
Abstract
Background
Assembly of heterologous metabolic pathways is commonly required to generate microbial cell factories for industrial production of both commodity chemicals (including biofuels) and high-value chemicals. Promoter-mediated transcriptional regulation coordinates the expression of the individual components of these heterologous pathways. Expression patterns vary during culture as conditions change, and this can influence yeast physiology and productivity in both positive and negative ways. Well-characterized strategies are required for matching transcriptional regulation with desired output across changing culture conditions.
Results
Here, constitutive and inducible regulatory mechanisms were examined to optimize synthetic isoprenoid metabolic pathway modules for production of trans-nerolidol, an acyclic sesquiterpene alcohol, in yeast. The choice of regulatory system significantly affected physiological features (growth and productivity) over batch cultivation. Use of constitutive promoters resulted in poor growth during the exponential phase. Delaying expression of the assembled metabolic modules using the copper-inducible CUP1 promoter resulted in a 1.6-fold increase in the exponential-phase growth rate and a twofold increase in productivity in the post-exponential phase. However, repeated use of the CUP1 promoter in multiple expression cassettes resulted in genetic instability. A diauxie-inducible expression system, based on an engineered GAL regulatory circuit and a set of four different GAL promoters, was characterized and employed to assemble nerolidol synthetic metabolic modules. Nerolidol production was further improved by 60% to 392 mg L−1 using this approach. Various carbon source systems were investigated in batch/fed-batch cultivation to regulate induction through the GAL system; final nerolidol titres of 4–5.5 g L−1 were achieved, depending on the conditions.
Conclusion
Direct comparison of different transcriptional regulatory mechanisms clearly demonstrated that coupling the output strength to the fermentation stage is important to optimize the growth fitness and overall productivities of engineered cells in industrially relevant processes. Applying different well-characterized promoters with the same induction behaviour mitigates against the risks of homologous sequence-mediated genetic instability. Using these approaches, we significantly improved sesquiterpene production in yeast.
Similar content being viewed by others

Background
Metabolic engineering and synthetic biology are now routinely used for the engineering of microorganisms for industrial production of desirable chemicals, including fuels and biochemicals [1–3]. In the first instance, metabolic pathway flux towards the desired product is optimized by introduction of enzymes with the best catalytic efficiency (which are often heterologous) [4–6]. Expression levels of these enzymes are then titrated for optimal pathway balance, in combination with other metabolic engineering strategies to redirect carbon in the metabolic network [7–9]. Complicating matters, the activities of synthetic pathways should be coordinated with the dynamic fermentation conditions and process stage [10]. Imbalance in pathway flux can have a negative effect on cell physiology (e.g. growth rate) and on product titre [11]. Coordination can be controlled at the transcriptional level to regulate gene expression (and hence, enzyme activity). However, there is only limited information available on the dynamic behaviour of promoters across the range of conditions that occur in industrial fermentation processes.
The budding yeast Saccharomyces cerevisiae is a common engineering platform for production of high-value plant terpenoids. Terpenoids are a diverse class of chemicals naturally synthesized from the universal 5-carbon precursors, isopentenyl pyrophosphate (IPP) and dimethylallyl pyrophosphate (DMAPP) [12]. The sesquiterpene sub-class of terpenes have 15 carbon atoms and are synthesized from farnesyl pyrophosphate (FPP), which is condensed from one molecule of DMAPP and two molecules of IPP. Sesquiterpenes have broad industrial applications, including as fragrances, flavours, pharmaceuticals, solvents and fuels. A generic set of metabolic engineering approaches can be used to improve sesquiterpene production (Fig. 1) [13–16]. First, high-level production of terpenoids requires improved flux from central carbon metabolism to the sesquiterpene precursor FPP [16–23]. This is typically achieved by augmenting the mevalonate (MVA) pathway through overexpression or heterologous expression of individual genes (including farnesyl pyrophosphate synthase, FPPS). Enhanced MVA pathway activity causes squalene accumulation [23–25] and it is necessary to constrain the flux-competing squalene synthase to redirect FPP flux away from sterol production and towards sesquiterpene production (Fig. 1). This can be achieved by decreasing activity of the FPP-consuming enzyme squalene synthase, either through engineered protein degradation [23] or transcriptional down-regulation [16, 26, 27]. These steps provide the basic principles of pathway optimization for sesquiterpene production in yeast.

Metabolic pathways for trans-nerolidol (sesquiterpene) production in yeast. ACS2 acetyl-CoA synthase, EfmvaE Enterococcus faecalis acetoacetyl-CoA thiolase/HMG-CoA reductase, EfmvaS E. faecalis HMG-CoA synthase, HMG2 HMG-CoA reductase 2, ERG12 mevalonate kinase, ERG8 phosphomevalonate kinase, MVD1 mevalonate pyrophosphate decarboxylase, IDI1 isopentenyl diphosphate:dimethylallyl diphosphate isomerase, ERG20 farnesyl pyrophosphate synthetase, AcNES1 Actinidia chinensis tran-nerolidol synthase, ERG9 squalene synthase, IPP isopentenyl pyrophosphate, DMAPP dimethylallyl pyrophosphate, FPP farnesyl pyrophosphate. Dashed arrow means multiple biochemical steps
An ideal microbial cell factory should simultaneously exhibit high specific production rate and high specific growth rate in a batch cultivation [28]. However, these two objectives are commonly incompatible due to the metabolic burden and/or metabolic imbalance found in the presence of engineered pathways [29–31]. An alternative option is to separate growth and production phases [31]. This can be achieved by induction of synthetic pathway genes upon an environmental stimulus occurring after sufficient biomass is accumulated [32].
Regulation of gene expression across a batch cultivation (expression pattern) is delicately controlled by the gene promoter, transcriptional regulatory networks and the environmental inputs (usually the cultivation conditions, including activator/repressor concentration in cultures) [33–36]. Native promoters and regulatory networks can readily be used for metabolic pathway construction and optimization if their response conditions are well characterized. Several promoters have been well characterized to achieve inducible gene expression systems in yeast; these include a copper-inducible promoter (P CUP1 , induced by high concentration of copper ion), galactose-inducible promoters (the bi-directional P GAL1 and P GAL10 promoters, de-repressed in the absence of glucose and induced when galactose is present), a sucrose-inducible promoter (P SUC1 , de-repressed in the absence of glucose and induced when sucrose is present), a high-affinity hexose transporter promoter (P HXT7 , induced when glucose levels are low) and heat shock transcriptional factor Hsf1p-mediated promoters (P SSA1 and P HSP26 ) [33, 36–42]. In addition, sophisticated synthetic regulatory circuits have been designed, including circuits that respond to cell density via an engineered quorum-sensing system [32] and circuits that are activated by product feedback [43]. These promoters and regulatory networks can be further explored for optimizing gene expression regulation in metabolic engineering.
Trans-nerolidol is a sesquiterpene alcohol with applications as fragrance, flavour, precursor for synthetic vitamin E/K1 and others [23]. Previously, we engineered a trans-nerolidol production pathway in yeast in concert with MVA pathway augmentation and a protein-mediated flux down-regulation strategy at squalene synthase [23]. We achieved a titre of ~100 mg L−1, but observed a decreased growth rate when using constitutive overexpression of genes. To attain high-level production of nerolidol without a growth defect, further metabolic engineering is required. In this work, we engineered transcriptional regulation module that responds to bioprocess conditions to optimize growth and production for improved nerolidol titre, in combination with metabolic pathway optimization.
