
Abstract
Temporal control of heterologous pathway expression is critical to achieve optimal efficiency in microbial metabolic engineering. The broadly-used GAL promoter system for engineered yeast (Saccharomyces cerevisiae) suffers from several drawbacks; specifically, unintended induction during laboratory development, and unintended repression in industrial production applications, which decreases overall production capacity. Eukaryotic synthetic circuits have not been well examined to address these problems. Here, we explore a modularised engineering method to deploy new genetic circuits applicable for expanding the control of GAL promoter-driven heterologous pathways in S. cerevisiae. Trans- and cis- modules, including eukaryotic trans-activating-and-repressing mechanisms, were characterised to provide new and better tools for circuit design. A eukaryote-like tetracycline-mediated circuit that delivers stringent repression was engineered to minimise metabolic burden during strain development and maintenance. This was combined with a novel 37 °C induction circuit to relief glucose-mediated repression on the GAL promoter during the bioprocess. This delivered a 44% increase in production of the terpenoid nerolidol, to 2.54 g L−1 in flask cultivation. These negative/positive transcriptional regulatory circuits expand global strategies of metabolic control to facilitate laboratory maintenance and for industry applications.
Similar content being viewed by others


Introduction
Metabolic burden from expression of heterologous pathways may dramatically inhibit normal cell proliferation in microbial cell factories1,2,3 and cause strain instability1,2,3,4,5. This delays laboratory strain development via the design-build-test-learn cycle and impedes industrial processes. It is critical to have robust control mechanisms that provide tight repression when optimal growth is required as well as boosted induction when maximal production is needed6. Importantly, these tools should not increase the cost for industrial processes.
The yeast Saccharomyces cerevisiae is a model Eukaryotic organism for understanding biological principles and a primary chassis organism used in manufacturing a variety of bio-products7,8. The endogenous yeast galactose-inducible (GAL) expression system has been engineered to respond to glucose starvation for automatic induction of heterologous pathway9,10. In the presence of glucose, the glucose-dependent repressor Mig1p represses expression of alternative carbon source catabolic genes (including the GAL genes for galactose utilisation). When the GAL repressor gene GAL80 is disrupted, GAL promoters are automatically induced upon glucose depletion in batch cultivation2,9,11. The low expression from GAL promoters in exponential growth phase on glucose prevents metabolic burden from causing growth inhibition. This engineered gal80Δ GAL induction circuit is now broadly used in metabolic engineering, as exemplified by high level production of terpenoids12 and flavanones13,14. However, the system has limitations: (i) unintended auto-induction during routine strain maintenance and development is problematic when induced pathways result in metabolic burden or cellular toxicity; (ii) in prolonged pulse-feeding high-cell-density glucose processes, Mig1-mediated repression on GAL promoters may disrupt expression patterns; (iii) an increase in promoter strength is highly desirable to improve expression of bottleneck enzymes. The current work aims to address these limitations by expanding control range of GAL promoters for better application in metabolic engineering.
One of the applications of synthetic biology is in development of artificial regulatory circuits for application in metabolic engineering15,16,17. Many synthetic transcriptional circuits have been reported for S. cerevisiae6,15,18,19,20. They include VP16 trans-activating sequence-mediated activation circuits19,20,21 and prokaryote-like bacterial repressor-mediated circuits6,15. A variety sensing mechanisms have been used to trigger the OFF/ON states of these circuits, i.e., small molecules6,15,18,19,20,22, light23,24,25, and cold -shock26. However, each circuit has specific down sides, which may make them non-ideal in application. For example, adding expansive small molecules is not economical for large-scale cultivation. In addition, cold-shock induction may not be possible in tropical regions, including Mackay, Queensland, one of Australia’s major cane sugar feedstock processing regions. Current prokaryote-like circuits require high-level expression of bacterial repressors and intensive optimisation of promoters to achieve ideal on/off response ratio6,15,27, which do not reach to the efficiency of natural eukaryotic regulatory mechanisms28.
The complexity and relative unpredictability of biological systems still hinders rational design of genetic circuitry6,15,29,30,31,32,33,34. Consequently, significant work has been devoted to development of robust, predictable genetic componentry in yeast6,35,36. Comprehensive characterisation of individual modular components in different growth states is required to improve design parameterisation for these circuits. In this study, we employed a modularised engineering strategy for synthesis of synthetic regulatory circuits to expand control mechanisms on GAL promoters for metabolic engineering applications. We investigated eukaryote-like mechanisms to deploy trans-activation modules and trans-repression modules. We integrated new control mechanisms to render non-natural regulatory properties to the GAL expression system, including using tetracycline to trigger repression on GAL promoters and elevated temperature to trigger de-repression of GAL promoters. We also investigated possibility of applying artificial trans-activators to enhance GAL promoter strength. The expanded GAL system was then validated for repression or upregulation of heterologous sesquiterpene production in yeast.
Results
Native transcription factor promoters express at moderate to low levels
A strong promoter, commonly used in overexpression of metabolic enzymes or primary cellular constitutive proteins, is often used to control the expression of artificial transcription factors (TFs) in synthetic circuits15,35,37. However, in natural eukaryotic systems, TFs are generally not expressed at high levels38. We wanted to explore whether we could achieve a better outcome by exploiting native TF promoters to mimic natural expression systems. We therefore characterised fourteen TF and regulatory protein promoters.
Yeast strains for promoter characterisation were obtained by transformation of an enhanced yeast green fluorescent protein (yEGFP) expression cassette under the control of promoter of interest via single-copy genome integration (Fig. 1a). Fluorescence was analysed in early exponential phase (when glucose was the carbon source) and in post-exponential phase (when ethanol was the carbon source).

a Schematic of the yEGFP reporter system used to examine the promoter strength. b The yEGFP expression levels from the TEF1 promoter using the reporter system in a. c The yEGFP expression levels from the promoters of regulatory genes using construct 3 in a. d The yEGFP expression levels from the DAL80 promoter under different carbon sources and nitrogen sources. b, c The yEGFP expression levels were characterised in the cells grown to the early exponential growth phase (EXP) or the ethanol-growth phase (ETH) in MES-buffered YNB media using 20 g L−1 glucose as the carbon source in 96-well plates. d the yEGFP expression levels were characterised in the cells grown to the exponential phase in YNB media with varied combination of 20 g L−1 glucose, 20 g L−1 sucrose, or 2 % (v/v) ethanol as the carbon source and 5 g L−1 ammonia sulphate or 0.46 g L−1 urea as the nitrogen source. GFP fluorescence is expressed as percentage of exponential-phase auto-fluorescence of the reference strain. Data for Construct 1 in b are extracted from previous study39. Mean values ± standard deviations are shown (N = 3 independent biological replicates). Source data in b–d are provided in Supplementary Data 1.
We tested three base constructs using the TEF1 promoter to identify the best one to use for promoter comparison (Fig. 1a). In our previous work39, we used a construct with a modified translation initiation region including a BamHI restriction enzyme site and a URA3 terminator (which also acts as a homologous recombination site to introduce the construct onto the genome). Insertion of a PGK1 terminator improved yEGFP fluorescence by >5-fold (Fig. 1b). removal of the BamHI site and reinstatement of the native translation sequence context (Fig. 1a, Construct 3) further improved yEGFP fluorescence by ~50%. This construct design was used to test regulatory protein promoters.
Compared to the TEF1 promoter (Fig. 1b, construct 3), the 14 tested endogenous regulatory protein promoters were weaker by 1–2 orders of magnitude (Fig. 1c). The promoters of the GCR1 and GCR2 genes, encoding transcriptional activators for glycolytic and ribosomal genes, were >39-fold weaker than the TEF1 promoter, and the promoter of the MIG1 gene, encoding an important repressor in the glucose repression signalling pathway, was further weaker. The promoters showing a similar or weaker strength include the promoters of GLN3 (encoding a transcriptional activator in nitrogen catabolite repression system), TOR1 (encoding a primary protein kinase in Target Of Rapamycin regulatory network), SNF1 (encoding a primary protein kinase in glucose repression regulatory networks), NRG1 (encoding a repressor mediating glucose repression), ROX1 (encoding a repressor in oxygen regulation), HAP4 (encoding a global regulator of respiration), and UPC2 (encoding a ergosterol-sensing activator). This shows that many primary components in natural regulatory networks are expressed at the levels much lower than the levels achievable from strong constitutive promoters.
