https://doi.org/10.1016/j.enzmictec.2006.12.014
L. N. Alexandrova, T. Th. Arshavskay, F. M. Dorfman, M. F. Lyuzin, and O. V. Yurlova, “Humus acids and their organo- mineral derivatives in soil,” Int. Soil Sci. Congr. Trans. 3 (9), 143–152 (1968).
R. Arakawa, M. Yamaguchi, H. Hotta, T. Osakai, and T. Kimoto, “Product analysis of caffeic acid oxidation by on-line electrochemistry/electrospray ionization mass spectrometry,” J. Am. Soc. Mass Spectrom. 15 (8), 1228–1236 (2004). https://doi.org/10.1016/j.jasms.2004.05.007
P. Baldrian, “Fungal laccases-occurrence and properties,” FEMS Microbiol. Rev. 30, 215–242 (2006). https://doi.org/10.1111/j.1574-4976.2005.00010.x
P. Baldrian and J. Snajdr, “Lignocellulose-degrading enzymes in soil,” in Soil Enzymology, Soil Biol. Ser., vol. 22 (Springer-Verlag, Berlin, 2011), pp. 167–186.
S. Bao, Z. Teng, and S. Ding, “Heterologous expression and characterization of a novel laccase isoenzyme with dyes decolorization potential from Coprinus comatus,” Mol. Biol. Rep. 40 (2), 1927–1936 (2012). https://doi.org/10.1007/s11033-012-2249-9
N. H. Batjes, “Total carbon and nitrogen in the soils of the world,” Eur. J. Soil Sci. 65, 4–21 (2014). https://doi.org/10.1111/ejss.12114_2
J.-M. Bollag, J. Dec, and P. M. Huang, “Formation mechanisms of complex organic structures in soil habitats,” in Advances in Agronomy (Elsevier, Amsterdam, 1997), Vol. 63, pp. 237–265. https://doi.org/10.1016/S0065-2113(08)60245-X
J.-M. Bollag, C. Meyers, S. Pal, and P. M. Huang, “Role of abiotic and biotic catalysts in the transformation of phenolic compounds,” in Environmental Impact of Soil Component Interactions (CRC Press, Boca Raton, 1995), pp. 299–310.
M. Bunzel, J. Ralph, P. Brüning, and H. Steinhart, “Structural identification of dehydrotriferulic and dehydrotetraferulic acids isolated from insoluble maize bran fiber,” J. Agric. Food Chem. 5 (17), 6409–6418 (2006). https://doi.org/10.1021/jf061196a
R. G. Burns, J. I. DeForest, J. Marxsen, R. I. Sinsabaugh, M. E. Stromberger, M. D. Wallenstein, M. N. Weintraub, and A. Zoppini, “Soil enzymes in a changing environment: current knowledge and future directions,” Soil Biol. Biochem. 58, 216–234 (2013). https://doi.org/10.1016/j.soilbio.2012.11.009
A. I. Cañas and S. Camarero, “Laccases and their natural mediators: biotechnological tools for sustainable eco-friendly processes,” Biotechnol. Adv. 28, 694–705 (2010). https://doi.org/10.1016/j.biotechadv.2010.05.002
X. Cao, Q. Ma, C. Zhong, X. Yang, L. Zhu, J. Zhang, et al., “Elevational variation in soil amino acid and inorganic nitrogen concentrations in Taibai Mountain, China,” PLoS One 11, e0157979 (2016). https://doi.org/10.1371/journal.pone.0157979
A. Chiorcea-Paquim, T. A. Enache, E. De Souza Gil, and A. M. Oliveira-Brett, “Natural phenolic antioxidants electrochemistry: towards a new food science methodology,” Compr. Rev. Food Sci. Food Saf. 19 (4), 1680–1726 (2020). https://doi.org/10.1111/1541-4337.12566
A. Christenson, N. Dimcheva, E. E. Ferapontova, et al., “Direct electron transfer between ligninolytic redox enzymes and electrodes,” Electroanalysis 16 (13–14), 1074–1092 (2004). https://doi.org/10.1002/elan.200403004
S. Dou, J. Shan, X. Song, R. Cao, M. Wu, C. Li, and S. Guan, “Are humic substances soil microbial residues or unique synthesized compounds? A perspective on their distinctiveness,” Pedosphere. 30 (2), 159–167 (2020). https://doi.org/10.1016/S1002-0160(20)60001-7
M. De Nobili, C. Bravo, and Y. Chen, “The spontaneous secondary synthesis of soil organic matter components: a critical examination of the soil continuum model theory,” Appl. Soil Ecol. 154, 10365 (2020). https://doi.org/10.1016/j.apsoil.2020.103655
I. Eichlerová, J. Šnajdr, and P. Baldrian, “Laccase activity in soils: considerations for the measurement of enzyme activity,” Chemosphere 88, 1154–1160 (2012). https://doi.org/10.1016/j.chemosphere.2012.03.019