Results
Constitutive expression of genes results in decreased growth rates and constrains product titres
In our previous work, nerolidol production was improved by heterologously expressing more efficient upper MVA pathway genes from Enterococcus faecalis (EfmvaS and EfmvaE), overexpressing the yeast lower MVA pathway genes and FPP synthase and destabilizing squalene synthase (Erg9p) [23]. The resulting strain (Table 1) produced 104 ± 35 mg L−1 nerolidol over 72 h in batch cultivation on minimal medium with 20 g L−1 glucose. All of the genes, including nerolidol synthase (AcNES1) were overexpressed from plasmids using promoters with constitutive activity (P RPL4A for EfmvaS, P RPL15A for EfmvaE, P RPL8B for ERG12, P SSB1 for ERG8, P RPL3 for MVD1, P YEF3 for IDI1, P TEF2 for ERG20 and P TEF1 for AcNES1; Tables 1 and 2). These promoters exhibit high-level activities in the exponential phase when glucose is available, but dramatically decreased activities when glucose is depleted and cells have shifted to the ethanol growth phase [33]. Strain N6D exhibited half the specific growth rate (μmax of 0.16 ± 0.03 h−1) of the wild-type CEN.PK reference strain (μmax of 0.30 ± 0.01 h−1) [23]. We presume that this is due to the metabolic burden from high expression levels of the heterologous synthetic pathway in the exponential phase. Furthermore, the decreased expression level after the diauxic shift might lead to low productivities in the post-exponential phase. To examine productivity across the fermentation period, 24- and 72-h samples were re-analysed using a new HPLC–UV method. Consistent with the GC–MS data obtained previously [23], a titre of 125 ± 30 mg L−1 was measured at 72 h. At 24 h, the titre was 82 ± 37 mg L−1 (Fig. 2b) and the productivity was calculated to 5.7 mg g−1 biomass h−1 in the exponential phase (from 0 to 24 h) compared to only 0.18 mg g−1 biomass h−1 in the post-exponential phase (from 24 to 72 h).

Characterizing strains N6D (with “constitutive” gene overexpression constructs) [23] and NC1D (with copper-inducible gene overexpression constructs): a the copper-inducible expression cassettes in plasmids pPMVAugw, pPMVAd36 and pJT3; b nerolidol titre at 24 and 72 h; c growth curves over batch cultivation; d mRNA levels in strain N6D in the exponential phase (EXP) and the ethanol phase (ETH, at 48 h); e mRNA levels in the strain NC1D in the pre-culture (without adding copper; un-induced) and in two-phase cultivation (with copper added; induced; at 10 or 36 h). Two-phase flask cultivation on 20 g L−1 glucose was employed; for the cultivation of NC1D, 100, 100 and 200 μM (final concentration) copper sulphate was added sequentially at 5, 10 and 24 h. mRNA levels were measured by quantitative real-time PCR. Mean values ± standard deviations are shown (N ≥ 3)
The decreased nerolidol production rate may be attributed to the lower promoter activity we previously reported for these constitutive promoters during the ethanol phase [33]. Indeed, the mRNA levels of the genes for nerolidol production (EfmvaS, EfmvaE, ERG12, ERG8, MVD1, IDI1, ERG20 and AcNES1) decreased 3- to 10-fold in the ethanol growth phase compared to the exponential growth phase (Fig. 2d). As noted previously [23], the mRNA levels of the reference genes ACT1 and PDC1 also decreased. Interestingly, in contrast to the classical ‘housekeeping’ genes ACT1 and PDC1, acetyl-CoA synthase (ACS2) exhibited similar transcriptional levels in the exponential/ethanol growth phases.
Copper-inducible expression improves nerolidol production
To avoid both the metabolic imbalances causing decreased growth rate in the exponential phase and the decreased productivities in the post-exponential phase, gene expression can be controlled using an induction system. The CUP1 promoter is responsive to copper ion concentration [38]. In the absence of additional copper, the CUP1 promoter exhibits a moderate expression level; with addition of high-concentration copper (above 100 μM), activity is induced to a level comparable to the strong TEF1 promoter in exponential phase [33]. In contrast to the TEF1 promoter, the CUP1 promoter can maintain high expression activity in the ethanol phase (in the presence of 300 μM copper; [33]). These characteristics make the CUP1 promoter an potentially useful candidate to address both of the aforementioned problems. To test the effect on sesquiterpene production, a new set of plasmids were constructed (Table 2): an E. faecalis upper MVA pathway plasmid (pPMVAgw) with EfmvaS and EfmvaE controlled by two divergent CUP1 promoters; a yeast lower MVA pathway plasmid (pPMVAd36) with the mevalonate kinase gene ERG12 controlled by the CUP1 promoter and the other three genes (ERG8-phosphomevalonate kinase, MVD1-mevalonate pyrophosphate decarboxylase and IDI1-Isopentenyl diphosphate:dimethylallyl diphosphate isomerase) controlled by a TEF1 promoter and two TEF2 promoters, respectively; and finally, a nerolidol synthetic plasmid (pJT3) with ERG20 and AcNES1 controlled by two divergent CUP1 promoters (Fig. 2a). The three plasmids were co-transformed into the Erg9p-destabilized strain oH5 [23] to generate strain NC1D (Table 1).
While the wild-type growth rate (μmax = 0.30 ± 0.01 h−1) was not fully recovered, strain NC1D exhibited a 1.6-fold faster growth rate (μmax = 0.25 ± 0.01 h−1) than strain N6D (Table 3), demonstrating that the metabolic imbalance in the exponential phase was partially relieved. Furthermore, higher nerolidol production was achieved, with a titre of 111 ± 26 mg L−1 at 24 h and 245 ± 8 mg L−1 at 72 h (Fig. 2b, c). This equates to production rates of 5.1 mg g−1 biomass h−1 in the period from 0 to 24 h, slightly lower than the rate in strain N6D; and 0.77 mg g−1 biomass h−1 in the post-exponential phase (from 24 to 72 h), fourfold higher than the rate in strain N6D (Table 3).
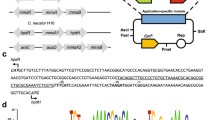
To verify the relationship between gene expression levels and nerolidol production, the mRNA levels in strain NC1D were also analysed. Unexpectedly, the mRNA levels for four genes from the ‘lower’ MVA pathway (ERG12, ERG8, MVD1 and IDI1; Fig. 2) were similar compared to those in the wild-type CEN.PK reference strain [23]. Furthermore, the overexpression cassettes could not be amplified from the genomic DNA (data not shown). The 2 μ plasmid originally containing the yeast lower MVA pathway was recovered from NC1D. Restriction mapping and DNA sequencing showed that DNA recombination occurred between pPMVAd36 and pJT3; the result of this recombination was a plasmid where the four lower MVA pathway gene expression cassettes from pPMVAd36 were replaced by the nerolidol synthase cassette from pJT3 (Additional file 1: Figure S1). This replacement pattern is consistent with two homologous recombination events occurring, one between the CUP1 promoters for ERG12 and AcNES1, and one between homologous sequences on the plasmid backbones. Despite this undesiriable recombination event, strain NC1D performed better than N6D (see above). Very low variability was observed between biological replicates for the mRNA levels (Fig. 2e) and nerolidol production (Fig. 2b), demonstrating that the plasmid recombination and resulting strain were stable.