Among the tested promoters, the promoters of YPK2 (encoding a protein kinase required for cell growth), ADR1 (encoding a transcriptional activator in glucose repression), and HAC1 (encoding a transcriptional activator regulating the unfolded protein response in endoplasmic reticulum) showed a moderate strength, around 10-fold lower than the TEF1 promoter. The promoter of DAL80 (encoding a negative repressor responsive to nitrogen levels) showed induced activities on ethanol (Fig. 1c, d), but not on sucrose (Fig. 1d). The DAL80 promoter could be further induced to a level higher the TEF1 promoter, when yeast was grown on urea, a poor nitrogen source (Fig. 1d). Due to the HAC1 showing a consistent expression output on the exponential growth phase and the ethanol-growth phase and its moderate activity, we chose it to control artificial TFs for the following modular characterisation.
Trans-activating modules have variable effects on transcriptional activity from different hybrid promotes
The trans-activating domain (TAD) binds to transcriptional co-regulator proteins to refine and determine the magnitude of the response. Ottoz and colleagues19 used a chimeric construct approach to characterise four TADs in yeast: VP16 (the alpha-gene-transactivating factor (α-TIF) from the alphaherpesviruses gene UL48 promoter), B112, B42, and the Gal4 activation domain. VP16A showed the strongest activation capacity19. To expand the pool of available elements, we characterised two more TAD modules, a ten-WD-repeat sequence (10*WD)40 and a Gcn4p trans-activating domain (Gcn4 aa107-144; Gcn4A). Mediators are evolutionarily conserved and form the coactivation complex generally required for eukaryotic RNA polymerase II-dependent transcription41. Recruiting mediators to upstream activation sequences in promoters may allow transcription, even under inhibitory conditions42. To better understanding transactivation effects of mediators, we included the tail mediator 3 (Med3)42 and the tail mediator 15 (Med15)42 in the comparison.
We expressed synthetic chimeric TFs consisting of an N-terminal zinc-finger (ZIF) DNA binding domain (Zif268), a linker harbouring a SV40 nuclear localisation sequence (NLS)43, and TAD (or mediator). VP16A was included as a reference. We selected the HAC1 promoter, which delivers moderate and stable expression during both the exponential and ethanol growth phases (Fig. 1c), to control the expression of synthetic TFs (Fig. 1a). This provides a TF expression system that mimics natural expression levels. We used a single-copy centromeric plasmid pRS41444 to introduce TF expression cassettes into yeast. The reference was the empty vector plasmid pRS414.
Synthetic cis-regulatory reporter cassettes were constructed using yEGFP as the reporter gene and introduced into yeast via genome integration at the URA3 locus (Fig. 1a). For a better understanding of trans-activation effects of artificial TFs, either the weak core CYC1 promoter or the strong TEF1 were fused to a synthetic DNA sequence (truncated from the synthetic P15 promoter, published previously20) including four Zif268 binding elements (Fig. 2a, b; PCYC1+4×[Z268] and PTEF1+4×[Z268]). Reference promoters were constructed by inserting the synthetic sequence with Z268 elements removed (PCYC1(−) and PTEF1(−)).

a The trans-activation effects of synthetic trans-activators on the hybrid CYC1 core promoters. DBD, DNA-binding domain. TA, trans-activating domain. b The trans-activation effects of synthetic trans-activators on the hybrid TEF1 promoters. c The trans-activation effects of synthetic trans-activators on the hybrid GAL promoters. Trans-activators are expressed under the control of the HAC1 promoter on a single copy centromeric plasmid. The yEGFP fluorescence were characterised in the cells grown to the early exponential growth phase (EXP) or the ethanol-growth phase (ETH) in MES-buffered YNB media using 20 g L−1 glucose as the carbon source in 96-well plates. #, yeast cells were grown in test tubes. N.G., not growing. The dashed horizontal line indicates the output from the native TEF1 promoter (Fig. 1), and the solid line indicates the output from the native TDH3 promoter in the EXP-phase cells39. GFP fluorescence is expressed as percentage of exponential-phase auto-fluorescence of a GFP-negative strain. Mean values ± standard deviations are shown (N = 3 independent biological replicates) or mean values are shown (N = 3 independent biological replicates but one replicate was removed as outlier or no data available). Two-tailed Welch’s t-test was used for comparing two groups, and p values are shown above the bars. Source data in a–c are provided in Supplementary Data 1.
The cis-regulatory reporter cassettes and the trans-activator-expressing cassettes were sequentially transformed into the CEN.PK2-1C strain. The yEGFP fluorescence from cells in exponential and in ethanol growth phases was measured.
The CYC1 core promoter, PCYC1(−), showed a very low basal activity (Fig. 2a). Insertion of Z268 elements (PCYC1+4×[Z268]) increased basal expression mildly in the absence of an artificial TF. A variety of responses were observed in the presence of synthetic TFs with different TADs. Gcn4A showed insignificant activation effects in the exponential growth phase, and a weak effect in the ethanol growth phase. VP16A, 10*WD, Med3, and Med15 showed >3-fold activation in the cells at the exponential phase. Med3 and Med15 drove significantly higher expression on ethanol than VP16A.
A short synthetic linker sequence with no biological relevance was into the TEF1 promoter to provide a landing pad for additional cis-acting elements (Fig. 2b). Insertion of this sequence resulted in a decrease in yEGFP expression (PTEF1(−); Fig. 2b). Addition of the Z268 element sequence did not change expression in the exponential phase, but led to a two-fold increase in expression on ethanol (Fig. 2b). Very similar results were observed for expression modification in the presence of the different TAD/mediator-containing constructs (Fig. 2a, b), such that the data from the PTEF1+4×[Z268] promoter responses positively correlated with the effects observed in the PCYC1+4×[Z268] promoter (Supplementary Figure 1). The strongest trans-activation was observed with Med15, which drove 30–50% higher expression on both exponential and ethanol phases than VP16A. Gcn4A showed the weakest trans-activating capacity, with 10*WD being slightly stronger. VP16A and Med3 showed similar activating capacity, in the cells at the exponential growth phase. In contrast to the others, Med3 drove higher expression on ethanol than during the exponential growth phase.
In summary, the artificial trans-activators increased the expression from the hybrid CYC1 promoters by larger fold-changes than that from the hybrid TEF1 promoters, but because the core CYC1 promoter expression was so weak, the absolute expression increases from the hybrid TEF1 promoters were larger. Med3 drove an increased activity at the ethanol-growth phase and Med15 had the strongest trans-activating capacity. Overall, these results provide insight on the variation of trans-activation effects of different protein modules on different core promoters. The stronger activation from Med15 than that from VP16A might be because Med15 activates directly, whereas VP16A recruits Med15 as a co-mediator to activate45.
Additional trans-activation modules do not increase the maximal activities of the Sk.GAL2 promoter
Previously, we have shown that the GAL1 promoter is stronger than glycolytic promoters when induced on galactose39. We further characterised a set of GAL promoters from other Saccharomyces species, and found even stronger GAL promoters, including the GAL2 promoters from S. eubayanus (Se.GAL2) and S. kudriavzevii (Sk.GAL2)9. As shown above, promoter hybrids with cis-regulatory elements can be boosted by synthetic trans-activators (Fig. 2b). We applied the same mechanism to the SkGAL2 promoter. The PZ4 cis-element ‘TA GAG TGA GAC GTT’ is found naturally in the Sk.GAL2 promoter, but not in the Se.GAL2 promoter or in other GAL promoters we have previously characterised9.
To develop an integrated synthetic trans-activating module to boost activity of the GAL2 promoter, we designed three synthetic Zif domains using ToolGen’s Zinc Finger Module set46 (ZifPZ42 targeting ‘GAG TGA GAC GTT’, ZifPZ43 targeting ‘TGA GAC GTT’, and ZifPZ44 targeting ‘GAG TGA GAC’) and one TALE domain, TALPZ4 (recognising ‘TA GAG TGA GAC GTT’) based on the dHax3 scaffold47. These DNA-binding domains were separately cloned into the N-terminal of the Med3 or Med15 modules to generate synthetic trans-activators. Only ZifPZ42 and ZifPZ43 (Fig. 2b)-containing trans-activators exhibited trans-activation on the hybrid TEF1-PZ4 promoter (PTEF1+4×[PZ4]), with the ZifPZ43-containing trans-activators being stronger. However, by observing multi-well plate cultures, we saw that the strains expressing ZifPZ43-Med3 and ZifPZ43-Med15 grew slower compared to the reference. Strains expressing ZifPZ42-Med15 and ZifPZ43-Med15 also exhibited a non-homogeneous population (Supplementary Fig. 2). Zif268-Med3 showed a stronger activation on PTEF1+4×[Z268] on the ethanol growth phase; this was not seen for ZifP643-Med3 with PTEF1+4×[PZ4] (Fig. 2b). By analysing the sequences inserted into the promoters using the YEASTRACT web-service48, we found the sequences containing the Z268 and the PZ4 elements also included binding sites for different endogenous yeast TFs (Supplementary Fig. 3). This may indicate a synergistic activation effect of synthetic Zif268-Med3 with endogenous TFs on the hybrid TEF1 + [Z268] promoter, whereas such effect is not present for ZifPZ43-Med3 on the hybrid TEF1 + [PZ4] promoter.