M. Fernández-Fernández, M. Á. Sanromán, and D. Moldes, “Recent developments and applications of immobilized laccase,” Biotechnol. Adv. 31, 1808–1825 (2013). https://doi.org/10.1016/j.biotechadv.2012.02.013
W. T. Frankenberger Jr. and J. B. Johanson, “Effect of pH on enzyme stability in soils,” Soil Biol. Biochem. 14, 433–437 (1982). https://doi.org/10.1016/0038-0717(82)90101-8
P. Giardina, V. Faraco, C. Pezzella, A. Piscitelli, S. Vanhulle, and G. Sannia, “Laccases: a never-ending story,” Cell. Mol. Life Sci. 67 (3), 369–385 (2009). https://doi.org/10.1007/s00018-009-0169-1
C. Gu, F. Zheng, L. Long, J. Wang, and S. Ding, “Engineering the expression and characterization of two novel laccase isoenzymes from Coprinus comatus in Pichia pastoris by fusing an additional ten amino acids tag at N-terminus,” PLoS One 9 (4), e93912 (2014). https://doi.org/10.1371/journal.pone.0093912
K. Haider, L. R. Frederick, and W. Flaig, “Reactions between amino acid compounds and phenols during oxidation,” Plant Soil 22, 49–64 (1965). https:// www.jstor.org/stable/42932090.
K. Haider and A. Schäffer, Soil Biochemistry (CRC Press, Boca Raton, FL, 2009).
M. H. B. Hayes and R. S. Swift, “Vindication of humic substances as a key component of organic matter in soil and water,” in Advances in Agronomy (Elsevier, Amsterdam, 2020), Vol. 163, Ch. 1. https://doi.org/10.1016/bs.agron.2020.05.001
A. Heinfling, A. T. Martinez, M. J. Martinez, et al., “Purification and characterization of peroxidases from the dye-decolorizing fungus Bjerkandera adusta,” FEMS Microbiol. Lett. 428, 43–50 (1998). https://doi.org/10.1016/S0014-5793(98)00512-2
I. Hradkowa and V. Filip, “Antioxidant stability of phenolic acids and their esters,” Czech J. Food Sci. 27, 41–44 (2009).
P. H. Hsu, “Aluminum hydroxides and oxyhydroxides,” in Minerals in Soil Environments, SSSA Book Ser., no. 1 (Soil Science Society of America, Madison, WI, 1989), pp. 331–378.
P. M. Huang, “Abiotic catalysis,” in Handbook of Soil Science: Properties and Processes (CRC Press, Boca Raton, FL, 2000), pp. 303–334.
P. M. Huang, “The role of short-range ordered mineral colloids in abiotic transformation of organic compounds in the environment,” in Environmental Impacts of Soil Component Interactions, Vol. 1: Natural and Anthropogenic Organics (CRC Press, Boca Raton, FL, 1995), pp. 135–167.
P. M. Huang and A. G. Hardie, “Formation mechanisms of humic substances in the environment,” in Biophysico-Chemical Processes Involving Natural Nonliving Organic Matter in Environmental Systems (Wiley, Hoboken, NJ, 2009), Ch. 2, pp. 84–98.
International Humic Substances Society, What are humic substances? 2021. http://humic-substances.org/ what-arehumic-substances-2/.
G. Janusz, A. Pawlik, U. Świderska-Burek, J. Polak, J. Sulej, A. Jarosz-Wilkołazka, and A. Paszczyński, “Laccase properties, physiological functions, and evolution,” Int. J. Mol. Sci. 21 (3), 966 (2020). https://doi.org/10.3390/ijms21030966
D. L. Jones, A. G. Owen, and J. F. Farrar, “Simple method to enable the high resolution determination of total free amino acids in soil solutions and soil extracts,” Soil Biol. Biochem. 34, 1893–1902 (2002). https://doi.org/10.1016/S0038-0717(02)00203-1
J. Lehmann and M. Kleber, “The contentious nature of soil organic matter,” Nature 528, 60–68 (2015). https://doi.org/10.1038/nature16069
A. Leonowicz, R. U. Edgehill, and J.-M. Bollag, “The effect of pH on the transformation of syringic and vanillic acids by the laccases of Rhizoctonia praticola and Trametes versicolor,” Arch. Microbiol. 137, 89–96 (1984). https://doi.org/10.1007/BF00414446
A. Leonowicz, J. M. Sarkar, and J.-M. Bollag, “Improvement in stability of an immobilized fungal laccase,” Appl. Microbiol. Biotechnol. 29, 129–135 (1988).