Despite the loss of the four lower MVA pathway genes, the upper pathway genes EfmvaS, EfmvaE and ERG20 and AcNES1 controlled by CUP1 promoters, were still present in NC1D. Consistent with our previous findings [33], the mRNA levels for these genes increased by about fourfold after copper induction, and high transcript levels were maintained in the ethanol growth phase for these genes (Fig. 2e). In the exponential phase under un-induced conditions, mRNA levels for EfmvaS and EfmvaE were 2 and sevenfold lower (Fig. 2e) than transcript levels driven by constitutive promoters (Fig. 2d). The ‘lower’ pathway was not augmented at all, considering the loss of the ‘lower’ MVA pathway genes during the recombination event. Together, these data support the idea that high transcription of MVA pathway genes during the exponential phase (driven by constitutive promoters) results in a metabolic imbalance that decreases growth rate and nerolidol titre. Applying the CUP1 promoter to control synthetic pathway genes improved nerolidol production; however, plasmid recombination caused by repeated usage of the CUP1 promoter is undesirable in a metabolic engineering context. Therefore, an alternative inducible expression system was developed.
Δgal80 enables auto-induction of GAL promoters over/after diauxic shift
In a previous comparison of a set of commonly used promoters, the GAL1 promoter drove the highest expression level [33]. The GAL1 promoter and galactose-based cultivation have also been used in the initial laboratory development of several sesquiterpene-producing strains [46, 47]. However, it is not feasible to use galactose as a carbon source in industrial production due to its high cost. In the core galactose regulon, Gal80p binds the transcription activator Gal4p to inhibit Gal4p-mediated transcription initiation of the GAL1 promoter in the absence of galactose; in the presence of galactose, transcriptional factor Gal3p binds with Gal80p to relieve Gal80p repression on Gal4p transcription activation (Fig. 3a) [48]. Galactose-independent (gratuitous) activation of the GAL1 promoter can be achieved by disruption of the gal80 repressor [49]. Additionally, galactose-inducible expression driven by GAL1 promoter is co-regulated by Mig1p-mediated glucose-dependent repression: in the presence of glucose, Mig1p can bind to the GAL1, GAL3 and GAL4 promoters to inhibit gene transcription (Fig. 3a) [48, 50]. Mig1p-mediated repression can lead to the glucose-dependent repression of the GAL1 promoter in a gal80Δ strain [16, 49, 51]. Consequently, it has been shown that the GAL1 promoter can be automatically induced in a gal80Δ strain as the cells shift to the ethanol growth phase [51].

Schematic representing regulatory networks for the galactose-inducible promoter (P GAL1 ; a) and GFP fluorescence levels in gal80Δ background strains (b) with the yEGFP gene driven by P TEF1 (strain G89J3), P GAL1 (strain GB5J3), P GAL10 (strain GB6J3), P GAL2 (strain GQ3J3) and P GAL7 (strain GQ4J3). Cultures were grown on 20 g L−1 glucose. GFP fluorescence is expressed as percentage of exponential-phase auto-fluorescence of the reference strain (GH4J3). The growth curve (OD600) for GH4J3 is shown. Mean values ± standard deviations are shown (N = 2)
In the current study, to avoid repeated use of a single promoter for multiple genes, the promoters from the galactose metabolic genes (GAL1, GAL10, GAL2 and GAL7) and a reference TEF1 promoter were characterized in a gal80Δ strain (Tables 1, 2; Fig. 3b). GFP was used as a reporter to measure the promoter activities over an entire batch cultivation on 20 g L−1 glucose. Growth profiles were similar and showed a diauxic growth pattern in all strains (data for the gal80Δ control strain are shown in Fig. 3b; other data not shown). We showed previously that, under the same conditions, the diauxic shift pattern observed in the growth curve was coincident with glucose depletion and the start of ethanol consumption, and occurred at ~12 h [33]. Similar to our previous study in the wild-type strain [33], activity driven by the TEF promoter in the gal80Δ strain decreased dramatically during and after the diauxic shift. In contrast, the GAL promoters exhibited diauxie-inducible expression patterns: their activities started to increase at 12 h, peaking at 48 h. The strength of GAL promoters in the post-exponential phase was as follows: P GAL2 >P GAL1 >P GAL7 >P GAL10 . The auto-inducible expression pattern makes the GAL promoter in combination with gal80Δ very useful in strain development to simultaneously avoid the metabolic burden in the exponential phase and increase productivity in the ethanol growth phase.
Auto-inducible GAL promoter regulation drives efficient nerolidol production
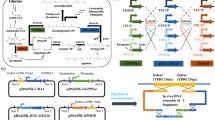
To apply the modified galactose-inducible system (GAL promoters in gal80Δ background), a new nerolidol-producing strain, N391DA, was constructed (Fig. 4a; Tables 1, 2). In the “upper” MVA module, genes EfmvaS (HMG-CoA synthase), ACS2 (native acetyl-CoA synthase) and EfmvaE (thiolase/HMG-CoA reductase) were controlled by P GAL1 , P GAL10 and P GAL2 , respectively. Overexpressing ACS2 has previously been shown to increase the intracellular concentration of acetyl-CoA [21], which is the precursor metabolite for the MVA pathway; hence, it was included in the ‘upper’ module. The three expression cassettes were integrated into the HMG2 (HMG-CoA reductase) promoter locus with the selection marker HIS3. At the same time, a P GAL7 promoter linked to a short sequence of the HMG2 gene including a K6R mutation that stabilizes Hmg2p from degradation [52, 53] was introduced as a fusion with the native HMG2. As a result, HMG2 K6R was expressed from the genome under the control of P GAL7 . To construct a “lower” MVA module, ERG12 (mevalonate kinase) was controlled by P GAL2 ; the other three genes (ERG8, MVD1, IDI1) were controlled by glucose-dependent “constitutive” promoters (P TEF2 , P ADH1 and P TEF1 , respectively). This construct was integrated with the TRP1 selection marker into the PDC5 locus, which encodes a weakly expressed pyruvate decarboxylase [54]. Dysfunction of PDC5 does not cause a major change in yeast metabolism, because of complementation by isoforms PDC1 and PDC6 [55]. For the nerolidol synthesis module, P GAL1 -controlled ERG20 and P GAL2 -controlled AcNES1 were introduced on a 2μ plasmid. Finally, squalene synthase (Erg9p) was destabilized by the addition of an endoplasmic-reticulum mediated protein degradation sequence to reduce its ability to compete for FPP with nerolidol synthase [23], and gal80 was disrupted to allow diauxic induction of GAL promoters. The resulting strain N391DA was evaluated through two-phase flask cultivation.

Characterizing strain N391DA with gal80Δ-GAL promoter constructs: a genetic modules/plasmid for the “upper” mevalonate pathway (pIMVAu1), the “lower” mevalonate pathway (pIMVAd39T) and the nerolidol synthetic genes (pJT9R); b, d metabolic and growth profiles (N = 2); c mRNA levels (N = 3) in the exponential phase (EXP) and the ethanol growth phase (ETH, at 36 h). Two-phase flask cultivation on 20 g L−1 glucose was employed. Mean values ± standard deviations are shown
Strain N391DA exhibited a normal exponential growth with μmax of 0.27 ± 0.01 h−1—1.7-fold faster than the strain N6D and slightly faster than strain NC1D. Consistent with the yeast diauxic growth model, N391DA consumed glucose and produced ethanol in the exponential growth phase, and ethanol was subsequently consumed in the secondary growth phase (Fig. 4b). N391DA produced 38 mg L−1 nerolidol at 24 h, production rates of 1.4 mg g−1 biomass h−1 in the period from 0 to 24 h, fourfold lower than the rate in strain N6D. Nerolidol reached 392 ± 2 mg L−1 at 72 h, which translates to a production rate of 2.1 mg g−1 biomass h−1 in the post-exponential phase (from 24 to 72 h)—12-fold higher than the rate in strain N6D (Table 3). The 72 h titre represents a 60% improvement relative to NC1D with copper-inducible constructs. N391DA exhibited the highest specific nerolidol production rate (r nerolidol ) between 24 and 48 h and 86% of nerolidol was produced after 24 h (Fig. 4d).