We chose the ZifPZ43 trans-activator to characterise potential combinatorial effects of additional trans-activation components on the GAL promoter. We modified the Sk.GAL2 promoter by inserting one to four additional PZ4 elements, and evaluated the expression outputs from the Sk.GAL2 promoter and its mutants in the presence of the synthetic trans-activators ZifPZ43-Med3 or ZifPZ43-Med15. The Se.GAL2 promoter (no PZ4 element) was used as a reference. Expression of a yEGFP reporter cassette was examined in the presence and absence of a synthetic trans-activator using a gal80Δ background strain in early exponential phase on glucose or ethanol (Fig. 2c). Expression of ZifPZ43-Med3 or ZifPZ43-Med15 did not appear to impact on growth rate in these strains.
Despite that GAL promoters and the expression of the Gal4 trans-activator are repressed by Mig1p in the presence of glucose, basal expression from GAL promoters was observed in strains grown on glucose (Fig. 2c). Consistent with previous observations9, the Sk.GAL2 promoter showed a higher basal expression on glucose than the Se.GAL2 promoter. As noted, the Se.GAL2 promoter does not have a PZ4 element, and expression of trans-activators ZifPZ43-Med3 or ZifPZ43-Med15 did not increase yEGFP expression from the Se.GAL2 promoter. However, expression of Zif268-VP16A led to a two-fold increase of yEGFP fluorescence, despite that the Se.GAL2 promoter does not contain Z268 element. Nevertheless, GFP fluorescence was still very low in this strain on glucose.
For strains harbouring the Sk.GAL2 promoter and its PZ4-element mutants growing on glucose, activators ZifPZ43-Med3 and ZifPZ43-Med15 increased yEGFP expression (Fig. 2c). There was a positive correlation between the number of PZ4 elements and the observed GFP fluorescence (Supplementary Fig. 4). In contrast to our previous findings with the TEF1 promoter (Fig. 2b), the Med3 mediator drove a stronger activation than the Med15 mediator. Zif268-VP16A drove a ~2-fold increase in the expression from Sk.GAL2 4×[PZ4] promoter and Sk.GAL2 5×[PZ4] promoter (Fig. 2c; but not from other ‘PZ4’-only Sk.GAL2 promoters). This may indicate that there is a non-specific trans-interaction between Zif268 and the sequences inserted into Sk.GAL2 4×PZ4 promoter and Sk.GAL2 5×[PZ4] promoter. The Sk.GAL2 5×[PZ4] promoter in the presence of ZifPZ43-Med3 showed ~3-fold stronger expression output, in comparison with the commonly-used TEF1 promoter with the same yEGFP-reporter construct (Construct 1, Fig. 1), and 1.7-fold stronger than the TDH3 promoter39. This shows the strong trans-activation effects from the artificial trans-activator-cis element interaction on the hybrid GAL promoter on glucose.
On ethanol, the Se.GAL2, Sk.GAL2, and Sk.GAL2 + [PZ4] promoters were activated to a very high level (Fig. 2c), at least seven-fold higher than the TEF1 promoter (Construct 1, Fig. 1). Neither ZifPZ43-Med3 nor ZifPZ43-Med15 significantly trans-activated the Sk.GAL2 + PZ4 promoters on ethanol.
In addition to the Sk.GAL2 + [PZ4] promoters, we also constructed a Sk.GAL2 + Z268 hybrid promoter, and tested the trans-activation effects of artificial trans-activators Zif268-VP16A, Zif268-Med3, and Zif268-Med15. Expression of these trans-activators increased the yEGFP activity from the Sk.GAL2 + [Z268] promoter by at least five-fold (Fig. 2c) on glucose. Zif268-Med3 showed the strongest activation effect, resulting in the expression of Sk.GAL2 + Z268 promoter on glucose being near to the levels on ethanol. These findings further confirmed the strong trans-activation capacity of artificial trans-activators.
However, by observing the cultures in multi-well plates, we saw that Sk.GAL2 + Z268 promoter-testing strain (gal80Δ background) with Zif268-Med3 and Zif268-Med15 grew slower compared to the reference, and the Zif268-Med15-expressing strain did not grow on ethanol. Such effects were not seen in the TEF1/CYC1 + [Z268] promoter-testing strains (GAL80-wildtype; Fig. 2). We postulated that the growth impairment might be caused by untargeted trans-activation from Zif268-Med3 and Zif268-Med15 in gal80Δ background.
In summary, artificial trans-activators can significantly activate the GAL promoter via the orthogonal cis-elements on glucose. However, they did not further increase the activities of the GAL promoters on ethanol, indicating that these activators do not significantly increase its maximum expression output of the GAL promoter.
Eukaryote-like trans-repression mechanisms can be used to build effective repression circuits in yeast
Trans-repression is another natural mechanism for transcriptional regulation. In bacteria, a repressor commonly functions by binding to the elements near the transcription start site to prevent transcription from the target promoter49,50. A similar mechanism can be reconstructed in yeast by the insertion of bacterial repressor-binding sites near to ‘TATA-box’ or transcription initiation sequence in the target promoter and the expression of the cognate bacterial repressor6,15,21. However, intensive effort, including construction and screening of large libraries, was required to obtain the ideal hybrid repressible promoter; in addition, a strong constitutive promoter is required to express bacterial repressors6,15. In contrast to bacterial repressors, transcriptional repressors in eukaryotes commonly mediate gene repression through a series of regulatory mechanisms including recruiting histone deacetylase complexes to direct nucleosome formation and blocking co-activators51,52. These mechanisms may be more relaxed about the position of cis-acting elements in target promoters and can potentially be exploited to develop a trans-repressing toolset.
We characterised the trans-repressing capacity of four protein modules, including Sin3 C-terminal domain (Sin3 aa600-1536; Sin3C)53, Tup154, Cyc854, and Mig1 C-terminal domain (Mig1 aa123-504; removal of DNA-binding domain; Mig1C)55. Sin3C mediates repression through its interactions with histone deacetylase complexes53, Tup1/Cyc8 through recruiting histone deacetylase complexes and masking binding of co-activators54, and Mig1C is thought to recruit the Tup1/Cyc8 complex55. Sin3C, Tup1, and Cyc8 have been previously tested in various studies by fusing them to a DNA-binding domain, but Mig1C has not53,54.
We first attempted to characterise the fusion of Zif268-SV40NLS with the C-terminal Sin3C, Tup1, or Mig1C. The HAC1 promoter was used to control the fusion expression. yEGFP controlled by PTEF1+4×[Z268] was used as the reporter (Fig. 3a). However, yeast transformants of Zif268-Sin3C and Zif268-Tup1 constructs grew extremely slowly on agar plates, and the colonies could not be picked after more than a week of incubation. We were able to characterise the strain expressing Zif268-Mig1C. We observed two distinct cell populations: in one population, the yEGFP expression was severely repressed, whereas in the other, no repression (relative to the control construct) was observed (Supplementary Fig. 5).

a The trans-regulating effects of TetR-repressors with or without tetracycline addition. The yEGFP fluorescence were characterised in the cells grown to the early exponential growth phase (EXP) or the ethanol-growth phase (ETH) in MES-buffered YNB media using 20 g L−1 glucose as the carbon source in 96-well plates. The dashed horizontal line indicates the output from the native TEF1 promoter (Fig. 1). b Schematic of tetracycline-mediated repression on the GAL promoter. c The effects of tetracycline on the GAL promoter in the cells integrated with tetracycline-repression module in b. YNB Ethanol Liquid: yeast cells were grown to OD600 ~ 1 in MES-buffered YNB media using 2% (v/v) ethanol as the carbon source at 30 °C in testing tubes and analysed. YNB Glucose Agar: yeast was streaked on YNB nutrient agar with 20 g L−1 glucose as the carbon source at 30 °C for 48 h, and single colonies were resuspended in water and analysed. The numbers on top of bars are the decrease fold-change at 37 °C to 30 °C. Mean values ± standard deviations are shown (N = 3 independent biological replicates). Source data in a & c are provided in Supplementary Data 1.
The unstable expression profiles in the Zif268-containing promoter constructs suggested off-target effects rendering the constructs unusable. We therefore developed an alternative approach using TetR elements as the DNA-binding domain for characterisation of the four trans-repressing modules. A cis-regulatory reporter cassette was constructed by replacing the Z268 elements in PTEF1+4×[Z268] with TetO elements. The control construct in the absence of a repressor module showed ~2-fold higher expression in the exponential phase than in the ethanol phase and did not respond to tetracycline addition (Fig. 3a).