N. Li, Q. Xia, M. Niu, Q. Ping, and H. Xiao, “Immobilizing laccase on different species wood biochar to remove the chlorinated biphenyl in wastewater,” Sci. Rep. 8, 13947 (2018). https://doi.org/10.1038/s41598-018-32013-0
A. Lisov, O. Belova, A. Zavarzina, A. Konstantinov, and A. Leontievsky, “The role of laccase from zygomycetous fungus Mortierella elasson in humic acids degradation,” Agronomy 11, 2169 (2021). https://doi.org/10.3390/agronomy11112169
S. Y. Liu, A. J. Freyer, R. D. Minard, and J.-M. Bollag, “Enzyme-catalyzed complex formation of amino acid esters and phenolic humus constituents,” Soil Sci. Soc. Am. J. 49, 337–342 (1985). https://doi.org/10.2136/sssaj1985.03615995004900020013x
I. Magario, F. S. García Einschlag, E. H. Rueda, J. Zygadlo, and M. L. Ferreira, “Mechanisms of radical generation in the removal of phenol derivatives and pigments using different Fe-based catalytic systems,” J. Mol. Cat. A: Chem. 352, 1–20 (2012). https://doi.org/10.1016/j.molcata.2011.10.006
O. L. Morozova, G. P. Shumakovich, M. A. Gorbacheva, S. V. Shleev, and A. I. Yaropolov, “Blue” laccases,” Biokhimiya 72 (10), 1396–1412 (2007). https://doi.org/10.1134/s0006297907100112
A. Naidja, P. M. Huang, J. Dec, and J.-M. Bollag, “Comparison of the reaction products from the transformation of catechol catalyzed by birnessite or tyrozinase,” Soil Sci. Soc. Am. J. 62, 188–195 (1998). https://doi.org/10.1016/j.soilbio.2005.08.016
D. C. Olk, P. R. Bloom, E. M. Perdue, et al., “Environmental and agricultural relevance of humic fractions extracted by alkali from soils and natural waters,” J. Environ. Qual. 48, 217–232 (2019). https://doi.org/10.2134/jeq2019.02.0041
M. A. Olofsson, S. H. Norström, and D. Bylund, “Evaluation of sampling and sample preparation procedures for the determination of aromatic acids and their distribution in a podzol soil using liquid chromatography-tandem mass spectrometry,” Geoderma 23, 373–380 (2014). https://doi.org/10.1016/j.geoderma.2014.06.005
S. Pal, J.-M. Bollag, and P. M. Huang, “Role of abiotic and biotic catalysts in the transformation of phenolic compounds through oxidative coupling reactions,” Soil Biol. Biochem. 26, 813–820 (1994). https://doi.org/10.1016/0038-0717(94)90297-6
A. Piccolo, “The supramolecular structure of humic substances: a novel understanding of humus chemistry and implications in soil sciences,” in Advances in Agronomy (Elsevier, Amsterdam, 2002), Vol. 75, pp. 57–134. https://doi.org/10.1016/S0065-2113(02)75003-7
Ł. Sęczyk, M. Świeca, I. Kapusta, and U. Gawlik-Dziki, “Protein-phenolic interactions as a factor affecting the physicochemical properties of white bean proteins,” Molecules 24 (3), 408 (2009). https://doi.org/10.3390/molecules24030408
A. Schaeffer, P. Nannipieri, M. Kästner, B. Schmidt, and J. Botterweck, “From humic substances to soil organic matter–microbial contributions. In honor of Konrad Haider and James P. Martin for their outstanding research contribution to soil science,” J. Soils Sediments 15, 1865–1881 (2015). https://doi.org/10.1007/s11368-015-1177-4
A. Simić, D. Manojlović, D. Šegan, and M. Todorović, “Electrochemical behavior and antioxidant and prooxidant activity of natural phenolics,” Molecules 12, 2327–2340 (2007). https://doi.org/10.3390/12102327
R. L. Sinsabaugh, “Phenol oxidase, peroxidase and organic matter dynamics of soil,” Soil Biol. Biochem. 42, 391–404 (2010). https://doi.org/10.1016/j.soilbio.2009.10.014
H. Shindo and P. M. Huang, “Catalytic effects of manganese(IV), iron(III), aluminum and silicon oxides on the formation of phenolic polymers,” Soil Sci. Soc. Am. J. 48, 927–934 (1984). https://doi.org/10.2136/sssaj1984.03615995004800040045x