To verify the expression pattern of the genes controlled by the modified galactose-inducible system, the mRNA levels were analysed in strain N391DA (Fig. 4c). In the exponential phase, the expression levels of the reference genes ACT1 and PDA1 in N391DA were similar (two-tailed t test p > 0.1) to those in strains N6D and NC1D. As observed previously, their mRNA levels decreased significantly in the ethanol growth phase; this was also the case for mRNAs levels from the genes controlled by TEF1, TEF2 and ADH1 promoters. In contrast, the genes controlled by GAL promoters exhibited increased transcriptional levels: twofold for ACS2, 11-fold for EfmvaS, 22-fold for EfmvaE, 27-fold for HMG2, ninefold for ERG12, 26-fold for ERG20 and fourfold for AcNES1 in the ethanol growth phase, compared to in the exponential growth phase. These fold-changes are consistent with the expression pattern of the four GAL promoters as characterized above (Fig. 3).
Sucrose as an alternative carbon source for nerolidol production
Sucrose from sugar cane/sugar beet is an alternative carbon source to glucose in industrial fermentation [56–58]. As for the GAL genes, the invertase gene for sucrose utilization (SUC2) is under Mig1p-mediated glucose repression, which is relieved when yeast is cultivated on sucrose [59, 60]. Considering this, it is reasonable to expect that the expression output from GAL promoter in gal80Δ background strain might be different on sucrose than on glucose. To investigate the effect of sucrose on nerolidol production, GAL promoter activities in gal80Δ background strains and nerolidol production for strain N391DA were characterized on sucrose. Yeast strains were pre-cultured on 40 g L−1 glucose, which minimized GAL promoter activities in gal80Δ strains (data not shown). A 6-h lag phase was exhibited after transferring to 20 g L−1 sucrose medium; this lag phase was seen in both gal80Δ strains and the GAL80 control strain (Fig. 5a; Additional file 1: Figure S2). During the lag phase on sucrose, glucose repression on GAL promoters was relieved. GFP expression driven by the GAL1 and GAL2 promoters increased sharply during the lag phase and plateaued during exponential growth (Fig. 5a); this expression level was similar to that observed in the ethanol growth phase of glucose batch cultivation (Fig. 3b). A further increase of ~twofold was observed after 24 h, presumably after the diauxic shift (Fig. 5a).

Effects of sucrose on GAL promoter activities in gal80Δ background and nerolidol production for strain N391DA: a the fluorescence levels of the yEGFP controlled by GAL1 (strain GB5J3) or GAL2 (strain GQ3J3) promoter over the batch cultivation on 20 g L−1 sucrose and the growth profile (OD600) of strain GH4J3; b, c metabolic and growth profile for strain N391DA in two-phase flask cultivation on 20 g L−1 sucrose. Vertical dashed lines indicate the end of lag phase. Mean values ± standard deviations are shown (N = 2)
To characterize nerolidol production on sucrose, strain N391DA was first pre-cultured on 40 g L−1 glucose and then cultivated on 20 g L−1 sucrose. N391DA exhibited a 48-h lag phase on sucrose (Fig. 5b), dramatically longer than that for the GFP strains (Fig. 5a). The sugar profile demonstrated that strain N391DA first fermented sucrose and its hydrolysate products (glucose and fructose) into ethanol; the diauxic shift occurred by 72 h, and the strain then re-used ethanol in the post-exponential phase (Fig. 5b). In sucrose batch cultivation, the final nerolidol titre was 632 ± 57 mg L−1, 1.6-fold higher than in glucose batch cultivations. In addition, N391DA exhibited the highest post-exponential-specific nerolidol production rate of 5 mg g−1 biomass h−1 (Fig. 5c compared to Fig. 4d).
Nerolidol production in fed-batch cultivation
To achieve high-titre nerolidol production for strain N391DA, fed-batch strategies were explored. We first explored a strategy designed to ensure that (a) fermentable sugars are catabolized through respiratory metabolism and (b) cultures are maintained under aerobic conditions. The initial feed rate was set to 1 mM glucose g−1 biomass h−1 with 600 g L−1 glucose feeding medium and then exponentially increased with a rate of 0.05 h−1; the feeding was switched off when dissolved oxygen (DO) was below 25% and maximum agitation and gassing were achieved, and the feeding was re-triggered when DO was above 30% (Additional file 1: Figure S3a). Two additional experiments were performed using volumetrically similar initial feed rates and feed solutions of 600 g L−1 sucrose and 400 g L−1 glucose/158 g L−1 ethanol, respectively.
In the three experiments, the respiration quotients fluctuated around 1 for glucose or sucrose feeding processes, and around 0.9 for glucose/ethanol feeding process (Additional file 1: Figure S4), demonstrating that the fermentable sugars were catabolized through respiration. All processes began in batch mode using 20 g L−1 glucose as a carbon source and proceeded through diauxie and into the ethanol growth phase until DO started in increase sharply, triggering the feed. In this batch period, the three cultures produced 406 ± 57 mg L−1 nerolidol at 30 h (Fig. 6a). In the subsequent feeding phase, the glucose/ethanol feed provided higher nerolidol production than glucose or sucrose feeding; >2 g L−1 nerolidol was achieved at 102 h for glucose/ethanol and this titre was not achieved until 150 h for glucose and sucrose feeding. The final titre for the glucose/ethanol feed was >3 g L−1 at 174 h. For all three fed-batch experiments, the specific nerolidol production rates during feeding were lower than the rate observed in the ethanol growth phase in the batch process (Fig. 6a). The C-mole yield at 102 h was 2.0 ± 0.4% in these three fed-batches.

Nerolidol production for strain N391DA in fed-batch cultivations: a nerolidol production (solid line) and specific production rate (r; dashed line) in carbon-source-restricted DO-triggered fed-batch cultivation with feeding carbon source of 600 g L−1 glucose (R-Glc), or 600 g L−1 sucrose (R-Suc), or 400 g L−1 glucose 158 g L−1 ethanol mixture (R-Glc/Eth); b nerolidol production (solid line) and specific production rate (r; dashed line) in carbon-source-overflowed/carbon-source-pulsing fed-batch cultivation with feeding carbon source of 600 g L−1 glucose (with 10 g L−1 glucose pulse, O10-Glc; with 20 g L−1 glucose pulse, O20-Glc) or 600 g L−1 sucrose (with 20 g L−1 sucrose pulse, O20-Suc). Vertical dashed line approximately indicated the start of feeding. Growth and process values refer to Additional file 1: Figures S4, S5. N = 1
Next, the feeding strategy was altered with the aim to maintain overflow metabolism and cycling between ethanol production and consumption (Additional file 1: Figure S3b). After the batch phase, exponential feeding with an initial feeding rate of 3 mM glucose g−1 biomass h−1 for 600 g L−1 glucose (or volumetrically the same for 600 g L−1 sucrose) feeding medium and a specific increasing rate of 0.05 h−1 were applied. Once 50 g L−1 sugar had been fed, the feeding was paused to allow cells to consume the ethanol produced during sugar fermentation. Subsequently, 10 or 20 g L−1 sugar pulse feeding was repeatedly triggered by sharp DO increases (Additional file 1: Figure S5). The production of ethanol was confirmed by the respiration quotient being over 2 when the sugar was fed (Additional file 1: Figure S5). In the batch phase for the three batches, 404 ± 11 mg L−1 nerolidol was produced (Fig. 6b), consistent with the above results (Fig. 6a). In these three fed-batch processes (Fig. 6b), >4 g L−1 nerolidol was produced at 96 h, and the specific nerolidol production rates in the early feeding phase (t < 96 h) were noticeably higher than those in the carbon-restricted processes (Fig. 6a). At 96 h, the C-mole yield was 3.8 ± 0.1% in the two glucose-overflow fed-batches and 4.5% in the sucrose-overflowed fed-batch (Fig. 6b).