Yeast expressing TetR-derivative repressors (TetR-Sin3C, TetR-Tup1, TetR-Cyc8, and TetR-Mig1C) grew normally on plates and showed strong repression on the TEF1 + 4×[TetO] promoter in the absence of tetracycline. TetR-Tup1 was most efficient, showing >100-fold repression and eliminating yEGFP fluorescence in > 90% cell population (Fig. 3a; Supplementary Fig. 5). TetR-Cyc8, and TetR-Mig1C showed the repression similar to TetR-Tup1. The repression was maintained in the cells at the ethanol-growth phase, even for the cells expressing TetR-Mig1C, although Mig1 itself is a glucose-dependent repressor. Repression on ethanol by TetR-Mig1C might be due to the inclusion of a nuclear localisation sequence (Fig. 1a), which dominates the glucose-dependent nuclear localisation of Mig156. Addition 50 μM tetracycline can de-repress yEGFP expression in the whole cell population (Supplementary Fig. 6). TetR-Sin3C was less efficient, not eliminating yEGFP fluorescence in the whole population (Fig. 3a; Supplementary Fig. 6). Yeast expressing TetR-repressors did not show a homogenous population: ~ 8% population deviated from the major population and showed higher yEGFP fluorescent levels under the conditions without tetracycline additions (TetR-Tup1 in Supplementary Fig. 5). We postulated that this might be caused by the loss of repressor-expressing plasmids: centromeric plasmids show a 3% loss rate per generation57.
The repression was maintained in the cells at the ethanol-growth phase, even for the cells expressing TetR-Mig1C, although Mig1 itself is a glucose-dependent repressor. Repression on ethanol by TetR-Mig1C might be due to the inclusion of a nuclear localisation sequence (Fig. 1a), which dominates the glucose-dependent nuclear localisation of Mig156.
In summary, repression capacities of the four trans-repressing domains were sequenced as Tup1 ~ Mig1C ~ Cyc8 » Sin3C, and a low-level expression of a TetR fusion with Tup1, Mig1C, or Cyc8 repressor can fully repress the expression of a strong promoter inserted with TetO elements.
Tetracycline-mediated repression of GAL promoters
To develop a mechanism that can fully repress GAL promoters-controlled metabolic pathways on demand, we used the tetracycline de-repressible circuit to control the expression of the GAL repressor Gal80p. In the resulting strain, TetR-Tup1 was expressed under the control of the HAC1 promoter, the TEF1 + [TetO] promoter was integrated upstream of the GAL80 gene to control its expression, and a yEGFP gene was introduced under the control of the GAL1 promoter to report the regulatory effects of tetracycline (Fig. 3b). The strain was first grown on ethanol to relieve Mig1p-mediated glucose repression on GAL promoters2,9, in the absence or presence of tetracycline. In the absence of tetracycline, a very high level of yEGFP fluorescence was detected (Fig. 3c and Supplementary Fig. 6), showing a fully de-repression state of the GAL1 promoter2,39. In the presence of 125 μM tetracycline, yEGFP fluorescence was repressed by >200 fold to very minimal fluorescence.
We then grew the strain on nutrient agar containing 20 g L−1 glucose as the carbon source. Consistent with our previous observation that 20 g L−1 glucose in agar is not sufficient to repress the expression from GAL promoters9, the GAL1 promoter was moderately de-repressed in the absence of tetracycline (Fig. 3c and Supplementary Fig. 6). Addition of 125 μM tetracycline repressed the yEGFP expression by >200 fold.
In summary, by integrating tetracycline-inducible expression circuit to control GAL80 expression, we successfully deployed a tetracycline-mediated mechanism to repress GAL promoters.
A heat-mediated response circuit for 37 °C induction of GAL promoters
Temperature is a primary parameter to control during yeast cultivation. Although the preferred temperature for S. cerevisiae is 30 °C, growth at higher temperature can potentially reduce the cooling costs for exothermic fermentation processes58, and increased temperatures are well tolerated. Transition to an increased bioprocess temperature upon entry into the production phase (when fast growth is no longer required) would therefore make a convenient tool for regulatory control and may make the bioprocess more economical. To exploit this potential, we designed a heat-inducible circuit to modulate expression from GAL promoters in strains growing on glucose. Our model employs a heat-inducible degron59 to regulate the stability of the glucose-dependent repressor Mig1p (Fig. 4b).

a Schematic of expression system of heat-inducible degradation of yEGFP. b Schematic of genetic circuits of 37 °C-induction of the GAL promoter. c Degradation efficiency of heat-inducible degron in a at 37 °C. d Induction response of the GAL promoter at 37 °C. H. Degron, Ubi4-RHGSGTMV-DHFR*. Yeast cells were grown to the early exponential growth phase in MES-buffered YNB media with 20 g L−1 glucose as the carbon source in testing tubes. The numbers on top of bars are the decrease fold-change at 37 °C to 30 °C. Mean values ± standard deviations are shown (N = 3 or 4 independent biological replicates). Source data in c and d are provided in Supplementary Data 1.
We first used the yEGFP reporter to characterise the previously-developed heat-inducible degron (H.degron)59 in our system. The full heat-inducible degron module comprises four parts: a ubiquitin moiety (Ubi) that is cleaved off by ubiquitin C-terminal hydrolase after translation, an ‘N-end rule’ residue Arg, a linker ‘HGSGTMV’, and mouse dihydrofolate reductase P66L temperature sensitive mutant (DHFR*). The heat-inducible degron Ubi4-RHGSGTMV-DHFR* was fused to the N-terminus of yEGFP, and the fusion (UBI4-RHGSGTMV-DHFR*-yEGFP) was expressed under the control of TEF1 promoter and the PGK1 terminator (Fig. 4a). yEGFP was characterised as the control. Two resulting yeast strains, expressing yEGFP and UBI4-RHGSGTMV-DHFR*-yEGFP, were grown at 30 °C or at 37 °C. Consistent with previous studies59,60, elevation of growth temperature to 37 °C did not slow the growth. In the yEGFP control, fluorescence was 1.2-fold lower at 37 °C (Fig. 4c), indicating that elevated temperature did not dramatically affect yEGFP abundance.
Tagging Ubi4-RHGSGTMV-DHFR* decreased fluorescence at 30 °C by 4.5-fold compared to the control yEGFP (Fig. 4c), suggesting either disruption of transcription/translation or basal degradation by the degron. At 37 °C, fluorescence decreased by 16.2-fold. These data demonstrate the functionality of Ubi4-RHGSGTMV-DHFR* as an effective heat-inducible degron (H.Degron).
In summary, the basal degradation (or impaired expression) at 30 °C caused by Ubi4-RHGSGTMV-DHFR* (H.Degron) is significant; moreover, heat-inducible degradation by this degron at 37 °C, although dramatically decreasing yEGFP abundance, did not result in a full depletion of target yEGFP (Fig. 4c). Incomplete degradation might be problematic for application to control the abundance of a transcription factor, because transcription factors can function effectively at a relatively low expression level. To apply the H.Degron to regulate the abundance of Mig1p for the proposed 37 °C-mediated de-repression of GAL promoters, it will be important to overcome these two limitations, otherwise a sharp temperature-induced boundary effect cannot be achieved.
To deploy the 37 °C-mediated de-repression module on GAL promoters, we used a gal80Δ strain expressing yEGFP under the control of the GAL1 promoter and the URA3 terminator. In this strain, we modified the MIG1 locus by fusing the H.Degron to the N-terminus of Mig1p and introducing an alternative promoter to control the expression strength. We tested three promoters to provide variable expression strength with the aim of titrating the transcription of H.Degron-MIG1 to obtain a sharp boundary effect: the TEF2 promoter (strong)39, the HAC1 promoter (medium-weak; Fig. 1c), and the NRG1 promoter (weak; Fig. 1c). The resulting yeast strains were grown on glucose at 30 °C or 37 °C to examine yEGFP expression from the GAL1 promoter (Fig. 4d). In these cases, cultivation at 37 °C did not result in slowed growth. The strain with the TEF2 promoter construct showed a very low level of yEGFP fluorescence at 37 °C, indicating the expression from the TEF2 promoter was too high and the H.Degron was not sufficient to deplete Mig1p. The strain with the NRG2 promoter showed an induced level of yEGFP fluorescence at 30 °C, indicating that the expression from the NRG2 promoter was not sufficient and the basal degradation of H.Degron caused the depletion of Mig1p. Fortunately, the strain with the HAC1 promoter construct showed very low level yEGFP fluorescence at 30 °C and good induction 37 °C (Fig. 4d and Supplementary Fig. 6). This confirmed the functionality of 37 °C-mediated de-repression of GAL promoters.