F. J. Stevenson, Humus Chemistry: Genesis, Composition, Reactions (Wiley, Chichester, 1994).
R. Sutton and G. Sposito, “Molecular structure in humic substances: the new view,” Environ. Sci. Technol. 39, 9009–9015 (2005). https://doi.org/10.1021/es050778q
M. A. Tadesse, A. D’Annibale, C. Galli, P. Gentili, and F. Sergi, “An assessment of the relative contributions of redox and steric issues to laccase specificity towards putative substrates,” Org. Biomol. Chem. 6, 868–878 (2008). https://doi.org/10.1039/B716002J
C. R. Warren and M. T. Taranto, “Temporal variation in pools of amino acids, inorganic and microbial N in a temperate grassland soil,” Soil Biol. Biochem. 42, 353–359 (2010). https://doi.org/10.1016/J.SOILBIO.2009.11.017
N. R. Werdin-Pfisterer, K. Kielland, and R. D. Boone, “Soil amino acid composition across a boreal forest successional sequence,” Soil Biol. Biochem. 41, 1210–1220 (2009). https://doi.org/10.1016/j.soilbio.2009.03.001
F. Xu, “Oxidation of phenols, anilines, and benzenethiols by fungal laccases: correlation between activity and redox potentials as well as halide inhibition,” Biochemistry 35, 7608–7614 (1996). https://doi.org/10.1021/bi952971a
F. Xu, “Effects of redox potential and hydroxide inhibition on the pH activity profile of fungal laccases,” J. Biol. Chem. 272, 924–928 (1997). https://doi.org/10.1074/jbc.272.2.924
J. Yang, W. Li, T. B. Ng, X. Deng, J. Lin, and X. Ye, “Laccases: production, expression regulation, and applications in pharmaceutical biodegradation,” Front. Microbiol. 8, 832 (2017). https://doi.org/10.3389/fmicb.2017.00832
I. S. Yavmetdinov, E. V. Stepanova, V. P. Gavrilova, B. V. Lokshin, I. V. Perminova, and O. V. Koroleva, “Isolation and characterization of humin-like substances produced by wood-degrading white-rot fungi,” Appl. Biochem. Microbiol. 39, 257–264 (2003).
Z. Yu, Q. Zhang, T. E. C. Kraus, R. A. Dahlgren, C. Anastasio, and R. J. Zasoski, “Contribution of amino compounds to dissolved organic nitrogen in forest soils,” Biogeochemistry 61, 173–198 (2002). https://www.jstor.org/stable/1469810
A. G. Zavarzina, “A mineral support and biotic catalyst are essential in the formation of highly polymeric soil humic substances,” Eurasian Soil Sci. 39, S48–S53 (2006). https://doi.org/10.1134/S1064229306130096
A. Zavarzina, “Heterophase synthesis of humic acids in soils by immobilized phenol oxidases,” in Soil Enzymology, Soil Biol. Ser., vol. 22 (Springer-Verlag, Berlin, 2011), pp. 207–228. https://doi.org/10.1007/978-3-642-14225-3_10
A. G. Zavarzina, A. V. Lisov, and A. A. Leontievsky, “The role of ligninolytic enzymes laccase and a versatile peroxidase of the white-rot fungus Lentinus tigrinus in biotransformation of soil humic matter: comparative in vivo study,” J. Geophys. Res.: Biogeosci. 123, 1–16 (2018). https://doi.org/10.1029/2017JG004309
A. Zavarzina, A. Lisov, A. Leontievsky, and A. Zavarzin, “Fungal oxidoreductases and humification in forest soils,” in Soil Enzymology, Soil Biol. Ser., vol. 22 (Springer-Verlag, Berlin, 2011), pp. 187–205. https://doi.org/10.1007/978-3-642-14225-3_11
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
CONFLICT OF INTEREST
The authors declare that they have no conflict of interest.
FINANCIAL SUPPORT
The financial support from the Russian Science Foundation grant № 17-14-01207 (experiments with laccase) and from the State Budget № 121040800154-8 (sorption of phenolic acids by mineral with inactivated laccase) are gratefully acknowledged. The work was conducted in the framework of Interdisciplinary school of MSU “Future planet and global environmental change”.
Additional information
Translated by A. Zavarzina
Rights and permissions
Open Access. This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zavarzina, A.G., Demin, V.V., Belova, O.V. et al. Heterophase Synthesis of Humic Substances at Low Substrate Concentrations and Flow-Through Conditions. Eurasian Soil Sc. 55, 911–925 (2022). https://doi.org/10.1134/S1064229322070146
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S1064229322070146