Discussion
An efficient sesquiterpene-producing yeast platform is of broad industrial interest, because it can be applied for the production of various high-value sesquiterpenes as well as FPP-derived biofuels by simply substituting a specific terpenoid production pathway (Fig. 1). There are two key principles for increasing FPP availability for sesquiterpene production in yeast: enhancing the MVA pathway to increase precursor supply, and reducing squalene synthase activity to decrease its consumption of FPP [14, 17–19, 23, 26, 27, 51, 61]. This study aimed to couple the transcriptional regulation pattern of heterologously expressed pathway genes to bioprocess conditions to minimize metabolic imbalance and optimize heterologous sesquiterpene production.
In previous studies, constitutive promoters were most often used to control the expression of heterologous genes, whereas the optimization of expression strength over the different fermentation stages has not been well investigated [7, 8]. In the current study, three categories of transcription regulation patterns were applied to assemble synthetic pathways: constitutive, copper-inducible and diauxie-induced. Using reporter gene systems, we showed previously that expression outputs from constitutive promoters (including “classical” translational elongation factor promoters, P TEF1 and P TEF2 ; glycolytic promoter, P ADH1 ; ribosome biogenesis promoters, P RPL3 , P RPL15A ) dramatically decrease after the diauxic shift [33]. Our transcription data (Fig. 2d) and nerolidol production data (Fig. 2b) confirmed this. Nerolidol is not toxic and does not cause dramatic growth inhibition (Additional file 1: Figure S6), in contrast to monoterpenes [62]. But we also observed a decreased growth rate (Fig. 2c), which was consistent with metabolic burden from high-level expression in the exponential phase.
Using a copper-inducible promoter (P CUP1 ), gene expression was shifted from exponential to the ethanol growth phase (Fig. 2e). This resulted in improved growth rate during the exponential phase (Fig. 2c) and improved productivities after the diauxic shift (Fig. 2b). However, the repeated use of the CUP1 promoter sequence led to undesirable homologous recombination and loss of introduced DNA fragments (Additional file 1: Figure S2). It is likely that similar losses have occurred previously when repeated promoter sequences have been used, for example, Beekwilder et al. [63] observed that almost half of colonies did not produce a carotenoid reporter product when they used the TDH3 promoter to control several carotogenic pathway genes. Genetic instability at repeat sequences has been documented in yeast [64], and provides a challenge for large-scale modification/introduction of metabolic pathways due to limited availability of appropriate promoters [33].
To address this problem, we characterized four native GAL promoters (P GAL1 , P GAL10 , P GAL2 and P GAL7 ) in a gal80Δ genetic background to engineer a diauxie-inducible system [16, 65], and applied them to assemble the MVA pathway modules (Figs. 3, 4a). While these promoters in gal80Δ background have been used to control the expression of the genes from the MVA pathway and amorphadiene/isoprene synthetic pathway previously [16, 65], they have not been characterized in terms of behaviour throughout the fermentation nor have they been ranked. The relative strength of P GAL1 and P GAL10 has been examined previously with conflicting results. One study [41] observed P GAL10 to be stronger than P GAL1 ; in contrast, our study and two others [39, 66] showed P GAL1 to be stronger than P GAL10 . It is possible that differences in experimental conditions caused this discrepancy. Using the GAL promoters, a further shift in gene expression from exponential to ethanol growth phase was achieved (Figs. 2e, 4c), which resulted in further improved growth during the exponential phase (Fig. 4b) and further improved productivities after the diauxic shift (Figs. 2b, 4d). Increased expression from the GAL promoters and improved nerolidol productivities were consistently observed when the engineered cells were cultivated on sucrose (Fig. 5). Together, these data demonstrate the importance of appropriate transcriptional control modules for optimized strain engineering. There are several genotypic differences between strains NC1D and N391DA, including promoter identity (P CUP1 vs. P GAL ), gene location (plasmid vs. genome integration) and gene expression level; and we have not examined each in detail to determine relative contribution to the phenotype.
In a previous study using the gal80Δ background, the sesquiterpene amorphadiene was produced at >40 g L−1 titre in ethanol fed-batch cultivation, whereas it reached only ~3 g L−1 in glucose fed-batch cultivation [16]. Here, nerolidol production in fed-batch cultivation was also investigated in the gal80Δ background; in contrast to the previous study, two substrate-feeding strategies, sugar-restricted and sugar-surplus, were compared (Fig. 6). In the sugar-restricted feeding processes, sugar was expected to be catabolized through respiratory metabolism; in the sugar-surplus feeding processes, ethanol was expected to accumulate in the sugar respiro-fermentative metabolism after each sugar feed and to be re-consumed when sugar was depleted. The two feeding strategies resulted in similar growth profiles (comparing OD600 when t ≤ 102 or 96 h; Additional file 1: Figures S4, S5). However, in the carbon-overflow fed-batches, higher titre/yield/rate of nerolidol were achieved, compared to the substrate-restricted fed-batches (Fig. 6). In addition to different promoter behaviours throughout the fed-batch cultivation, the improvement in nerolidol production might be due to a MVA pathway response to “overflow” metabolism, as is seen in ethanol/acetate/glycerol production through glycolysis [67–69]. In support of this possibility, the concentrations of intracellular pyruvate, acetyl-CoA, acetoacetyl-CoA and FPP were shown to increase as the dilution rate increased in carbon-limited chemostat processes [70, 71]. The pyruvate node is a key metabolic node for regulation of carbon flux in yeast; specifically, it determines flux towards mitochondrial pyruvate dehydrogenase or the cytosolic pyruvate dehydrogenase bypass (and ultimately, acetyl-CoA, which provides substrate for the MVA pathway) [72, 73]. Moreover, flux distribution into the cytosolic pyruvate dehydrogenase bypass decreases with decreased glucose feeding rate [74]. This can explain the decreased flux through the MVA pathway for nerolidol production under the carbon-limited fed-batch.
Conclusion
Here, we present an expanded modular genetic regulatory system to co-ordinate expression outputs with physiological behaviour of engineered cells under industrially relevant cultivation conditions. The gene transcriptional regulatory pattern for assembled metabolic pathways is critically important for metabolic functionality and optimal productivity in engineered strains. This principle should be considered for de novo assembly of heterogeneous metabolic pathways in the “design-build” construction cycle for industrial microbial producers. The diauxie-induction system, including the four characterized GAL promoters, is simple and efficient for developing a yeast strain for high-level sesquiterpene production. Further optimization to balance sugar and ethanol growth phases might significantly increase titres.