Expanded regulatory circuits for conditional control of sesquiterpene (nerolidol) production
Previously, we developed yeast strains for high-level sesquiterpene production through metabolic engineering17,61,62. To develop these strains, GAL promoters, coupled with the deletion of GAL repressor gene GAL80, were used to control the expression of the genes from the mevalonate pathway and the genes for production of the sesquiterpene trans-nerolidol. The resulting strain NLD401 produced ~1.7 g L−1 nerolidol in two-phase flask cultivation at 30 °C17. In the current work, we developed an isogenetic yeast strain (N9R5HIRBU), in which the synthetic genetic modules for tetracycline-mediated repression and 37 °C-mediated de-repression of GAL promoters were also established (Fig. 5a). In this strain, Yellow Fluorescence-Activating and absorption-Shifting Tag (YFAST) and Actinidia chinensis (kiwifruit) nerolidol synthase (AcNES1) were expressed through a 2A sequence-mediated bicistronic expression mechanism (YFAST-2A-AcNES1)17. The YFAST-2A-AcNES1 was under the control of the GAL2 promoter. This construct not only contributed to an improved nerolidol production, but also allowed us to monitor the GAL2 promoter activities by measuring YFAST fluorescence throughout the whole cultivation as a proxy for GAL2 promoter-driven expression. We evaluated the effects of tetracycline and elevated temperature on nerolidol production and the GAL promoter activities in this strain via two-phase dodecane-overlayed flask cultivation.
In flask cultivation at 30 °C and the absence of tetracycline, strain N9R5HIRBU (+ tetracycline-repressible/heat-inducible regulatory modules: Fig. 3b and Fig. 4b) showed a specific maximum growth rate (µmax) of 0.3 h−1 and a maximum cell density at OD600 of ~14 at 72 hr. N9R5HIRBU produced ~1.76 g L−1 nerolidol at 72 hr (Fig. 5e). The dynamics of YFAST fluorescence were also similar in both strains: a very low expression in the first 12 hr when glucose was the carbon source and a gradually induced expression after 12 hr when glucose was depleted (Fig. 5c). These physiological features were similar to the isogeneic strain NLD401 published previously (w/o tetracycline-repressible/heat-inducible regulatory modules; gal80Δ)17. These data show that the ‘OFF’ state of tetracycline-mediated repression on GAL promoters is tight, producing a gal80Δ-like genotype and phenotype.
Addition of 125 μM tetracycline at 30 °C did not significantly alter growth characteristics in the exponential phase (Fig. 5b and d), however much more biomass was produced during the post-exponential phase, resulting in a maximum OD600 of ~22 (Fig. 5b). Consistent with tetracycline-mediated repression on yEGFP expression from the GAL1 promoter (Fig. 3), YFAST expression in N9R5HIRBU was essentially fully repressed by 125 μM tetracycline throughout the whole cultivation (Fig. 5c). Only ~0.03 g L−1 nerolidol was produced at 72 hr, 60-fold lower than in the absence of tetracycline at 30 °C (Fig. 5e). These data demonstrate that the ‘ON’ state of tetracycline-mediated repression on GAL promoters provided a fine mechanism for nearly full repression on GAL promoter-controlled heterologous synthetic pathways. It also demonstrates that nerolidol production delivers a metabolic burden which decreases biomass production. The tetracycline repression module can facilitate strain maintenance during laboratory manipulation by relieving the metabolic burden caused by heterologous synthetic pathways.
We then characterised strain N9R5HIRBU at 37 °C in the absence of tetracycline to investigate the effects of elevated temperature on nerolidol production. Pre-culturing was performed at 37 °C to prepare the seed inoculum. A decrease in exponential growth rate and biomass accumulation was observed at 37 °C, with a maximum OD600 of ~11 (Fig. 5b and d). YFAST fluorescence at 0 h was ~20-fold higher at 37 °C than at 30 °C, and was continuously induced during the exponential and post-exponential growth phases (Fig. 5c). This suggests that heat-inducible protein degradation might not be efficient for complete depletion of Mig1p, but does provide a moderate relief of glucose repression. Reflecting the induced expression from the GAL promoter, strain N9R5HIRBU produced ~ 0.5 g L−1 nerolidol at 24 h and 37 °C, ~6-fold improvement compared to that at 30 °C; and produced ~2.54 g L−1 nerolidol at 72 h, a 45% improvement on the original NLD401 strain and on the N9R5HIRBU strain grown at 30 °C (Fig. 5e). Consistent with this, the specific nerolidol production rates in the cells grown at 37 °C were significantly higher than those in the cells grown at 30 °C, with a 9-fold increase in the first 24 h and a 49% increase from 24 hr to 72 h (Fig. 5f). These data demonstrate that the integration of 37 °C-mediated induction circuits for the GAL promoters resulted in improved production of nerolidol-producing strains at an elevated temperature.
Discussion
Genetic regulation tools should be applicable in biotechnology for development of microbial cell factories in metabolic engineering63,64. Such tools can be used in industrial processes to dynamically tune host cell metabolism for maximal productivities, or in the laboratory to facilitate strain maintenance and engineering. Development of these tools is often encumbered by unknown properties of basic genetic modules under variable conditions. Therefore, in this work, we started with characterisation of basic genetic circuits, including eukaryote-like trans-activating and trans-repressing modules, and then deployed new mechanisms to regulate GAL promoters aiming for application in metabolic engineering. Our approaches included: characterisation of different promoters from transcription factor genes across exponential and ethanol phases to better understand endogenous eukaryotic controls on regulatory proteins (Fig. 1), engineering promoters with cis-elements in combination with trans-acting factors to modulate transcriptional activity to constitutive and galactose-responsive promoters (Fig. 2), establishing repression circuits based on eukaryotic systems (Fig. 3), establishing a temperature-inducible circuit (Fig. 4), and applying selected circuits to production of nerolidol (Fig. 5).

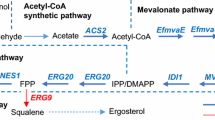
a Schematic of nerolidol synthetic pathway and GAL promoter regulation in strain N9R5MIRBU. Tetracycline/heat regulatory mechanisms on GAL promoters refer to Figs. 3b and 4b. IPP, isopentenyl pyrophosphate; DMAPP, dimethylallyl pyrophosphate; FPP, farnesyl pyrophosphate; ERG20, FPP synthase; YFAST-2A-AcNES1, 2A-mediated bicistronic gene of Yellow Fluorescence-Activating and absorption-Shifting Tag (YFAST) and Actinidia chinensis (kiwifruit) nerolidol synthase (AcNES1). b–f Characterisation of strain N9R5MIRBU in two-phase flask cultivations under the conditions: at 30 °C and in the absence of tetracycline (30 °C w/o tetracycline); at 30 °C and in the presence of 125 μM tetracycline (30 °C + tetracycline); or at 37 °C and in the absence of tetracycline (37 °C w/o tetracycline). The yeast cells were grown in 2-(N-morpholino)ethanesulfonic acid (MES)-buffered synthetic mineral salt medium (prepared from yeast nitrogen base, YNB) with 20 g L−1 glucose as the carbon source. YFAST fluorescence was measured after 4-hydroxy-3-methylbenzylidene rhodanine (HMBR) with final concentration 20 μM was added in yeast samples before flow cytometry assay. Mean values ± standard deviations are shown (N = 3 independent biological replicates) in b–d, e for 30 °C w/o tetracycline and 37 °C w/o tetracycline, and f. Mean value and absolute errors are shown in e for 30 °C + tetracycline (N = 2 independent biological replicates). n.d., not determined. Source data in b–f are provided in Supplementary Data 1.
In terms of basic promoter-mediated control, we observed that promoters from transcription factor genes expressed at 1-2 orders of magnitude lower than commonly used constitutive promoters. We used this information to identify preferred promoters for control of transcription factors in engineered circuits, so that regulation similar to endogenous systems could be achieved. We then applied this to regulate cis-element engineered promoters. We found that trans-activating modules have variable effects on transcriptional activity from different hybrid promoters; in particular, the weak CYC1 promoter demonstrated a much higher fold-change in response to trans-activating module engineering than the strong TEF1 promoter (Fig. 2). Our data suggest that (a) the activation effects of an artificial trans-activator varies depending on the core promoter used in the reporter system, and (b) threshold effects exist for trans-activating module activity in engineered promoters. In support of this, while there was generally a positive correlation between the number of cis-acting elements inserted in engineered promoters, additional trans-activation modules did not increase the maximal activities of the Sk.GAL2 promoter, which can already be activated to very high levels (Fig. 2c and supplementary Fig. 4). We also observed a potential syngenetic effect with endogenous TFs, shown by Zif268-Med3 on TEF1 + [Z268] promoter (Fig. 2b). Overall, these observations are consistent with the previous observation that core promoter sequence plays a major role determining expression level65. They further suggest that maximal activation by Gal4p on the GAL promoter under de-repression conditions had been achieved, and suggested that any further increases would require engineering of the core promoter.