Methods
Plasmid and strain construction
Strains used in this work are listed in Table 1 and plasmids are listed in Table 2. Primers used in polymerase chain reaction (PCR) and details of PCR performed in this work are listed in Additional file 1: Table S1. Plasmid construction processes are listed in Additional file 1: Table S2.
Strain NC1D was generated by co-transforming plasmids pPMVAugw, pPMVAd36 and pJT3 into the Erg9p-destabilized strain oH5 (ERG9-yEGFP-CLN2 PEST) [23]. Strain o391D was generated by sequentially transforming strain CEN.PK2-1C [44] with PmeI-digested pIMVAd39T, SwaI-digested pIMVAu1 and an Erg9p-destabilization fragment (ERG9 C-terminal-CLN2 PEST-T URA3 -loxP-KlURA3-loxP-T ERG9 ; Additional file 1: Table S1#21; [23]). The nerolidol production plasmid pJT9R was then transformed into strain o391D. Finally, gal80 was disrupted by integrating a LoxP-KanMX4-loxP marker (Additional file 1: Table S1#23) to generate strain N391DA. N391DA was selected on SC-glutamate-high-glucose (SCGHG) agar plate with 300 μg mL−1 G-418. The nutrient recipe of the SCGHG agar is as follows: 1.6 g L−1 uracil-drop-out amino acid mixture [75], 1.7 g L−1 yeast nitrogen base (YNB) without ammonium sulphate (Sigma-Aldrich#Y1251), 1 g L−1 glutamate, 200 g L−1 glucose. For strains NC1D and N391DA, at least 3 independent colonies were stored separately in 20% glycerol at −80 °C for subsequent evaluation.
To disrupt gal80, a LoxP-KanMX4-loxP PCR fragment (Additional file 1: Table S1#23) was transformed into CEN.PK113-5D; the resultant strain oJ3 was transformed with SwaI-digested pILGH4, pILG89S, pILGB5AS, pILGB6S, pILGQ3 and pILGQ4 to generate strain GH4J3, GB5J3, GB6J3, GQ3J3 and GQ4J3, respectively.
Two-phase flask cultivation and sampling
Nerolidol-producing strains were evaluated through two-phase flask cultivation using dodecane as a non-toxic organic extractant phase [76]. Synthetic minimal (SM) medium containing 6.7 g L−1 YNB (Sigma-Aldrich #Y0626; pH 6.0) with 20 g L−1 glucose as the carbon source was used (SM-glucose). 100 mM 2-(N-morpholino) ethanesulfonic acid (MES, Sigma-Aldrich#M8250) was used to buffer medium pH and the initial pH was adjusted to 6.0 by adding ammonium hydroxide. Strains were recovered from glycerol stocks by streaking on SM-glucose agar plates (for N6D and NC1D) or SCGHG agar plates (for N391DA) and pre-cultured in MES-buffered SM 20 g L−1 glucose (or 40 g L−1 glucose as indicated) medium to exponential phase (cell density OD600 between 1 and 4). Pre-cultured cells were collected by centrifugation and re-suspend in fresh media before initiating two-phase cultivations. Two-phase flask cultivation was initiated by inoculating pre-cultured cells to OD600 = 0.2 in 25 mL MES-buffered SM 20 g L−1 glucose medium (or 23 mL MES-buffered SM 20 g L−1 sucrose medium when cultures were not sampled for RNA extraction) in 250 mL flasks with solvent-resistant screw-caps [76]; 2 mL dodecane was added to extract nerolidol. Flask cultivation was performed at 30°C and 200 rpm. For all cultivations, about 3 mL culture was sampled before the end of exponential growth phase for OD measurement; meanwhile, dodecane and culture were sampled in 1:10 ratio for metabolite analysis (see below). For RNA extraction, 2 mL exponential-phase culture (OD600 = 1–1.5) and 1 mL ethanol growth-phase culture (at 36 or 48 h) were sampled, and cells were collected and stored at −80 °C.
Fed-batch cultivation
Fed-batch cultivation was performed in DASGIP 400-mL fermenters (DASGIP#SR0400SS, Jülich, Germany). The medium for fed-batch cultivation was modified from previous reports [16, 77, 78]. 1× trace element composition and 1× vitamin composition from previous report [77] were used. Seed and batch media contained 15 g L−1 (NH4)2SO4, 8 g L−1 KH2PO4, 3 g L−1 MgSO4, 10× trace element composition and 10× vitamin composition; additionally, seed medium contained 40 g L−1 glucose, and batch medium contained 20 g L−1 glucose. Feed medium contained 9 g L−1 KH2PO4, 2.5 g L−1 MgSO4, 3.5 g L−1 K2SO4, 0.28 g L−1 Na2SO4, 10× trace element composition and 10× vitamin composition, with carbon source of 600 g L−1 glucose, or 600 g L−1 sucrose, or a mixture of 400 g L−1 glucose and 158 g L−1 ethanol. Ammonium hydroxide (~10 M) was used to adjust pH value to 5. Dissolved oxygen (DO) was monitored using a PreSens minisensor oxygen metre (PreSens, Germany) and controlled with a proportional–integral (PI; DO set-point, 30%; P, 0.5; Ti, 50 s; PI output, 0–100%) agitation-gassing-cascade (PI input 0–60% ~agitation output 300–600 rpm; PI input 20–60% ~air gassing output 1.58–3.16 L h−1) controller (DASGIP control). Off-gas was dried using a 10 °C chiller and 50 mL self-indicating silica gel was analysed using the DASGIP off-gas analyser.
In batch cultivation, 130 mL batch medium and 25 ml dodecane were used initially; seed culture in exponential phase (OD600 = 5–10) was directly inoculated to OD600 = 0.2 in batch culture. Medium feeding was triggered when the DO increased sharply after carbon sources in batch medium were depleted. Medium feeding was programmed using the DASGIP VB.NET script feeding controller (for feeding script logic charts refer to Additional file 1: Figure S3).
Quantitation of mRNA level
Total RNA was extracted using a yeast RiboPure™ RNA Purification Kit (Ambion #AM1926) or a TRIzol® Plus RNA Purification Kit (Ambion #12183555). After DNase treatment, 0.1 or 0.2 μg total RNA was used for first-strand cDNA synthesis in a 10 μL reaction using ProtoScript® II Reverse Transcriptase (NEB #M0368). The diluted cDNA was used as the template for quantitative real-time (qRT) PCR (primers are listed in Additional file 1: Table S1#29 to #40). KAPA SYBR® FAST qPCR Kit (Kapa Biosystems#KP-KK4601) and CFX96 Touch™ Real-Time PCR Detection System (BIO-RAD) were used in qRT-PCR. Yeast genomic or plasmid DNA was used to prepare the standard curve. C t values were analysed using CFX Manager Software (Bio-Rad Laboratories, QLD Australia). The mRNA levels (N pg−1 total RNA) were calculated by referring to the standard curve equations.
GFP fluorescence assay
To monitor GFP fluorescence over the entire batch cultivation, cells were cultivated aerobically in MES-buffered 20 mL SM-glucose (SM-sucrose) medium in 100 mL flasks. Samples were taken at indicated time points, and GFP fluorescence in single cells was analysed immediately after sampling using a flow cytometer (BD Accuri™ C6; BD Biosciences, USA). Cultures were diluted after 12 h by 10 volume water for flow cytometer analysis. GFP fluorescence was excited by a 488 nm laser and monitored through a 530/20 nm band-pass filter (FL1.A); 10,000 events were counted per sample. The GFP fluorescence signal (FL1.A) was corrected according to the values of FSC.A (forward scatter detector) and SSC.A (side scatter detector) [33]. GFP fluorescence level was expressed as the percentage relative to the average background auto-fluorescence from exponential-phase cells of a GFP-negative reference strain GH4J3.
Metabolite analysis
Extracellular metabolites were analysed by the Metabolomics Australia Queensland Node. Trans-nerolidol was analysed using a novel HPLC method. Dodecane samples (in some cases, diluted with dodecane) were diluted in 40-fold volume of ethanol. Ethanol-diluted dodecane samples of 20 μL were injected into a Zorbax Extend C18 column (4.6 × 150 mm, 3.5 µm, Agilent PN: 763953-902) with a guard column (SecurityGuard Gemini C18, Phenomenex PN: AJO-7597). Analytes were eluted at 35 °C at 0.9 mL/min using the mixture of solvent A (high purity water, 18.2 kΩ) and solvent B (45% acetonitrile, 45% methanol and 10% water), with a linear gradient of 5–100% solvent B from 0 to 24 min, then 100% from 24 to 30 min and finally 5% from 30.1 to 35 min. Analytes of interest were monitored using a diode array detector (Agilent DAD SL, G1315C) at 196 and 202 nm wavelengths. Spectral scans were also performed on each of the compounds from 190 to 400 nm in steps of 2 nm to confirm their identity and purity. Trans-nerolidol primary pharmaceutical reference standard (93.7% purity; Sigma-Aldrich #04610590) was used to prepare the standard curve for quantification.
Glucose, ethanol, acetate, glycerol and mevalonate were analysed through ion-exclusion chromatography [79]. Ion-exclusion chromatography was performed using an Agilent 1200 HPLC system and an Agilent Hi-Plex H column (300 × 7.7 mm, PL1170-6830) with guard column (SecurityGuard Carbo-H, Phenomenex PN: AJO-4490). Analytes were eluted isocratically with 4 mM H2SO4 at 0.6 mL/min at 65 °C. Glucose, ethanol, glycerol and mevalonate were monitored using a refractive index detector (Agilent RID, G1362A); acetate and mevalonate were detected using an ultraviolet–visible light absorbance detector (Agilent MWD, G1365B) at 210 nm. For sucrose, glucose and fructose analysis, analytes were eluted isocratically using high purity water (18.2 MΩ cm) as the mobile phase, at 0.4 mL/min for 21 min, and sugars (sucrose, glucose and fructose) were monitored using a refractive index detector (Agilent RID, G1362A).
Physiological feature calculation
Physiological parameters were calculated as reported previously [80]: the maximum growth rates are the linear regression coefficients of the ln OD600 versus time during the exponential growth phase; one unit of OD600 equals 0.23 g L−1 biomass; the specific production rate of nerolidol (r nerolidol, mg g−1 biomass h−1) was calculated by dividing Δ nerolidol titre (mg L−1) with the integral of biomass (g L−1) in defined time (h).
Abbreviations
- IPP:
-
isopentenyl pyrophosphate
- DMAPP:
-
dimethylallyl pyrophosphate
- FPP:
-
farnesyl pyrophosphate
- MVA:
-
mevalonate
- P XXXX :
-
promoter for gene XXXX
References
Nielsen J, Keasling JD. Engineering cellular metabolism. Cell. 2016;164(6):1185–97.
Lee SY, Kim HM, Cheon S. Metabolic engineering for the production of hydrocarbon fuels. Curr Opin Biotechnol. 2015;33:15–22.
Sun J, Alper HS. Metabolic engineering of strains: from industrial-scale to lab-scale chemical production. J Ind Microbiol Biotechnol. 2015;42(3):423–36.
Castellana M, Wilson MZ, Xu Y, Joshi P, Cristea IM, Rabinowitz JD, Gitai Z, Wingreen NS. Enzyme clustering accelerates processing of intermediates through metabolic channeling. Nat Biotechnol. 2014;32(10):1011–8.
Johannes TW, Zhao H. Directed evolution of enzymes and biosynthetic pathways. Curr Opin Microbiol. 2006;9(3):261–7.
Peng B, Huang S, Liu T, Geng A. Bacterial xylose isomerases from the mammal gut Bacteroidetes cluster function in Saccharomyces cerevisiae for effective xylose fermentation. Microb Cell Fact. 2015;14:70.
Du J, Yuan Y, Si T, Lian J, Zhao H. Customized optimization of metabolic pathways by combinatorial transcriptional engineering. Nucleic Acids Res. 2012;40(18):e142.
Alper H, Fischer C, Nevoigt E, Stephanopoulos G. Tuning genetic control through promoter engineering. Proc Natl Acad Sci USA. 2005;102(36):12678–83.
Tyo KE, Kocharin K, Nielsen J. Toward design-based engineering of industrial microbes. Curr Opin Microbiol. 2010;13(3):255–62.
Pfleger BF, Pitera DJ, Smolke CD, Keasling JD. Combinatorial engineering of intergenic regions in operons tunes expression of multiple genes. Nat Biotechnol. 2006;24(8):1027–32.
Cakir T, Kirdar B, Onsan ZI, Ulgen KO, Nielsen J. Effect of carbon source perturbations on transcriptional regulation of metabolic fluxes in Saccharomyces cerevisiae. BMC Syst Biol. 2007;1:18.
Vickers CE, Behrendorff JB, Bongers M, Brennan TC, Bruschi M, Nielsen LK. Production of industrially relevant isoprenoid compounds in engineered microbes. Microorg Biorefineries. 2015:303–34.
Ro DK, Paradise EM, Ouellet M, Fisher KJ, Newman KL, Ndungu JM, Ho KA, Eachus RA, Ham TS, Kirby J, et al. Production of the antimalarial drug precursor artemisinic acid in engineered yeast. Nature. 2006;440(7086):940–3.
Scalcinati G, Partow S, Siewers V, Schalk M, Daviet L, Nielsen J. Combined metabolic engineering of precursor and co-factor supply to increase alpha-santalene production by Saccharomyces cerevisiae. Microb Cell Fact. 2012;11:117.
Tippmann S, Scalcinati G, Siewers V, Nielsen J. Production of farnesene and santalene by Saccharomyces cerevisiae using fed-batch cultivations with RQ-controlled feed. Biotechnol Bioeng. 2016;113(1):72–81.
Westfall P, Pitera D, Lenihan J, Eng D, Woolard F, Regentin R. Production of amorphadiene in yeast, and its conversion to dihydroartemisinic acid, precursor to the antimalarial agent artemisinin. Proc Natl Acad Sci USA. 2012;109:E111–8.
Ignea C, Cvetkovic I, Loupassaki S, Kefalas P, Johnson CB, Kampranis SC, Makris AM. Improving yeast strains using recyclable integration cassettes, for the production of plant terpenoids. Microb Cell Fact. 2011;10:4.
Yoon SH, Lee SH, Das A, Ryu HK, Jang HJ, Kim JY, Oh DK, Keasling JD, Kim SW. Combinatorial expression of bacterial whole mevalonate pathway for the production of beta-carotene in E. coli. J Biotechnol. 2009;140(3–4):218–26.
Tsuruta H, Paddon CJ, Eng D, Lenihan JR, Horning T, Anthony LC, Regentin R, Keasling JD, Renninger NS, Newman JD. High-level production of amorpha-4,11-diene, a precursor of the antimalarial agent artemisinin, in Escherichia coli. PLoS ONE. 2009;4(2):e4489.