Moreover, some synthetic TFs like Zif268-Tup1 and Zif268-Sin3C are not compatible with yeast, leading to the problems or failure in transformation, whereas expression of some TFs like Zif268-Med3 and Zif268-Med15 in gal80Δ background, but not in GAL80 background, decreased growth fitness. Similarly, toxicity of artificial TFs like PhlF-VP16A, CamR-VP16A, lexA-VP16A was previously reported19,33. Toxicities associated with Zif nucleases are known to be caused by off-target effects66. Similarly, such detrimental effects of these artificial TFs might be caused by off-target interactions that perturb genetic networks for growth fitness. It is noteworthy that Zif268-Med3/15 toxicity is apparently masked by the Gal80 repressor. Genetic profiling will be required to identify shared targets that result in toxic effects. Also, Zif268-VP16A, although summoning Med15 for trans-activation45, did not show off-target toxicity (which would be evidenced by growth defects) in the gal80Δ background. Similarly, Zif268-Mig1C, but not Zif268-Tup1, could be transformed into yeast, although Mig1C recruits the Tup1-Cyc8 repression complex55. These results suggest that adding a layer of protein-protein interaction may provide an equilibrium to the system, resulting in attenuation of off-target toxicity. On the other hand, such a multi-layer regulatory mechanism may contribute to the specificity of transcription factor regulation67.
Adding to the component toolbox, we went on to establish repression circuits based on eukaryotic trans-repressing modules, similarly to the approach used for the trans-activating modules (cis-element engineering on promoters with concurrent expression of trans-repressing transcription factors). This system was established using the TEF1 promoter; the established module was then applied to control the GAL80 repressor, providing a further level of control to the ‘on’ and ‘off’ switch and decrease leakiness of the GAL promoter.
For application in yeast metabolic engineering, we successfully deployed the circuit for tetracycline-mediated repression on GAL promoters and the circuit for 37 °C-mediated induction of GAL promoters. Improving on previous tetracycline regulatory circuits15,18,33, the TetR-derivative circuit developed in this study fully represses the TEF1 + [TetO] promoter in absence of tetracycline (Fig. 3a) and a weak expression of TetR-fusion repressor is efficient for such repression. Moreover, we used it to control GAL80 expression delivering an efficient binary ON/OFF control on GAL promoters (Fig. 3b, c). In contrast, the heat-inducible degron in isolation was not efficient to deplete the target protein. In this case, precise titration of the expression strength of Mig1p fused with heat-inducible degron was required to deliver a binary ON/OFF effect on GAL promoters (Fig. 4d). Combination of these two circuits provides novel operation principles: (1) heterologous pathways can be repressed in small scale laboratory development procedures by addition of a small molecule, which facilitates strain maintenance, rather than adding the molecule to induce these pathways in industrial processes; (2) heat released from large-scale fermentation can be hijacked for metabolic pathway induction, also decreasing the overall bioprocess cost. Although not fully de-repressing the GAL promoter on glucose (Fig. 5c), heat-inducible degradation of Mig1p led to improved nerolidol productivities at 37 °C (Fig. 5). This prototyped a process in S. cerevisiae featuring independence from expensive inducers and improvement in production. However, further validation is necessary for its application in real industrial processes, and to determine if this approach can reduce cooling costs.
In summary, proper characterisation of each modularised genetic module prior to deploying them in a circuit format was an essential element for success of this project. Although not all characterised genetic modules were eventually applied in development of metabolic engineering tools, such characterisation revealed basic features of these modules, which provides guidance for circuit design. Continuing to build up part libraries and even re-characterisation of known parts under varied conditions and/or through independent systems or procedure will improve our knowledge about their features and is important for precise synthetic circuit design. Capacities for biofoundry-based molecular laboratories68,69, model-driven design70, and directed evolution of module components33 will be important for symmetrical and modularised engineering for synthetic circuits. Fortunately, the two novel regulatory models were straightforward to develop, and are generally applicable in metabolic engineering. Isolated modules, either tetracycline-mediated repression or 37 °C-mediated induction of GAL promoters, can be integrated in previously developed strains where GAL promoters are used to control the expression of heterologous metabolic pathways. It should be also emphasised that synthetic regulatory circuits for metabolic engineering can be engineered towards both facilitating laboratory strain maintenance and bringing economic mechanisms for pathway control during production.
Methods
Plasmid and strain construction
Plasmids used in this work are listed in Supplementary Table 1, and strains are listed in Supplementary Table 2. Primers used in polymerase chain reaction (PCR) and PCR performed in this work are listed in Supplementary Table 3. Plasmid construction processes are listed in Supplementary Table 4. Yeast strain construction processes are listed in Supplementary Table 5. A LiAc/SS carrier DNA/PEG method71 was used for yeast transformation. Molecular Cloning Designer Simulator was used to manage DNA sequence design72. Promoters inserted with synthetic cis-elements and synthetic protein domains are listed in Supplementary Table 6.
Yeast cultivation
Yeast nitrogen base without amino acids (YNB, FORMEDIUM#CYN0402; 6.9 g L−1) was used to prepare the media with ammonium as the nitrogen source. When urea was used as the nitrogen source, 1.7 g L−1 yeast nitrogen base without amino acids and without ammonium sulphate (Sigma Aldrich) and 1 g L−1 urea were used to prepare the base media, and sterilised by filtration. Amino acids (leucine, histamine, tryptophan) were supplemented in YNB media to grow auxotrophic strains. For the YNB media without additional buffer, pH was adjusted to 6.0 using sodium hydroxide solution; for the YNB media with 2-(N-morpholino)ethanesulfonic acid (MES) buffer, 19.5 g L−1 MES was used, and pH was adjusted to 6.0 with ammonia hydroxide solution. Glucose (20 g L−1) or ethanol (2% v/v) was used as the carbon source.
For characterisation of yEGFP-expressing strains, yeast cells from glycerol stocks were streaked on YNB-glucose agar. For the growth in 96-well microplates, yeast cells were grown in YNB-glucose medium for about 20 h to stationary phase in a 350 rpm 30 °C incubator to prepare seed culture. Seed culture (5 μl) was inoculated into 100 μl MES-buffered YNB-glucose medium to prepare Culture 1. Culture 1 (2 μl) was inoculated into 100 μl MES-buffered YNB-glucose medium to prepare Culture 2. Culture 2 was incubated in a 350 rpm 30 °C incubator overnight for analysis of yEGFP fluorescent in the cells grown to the exponential growth phase, and Culture 1 for ~36 h for analysis in the cells grown to the ethanol growth phase. Seed culture (2 μl) was inoculated into 100 μl YNB-ethanol medium, and the culture was incubated in a 350 rpm 30 °C incubator for ~48 h for analysis in the cells grown grow on ethanol. For the growth in 10 ml test tubes, yeast cells were inoculated into 500 μl MES-buffered YNB-glucose medium and grown overnight in a 200 rpm 30 °C or 37 °C incubator for analysis of yEGFP fluorescent in the cells grown to the exponential growth phase. Stock tetracycline solutions (125 mM) was prepared in DMSO-ethanol (1:1) solution and supplemented to prepare the medium with 125 μM or 250 μM tetracycline, and 25 mM stock solution for the medium with 50 μM tetracycline, and 2.5 mM stock solution for the medium with 5 μM tetracycline.
For characterisation of nerolidol-producing strains, dodecane-overlayed two-phase flask cultivation was used. Yeast cells from glycerol stocks were streaked on YNB -glucose agar containing 125 μM tetracycline. Before initiating the two-phase flask cultivation, cells were pre-cultured in MES-buffered YNB-glucose to exponential phase (OD600 between 1 to 4) and collected by centrifugation. Collected cells were then resuspended in fresh fermentation medium. To initiate the cultivation, appropriate volumes of pre-cultured cells were transferred to MES-buffered YNB medium with 20 g L−1 glucose to an initial OD600 of 0.2 in a total volume of 23 mL medium in a 250 mL flask, and 2 mL sterile dodecane was added after inoculation. Seed culture and test culture were grown in a 200 rpm 30 °C or 37 °C incubator. In the first 12 h of cultivation, 3 ml culture was sampled for growth curve measurement. Dodecane was sampled and stored at −80 °C for terpene analysis.
Flow cytometry
Fluorescence in single cells was analysed using a BD Accuri™ C6 flow cytometer (BD Biosciences, USA)17,39. For analysis of yEGFP fluorescence, cells sampled from characterisations were directly used for flow cytometry analysis. For analysis of YFAST fluorescence, 100-time-concentrated HMBR was added to the samples to 20 μM final concentration and the sample was mixed before analysis17. FSC.H threshold was set at the value of 250,000 for exclusion of debris particles. GFP and/or YFAST fluorescence was excited by a 488 nm laser and monitored through a 530/20 nm bandpass filter (FL1.A), with 10,000 events recorded per sample. Mean values of FSC.A, SSC.A, and FL1.A for all detected events were extracted using a BD Csampler software (BD Accuri C6 software version 1.0.264.21). GFP or YFAST fluorescence level was expressed as the percentage of the average background auto-fluorescence from the exponential-phase cells of GFP-negative reference strain GH4 as described previously39.
Metabolite analysis
The Metabolomics Australia Queensland Node analysed extracellular metabolites. Nerolidol in dodecane samples were analysed as previously described62. Dodecane samples (in some cases, diluted with dodecane) were diluted in 40-fold volume of ethanol. The ethanol-diluted samples (20 µL) were injected. A Zorbax Extend C18 column (4.6 × 150 mm, 3.5 µm, Agilent PN: 763953-902) equipped with a guard column (SecurityGuard Gemini C18, Phenomenex PN: AJO-7597) was used. Analytes were eluted at 35 °C at 0.9 mL/min using the mixture of solvent A (water) and solvent B (45% acetonitrile, 45% methanol, and 10% water), with a linear gradient of 5–100% solvent B from 0–24 min, then 100% from 24–30 min, and finally 5% from 30.1–35 min. Analytes of interest were monitored using a diode array detector (Agilent DAD SL, G1315C) at 202 nm wavelength.
Statistics and reproducibility
Two-tailed Welch’s t-test was used for comparing two groups. Data generated from two-to-four biological replicates are presented. Construction-in-processing strains and strain construction procedures are not replicated.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
Source data are provided in Supplementary Data 1. Any remaining information can be obtained from the corresponding author upon reasonable request.
Materials availability
Plasmids used in this study are available on Addgene (under submission).
Code availability
Custom code or mathematical algorithm was not used in this study.
References
Rugbjerg, P. & Sommer, M. O. A. Overcoming genetic heterogeneity in industrial fermentations. Nat. Biotechnol. 37, 869–876 (2019).
Peng, B., Plan, M. R., Carpenter, A., Nielsen, L. K. & Vickers, C. E. Coupling gene regulatory patterns to bioprocess conditions to optimize synthetic metabolic modules for improved sesquiterpene production in yeast. Biotechnol. Biofuels 10, 43 (2017).
Wu, G. et al. Metabolic burden: cornerstones in synthetic biology and metabolic engineering applications. Trends Biotechnol. 34, 652–664 (2016).
Couto, J. M., McGarrity, A., Russell, J. & Sloan, W. T. The effect of metabolic stress on genome stability of a synthetic biology chassis Escherichia coli K12 strain. Microb. Cell Factories 17, 8 (2018).
Lv, Y., Gu, Y., Xu, J., Zhou, J. & Xu, P. Coupling metabolic addiction with negative autoregulation to improve strain stability and pathway yield. Metab. Eng. 61, 79–88 (2020).
Chen, Y. et al. Genetic circuit design automation for yeast. Nat. Microbiol 5, 1349 (2020).
Li, M. & Borodina, I. Application of synthetic biology for production of chemicals in yeast Saccharomyces cerevisiae. FEMS Yeast Res. 15, 1–12 (2015).
Krivoruchko, A. & Nielsen, J. Production of natural products through metabolic engineering of Saccharomyces cerevisiae. Curr. Opin. Biotechnol. 35, 7–15 (2015).
Peng, B., Wood, R. J., Nielsen, L. K. & Vickers, C. E. An expanded heterologous GAL promoter collection for diauxie-inducible expression in Saccharomyces cerevisiae. ACS Synth. Biol. 7, 748–751 (2018).
Hayat, I. F. et al. Auxin-mediated induction of GAL promoters by conditional degradation of Mig1p improves sesquiterpene production in Saccharomyces cerevisiae with engineered acetyl-CoA synthesis. Microb. Biotechnol. 14, 2627–2642 (2021).
Shi, B. et al. Systematic metabolic engineering of Saccharomyces cerevisiae for lycopene overproduction. J. Agric. Food Chem. 67, 11148–11157 (2019).
Vickers, C. E., Williams, T. C., Peng, B. & Cherry, J. Recent advances in synthetic biology for engineering isoprenoid production in yeast. Curr. Opin. Chem. Biol. 40, 47–56 (2017).
Ma, J., Gu, Y. & Xu, P. A roadmap to engineering antiviral natural products synthesis in microbes. Curr. Opin. Biotechnol. 66, 140–149 (2020).
Lyu, X., Ng, K. R., Lee, J. L., Mark, R. & Chen, W. N. Enhancement of naringenin biosynthesis from tyrosine by metabolic engineering of Saccharomyces cerevisiae. J. Agric. Food Chem. 65, 6638–6646 (2017).
Ellis, T., Wang, X. & Collins, J. J. Diversity-based, model-guided construction of synthetic gene networks with predicted functions. Nat. Biotechnol. 27, 465–471 (2009).
Khalil, A. S. & Collins, J. J. Synthetic biology: applications come of age. Nat. Rev. Genet. 11, 367–379 (2010).
Lu, Z., Peng, B., Ebert, B. E., Dumsday, G. & Vickers, C. E. Auxin-mediated protein depletion for metabolic engineering in terpene-producing yeast. Nat. Commun. 12, 1051 (2021).
Belli, G., Gari, E., Piedrafita, L., Aldea, M. & Herrero, E. An activator/repressor dual system allows tight tetracycline-regulated gene expression in budding yeast. Nucleic Acids Res. 26, 942–947 (1998).
Ottoz, D. S., Rudolf, F. & Stelling, J. Inducible, tightly regulated and growth condition-independent transcription factor in Saccharomyces cerevisiae. Nucleic Acids Res. 42, e130 (2014).
McIsaac, R. S., Gibney, P. A., Chandran, S. S., Benjamin, K. R. & Botstein, D. Synthetic biology tools for programming gene expression without nutritional perturbations in Saccharomyces cerevisiae. Nucleic Acids Res. 42, e48 (2014).
Rantasalo, A., Kuivanen, J., Penttila, M., Jantti, J. & Mojzita, D. Synthetic toolkit for complex genetic circuit engineering in Saccharomyces cerevisiae. ACS Synth. Biol. 7, 1573–1587 (2018).
Khakhar, A., Bolten, N. J., Nemhauser, J. & Klavins, E. Cell-cell communication in yeast using auxin biosynthesis and auxin responsive CRISPR transcription factors. ACS Synth. Biol. 5, 279–286 (2016).
An-Adirekkun, J. M. et al. A yeast optogenetic toolkit (yOTK) for gene expression control in Saccharomyces cerevisiae. Biotechnol. Bioeng. 117, 886–893 (2020).
Renicke, C., Schuster, D., Usherenko, S., Essen, L.-O. & Taxis, C. A LOV2 domain-based optogenetic tool to control protein degradation and cellular function. Chem. Biol. 20, 619–626 (2013).
Salinas, F., Rojas, V., Delgado, V., Agosin, E. & Larrondo, L. F. Optogenetic switches for light-controlled gene expression in yeast. Appl. Microbiol. Biotechnol. 101, 2629–2640 (2017).
Shen, B. et al. Fermentative production of Vitamin E tocotrienols in Saccharomyces cerevisiae under cold-shock-triggered temperature control. Nat. Commun. 11, 5155 (2020).
Bashor, C. J. & Collins, J. J. Understanding biological regulation through synthetic biology. Annu. Rev. Biophys. 47, 399–423 (2018).
Struhl, K. Fundamentally different logic of gene regulation in eukaryotes and prokaryotes. Cell 98, 1–4 (1999).
Grunberg, T. W. & Vecchio, Del D. Modular analysis and design of biological circuits. Curr. Opin. Biotechnol. 63, 41–47 (2020).
Zorzan, I., López, A. R., Malyshava, A., Ellis, T. & Barberis, M. Synthetic designs regulating cellular transitions: fine-tuning of switches and oscillators. Curr. Opin. Syst. Biol. 25, 11–26 (2021).
Nielsen, A. A. K. et al. Genetic circuit design automation. Science 352, aac7341 (2016).
Szymanski, E. & Calvert, J. Designing with living systems in the synthetic yeast project. Nat. Commun. 9, 2950 (2018).
Tominaga, M., Nozaki, K., Umeno, D., Ishii, J. & Kondo, A. Robust and flexible platform for directed evolution of yeast genetic switches. Nat. Commun. 12, 1846 (2021).
Shaw, W. M. et al. Engineering a model cell for rational tuning of GPCR signaling. Cell 177, 782–796 e727 (2019).
Aranda-Díaz, A., Mace, K., Zuleta, I., Harrigan, P. & El-Samad, H. Robust synthetic circuits for two-dimensional control of gene expression in yeast. ACS Synth. Biol. 6, 545–554 (2017).
Scott, L. H., Mathews, J. C., Flematti, G. R., Filipovska, A. & Rackham, O. An artificial yeast genetic circuit enables deep mutational scanning of an antimicrobial resistance protein. ACS Synth. Biol. 7, 1907–1917 (2018).
Camara, E., Lenitz, I. & Nygard, Y. A CRISPR activation and interference toolkit for industrial Saccharomyces cerevisiae strain KE6-12. Sci. Rep. 10, 14605 (2020).
Ding, C. et al. Proteome-wide profiling of activated transcription factors with a concatenated tandem array of transcription factor response elements. Proc. Natl Acad. Sci. USA 110, 6771–6776 (2013).
Peng, B., Williams, T., Henry, M., Nielsen, L. & Vickers, C. Controlling heterologous gene expression in yeast cell factories on different carbon substrates and across the diauxic shift: a comparison of yeast promoter activities. Microb. Cell Factories 14, 91 (2015).
Ravarani, C. N. et al. High-throughput discovery of functional disordered regions: investigation of transactivation domains. Mol. Syst. Biol. 14, e8190 (2018).
Saleh, M. M., Jeronimo, C., Robert, F. & Zentner, G. E. Connection of core and tail Mediator modules restrains transcription from TFIID-dependent promoters. PLoS Genet. 17, e1009529–e1009529 (2021).
Young, E. T. et al. Artificial recruitment of mediator by the DNA-binding domain of Adr1 overcomes glucose repression of ADH2 expression. Mol. Cell. Biol. 28, 2509–2516 (2008).
Ray, M., Tang, R., Jiang, Z. W. & Rotello, V. M. Quantitative tracking of protein trafficking to the nucleus using cytosolic protein delivery by nanoparticle-stabilized nanocapsules. Bioconjugate Chem. 26, 1004–1007 (2015).
Sikorski, R. S. & Hieter, P. A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics 122, 19–27 (1989).
Majmudar, C. Y., Lum, J. K., Prasov, L. & Mapp, A. K. Functional specificity of artificial transcriptional activators. Chem. Biol. 12, 313–321 (2005).
Bhakta, M. S. & Segal, D. J. The generation of zinc finger proteins by modular assembly. Methods Mol. Biol. 649, 3–30 (2010).
Gaj, T., Gersbach, C. A. & Barbas, C. F. 3rd ZFN, TALEN, and CRISPR/Cas-based methods for genome engineering. Trends Biotechnol. 31, 397–405 (2013).
Monteiro, P. T. et al. YEASTRACT+: a portal for cross-species comparative genomics of transcription regulation in yeasts. Nucleic Acids Res. 48, D642–D649 (2020).
Sanchez, A., Osborne, M. L., Friedman, L. J., Kondev, J. & Gelles, J. Mechanism of transcriptional repression at a bacterial promoter by analysis of single molecules. EMBO J. 30, 3940–3946 (2011).
Belliveau, N. M. et al. Systematic approach for dissecting the molecular mechanisms of transcriptional regulation in bacteria. Proc. Natl Acad. Sci. USA 115, E4796–E4805 (2018).
Kingston, R. E., Bunker, C. A. & Imbalzano, A. N. Repression and activation by multiprotein complexes that alter chromatin structure. Genes Dev. 10, 905–920 (1996).
Hahn, S. & Young, E. T. Transcriptional regulation in Saccharomyces cerevisiae: transcription factor regulation and function, mechanisms of initiation, and roles of activators and coactivators. Genetics 189, 705–736 (2011).
Wang, H. M. & Stillman, D. J. Transcriptional repression in Saccharomyces cerevisiae by a Sin3-Lexa fusion protein. Mol. Cell. Biol. 13, 1805–1814 (1993).
Wong, K. H. & Struhl, K. The Cyc8-Tup1 complex inhibits transcription primarily by masking the activation domain of the recruiting protein. Genes Dev. 25, 2525–2539 (2011).
Ostling, J., Carlberg, M. & Ronne, H. Functional domains in the Mig1 repressor. Mol. Cell. Biol. 16, 753–761 (1996).
DeVit, M. J., Waddle, J. A. & Johnston, M. Regulated nuclear translocation of the Mig1 glucose repressor. Mol. Biol. Cell 8, 1603–1618 (1997).
Dani, G. M. & Zakian, V. A. Mitotic and meiotic stability of linear plasmids in yeast. Proc. Natl Acad. Sci. USA 80, 3406–3410 (1983).
Caspeta, L. et al. Altered sterol composition renders yeast thermotolerant. Science 346, 75–78 (2014).
Dohmen, R. J., Wu, P. & Varshavsky, A. Heat-inducible degron: a method for constructing temperature-sensitive mutants. Science 263, 1273–1276 (1994).
Costa, D. A. et al. Physiological characterization of thermotolerant yeast for cellulosic ethanol production. Appl. Microbiol. Biotechnol. 98, 3829–3840 (2014).
Peng, B., Nielsen, L. K., Kampranis, S. C. & Vickers, C. E. Engineered protein degradation of farnesyl pyrophosphate synthase is an effective regulatory mechanism to increase monoterpene production in Saccharomyces cerevisiae. Metab. Eng. 47, 83–93 (2018).
Peng, B. et al. A squalene synthase protein degradation method for improved sesquiterpene production in Saccharomyces cerevisiae. Metab. Eng. 39, 209–219 (2017).
Stephanopoulos, G. Synthetic biology and metabolic engineering. ACS Synth. Biol. 1, 514–525 (2012).
Nielsen, J. & Keasling, J. D. Synergies between synthetic biology and metabolic engineering. Nat. Biotechnol. 29, 693–695 (2011).
Lubliner, S. et al. Core promoter sequence in yeast is a major determinant of expression level. Genome Res. 25, 1008–1017 (2015).
Szczepek, M. et al. Structure-based redesign of the dimerization interface reduces the toxicity of zinc-finger nucleases. Nat. Biotechnol. 25, 786–793 (2007).
Eeckhoute, J., Metivier, R. & Salbert, G. Defining specificity of transcription factor regulatory activities. J. Cell Sci. 122, 4027–4034 (2009).
Holowko, M. B., Frow, E. K., Reid, J. C., Rourke, M. & Vickers, C. E. Building a biofoundry. Synth. Biol. 6, ysaa026 (2021).
Hillson, N. et al. Building a global alliance of biofoundries. Nat. Commun. 10, 2040 (2019).
Kotopka, B. J. & Smolke, C. D. Model-driven generation of artificial yeast promoters. Nat. Commun. 11, 2113 (2020).
Gietz, R. D. & Schiestl, R. H. Large-scale high-efficiency yeast transformation using the LiAc/SS carrier DNA/PEG method. Nat. Protoc. 2, 38–41 (2007).
Shi, Z. & Vickers, C. E. Molecular Cloning Designer Simulator (MCDS): all-in-one molecular cloning and genetic engineering design, simulation and management software for complex synthetic biology and metabolic engineering projects. Metab. Eng. Commun. 3, 173–186 (2016).
Acknowledgements
BP and this research were supported by a CSIRO Synthetic Biology Future Science Fellowship and the University of Queensland. BP and CEV acknowledge current support from ARC Centre of Excellence in Synthetic Biology and Queensland University of Technology. Metabolite analysis was performed by Dr Manual Plan at Metabolomics Australia Queensland Node. Metabolomics Australia is supported by BioPlatforms Australia through the Commonwealth government’s National Collaborative Research Infrastructure Strategy (NCRIS). Yeast strains in this study derive from CEN.PK background strains, which were provided by EUROSCARF (Scientific Research and Development GmbH, Germany) under a non-commercial licence.
Author information
Authors and Affiliations
Contributions
B.P., C.E.V. and G.D. contributed to early-stage conception of the project. C.S., M.T. and C.B.H. provided advisory opinions on the project. C.E.V., G.D. and M.T. supported and coordinated the running of the project. B.P. performed the experiments. N.C.B. and Z.L. participated in strain construction and characterisation. B.P. prepared the draft of the manuscript. C.E.V. revised manuscript. G.D., C.S., M.T. and C.B.H. participated in manuscript revision. All authors contributed to result analysis and discussion.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Biology thanks the anonymous reviewers for their contribution to the peer review of this work. Primary Handling Editor: Anam Akhtar. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Peng, B., Bandari, N.C., Lu, Z. et al. Engineering eukaryote-like regulatory circuits to expand artificial control mechanisms for metabolic engineering in Saccharomyces cerevisiae. Commun Biol 5, 135 (2022). https://doi.org/10.1038/s42003-022-03070-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42003-022-03070-z
- Springer Nature Limited
This article is cited by
-
An in vivo gene amplification system for high level expression in Saccharomyces cerevisiae
Nature Communications (2022)