Choi SY, Lee HJ, Choi J, Kim J, Sim SJ, Um Y, Kim Y, Lee TS, Keasling JD, Woo HM. Photosynthetic conversion of CO2 to farnesyl diphosphate-derived phytochemicals (amorpha-4,11-diene and squalene) by engineered cyanobacteria. Biotechnol Biofuels. 2016;9(1):202.
Chen F, Zhou J, Shi Z, Liu L, Du G. Effect of acetyl-CoA synthase gene overexpression on physiological function of Saccharomyces cerevisiae. Wei sheng wu xue bao=Acta Microbiol Sinica. 2010;50(9):1172–9.
Meadows AL, Hawkins KM, Tsegaye Y, Antipov E, Kim Y, Raetz L, Dahl RH, Tai A, Mahatdejkul-Meadows T, Xu L, et al. Rewriting yeast central carbon metabolism for industrial isoprenoid production. Nature. 2016;537(7622):694–7.
Peng B, Plan MR, Chrysanthopoulos P, Hodson MP, Nielsen LK, Vickers CE. A squalene synthase protein degradation method for improved sesquiterpene production in Saccharomyces cerevisiae. Metab Eng. 2017;39:209–19.
Liu J, Zhang W, Du G, Chen J, Zhou J. Overproduction of geraniol by enhanced precursor supply in Saccharomyces cerevisiae. J Biotechnol. 2013;168(4):446–51.
Polakowski T, Stahl U, Lang C. Overexpression of a cytosolic hydroxymethylglutaryl-CoA reductase leads to squalene accumulation in yeast. Appl Microbiol Biotechnol. 1998;49(1):66–71.
Asadollahi MA, Maury J, Moller K, Nielsen KF, Schalk M, Clark A, Nielsen J. Production of plant sesquiterpenes in Saccharomyces cerevisiae: effect of ERG9 repression on sesquiterpene biosynthesis. Biotechnol Bioeng. 2008;99(3):666–77.
Scalcinati G, Knuf C, Partow S, Chen Y, Maury J, Schalk M, Daviet L, Nielsen J, Siewers V. Dynamic control of gene expression in Saccharomyces cerevisiae engineered for the production of plant sesquitepene alpha-santalene in a fed-batch mode. Metab Eng. 2012;14(2):91–103.
Van Hoek P, Van Dijken JP, Pronk JT. Effect of specific growth rate on fermentative capacity of baker’s yeast. Appl Environ Microbiol. 1998;64(11):4226–33.
Nevoigt E. Progress in metabolic engineering of Saccharomyces cerevisiae. Microbiol Mol Biol Rev. 2008;72(3):379–412.
Görgens JF, van Zyl WH, Knoetze JH, Hahn-Hägerdal B. The metabolic burden of the PGK1 and ADH2 promoter systems for heterologous xylanase production by Saccharomyces cerevisiae in defined medium. Biotechnol Bioeng. 2001;73:238–45.
Wu G, Yan Q, Jones JA, Tang YJ, Fong SS, Koffas MA. Metabolic burden: cornerstones in synthetic biology and metabolic engineering applications. Trends Biotechnol. 2016;34(8):652–64.
Williams TC, Nielsen LK, Vickers CE. Engineered quorum sensing using pheromone-mediated cell-to-cell communication in saccharomyces cerevisiae. ACS Synth Biol. 2013;2(3):136–49.
Peng B, Williams T, Henry M, Nielsen L, Vickers C. Controlling heterologous gene expression in yeast cell factories on different carbon substrates and across the diauxic shift: a comparison of yeast promoter activities. Microb Cell Fact. 2015;14(1):91.
Osterlund T, Nookaew I, Bordel S, Nielsen J. Mapping condition-dependent regulation of metabolism in yeast through genome-scale modeling. BMC Syst Biol. 2013;7:36.
Boer VM, de Winde JH, Pronk JT, Piper MD. The genome-wide transcriptional responses of Saccharomyces cerevisiae grown on glucose in aerobic chemostat cultures limited for carbon, nitrogen, phosphorus, or sulfur. J Biol Chem. 2003;278(5):3265–74.
Sun J, Shao Z, Zhao H, Nair N, Wen F, Xu JH, Zhao H. Cloning and characterization of a panel of constitutive promoters for applications in pathway engineering in Saccharomyces cerevisiae. Biotechnol Bioeng. 2012;109(8):2082–92.
Cao L, Tang X, Zhang X, Zhang J, Tian X, Wang J, Xiong M, Xiao W. Two-stage transcriptional reprogramming in Saccharomyces cerevisiae for optimizing ethanol production from xylose. Metab Eng. 2014;24:150–9.
Butt TR, Ecker DJ. Yeast metallothionein and applications in biotechnology. Microbiol Rev. 1987;51(3):351–64.
West RW Jr, Chen SM, Putz H, Butler G, Banerjee M. GAL1-GAL10 divergent promoter region of Saccharomyces cerevisiae contains negative control elements in addition to functionally separate and possibly overlapping upstream activating sequences. Genes Dev. 1987;1(10):1118–31.
Lee KM, DaSilva NA. Evaluation of the Saccharomyces cerevisiae ADH2 promoter for protein synthesis. Yeast (Chichester, England). 2005;22(6):431–40.
Partow S, Siewers V, Bjorn S, Nielsen J, Maury J. Characterization of different promoters for designing a new expression vector in Saccharomyces cerevisiae. Yeast (Chichester, England). 2010;27(11):955–64.
Williams TC, Espinosa MI, Nielsen LK, Vickers CE. Dynamic regulation of gene expression using sucrose responsive promoters and RNA interference in Saccharomyces cerevisiae. Microb Cell Fact. 2015;14:43.
McIsaac RS, Gibney PA, Chandran SS, Benjamin KR, Botstein D. Synthetic biology tools for programming gene expression without nutritional perturbations in Saccharomyces cerevisiae. Nucleic Acids Res. 2014;42(6):e48.
Entian K-D, Kötter P. 23 yeast mutant and plasmid collections. Methods Microbiol. 1998;26:431–49.
Christianson TW, Sikorski RS, Dante M, Shero JH, Hieter P. Multifunctional yeast high-copy-number shuttle vectors. Gene. 1992;110(1):119–22.
Shiba Y, Paradise EM, Kirby J, Ro DK, Keasling JD. Engineering of the pyruvate dehydrogenase bypass in Saccharomyces cerevisiae for high-level production of isoprenoids. Metab Eng. 2007;9(2):160–8.
Ro DK, Ouellet M, Paradise EM, Burd H, Eng D, Paddon CJ, Newman JD, Keasling JD. Induction of multiple pleiotropic drug resistance genes in yeast engineered to produce an increased level of anti-malarial drug precursor, artemisinic acid. BMC Biotechnol. 2008;8:83.
Bhat PJ. Galactose regulon of yeast. Genet Systs Biol. 2008.
Torchia TE, Hamilton RW, Cano CL, Hopper JE. Disruption of regulatory gene GAL80 in Saccharomyces cerevisiae: effects on carbon-controlled regulation of the galactose/melibiose pathway genes. Mol Cell Biol. 1984;4(8):1521–7.
Nehlin JO, Carlberg M, Ronne H. Control of yeast GAL genes by MIG1 repressor: a transcriptional cascade in the glucose response. EMBO J. 1991;10(11):3373–7.
Xie W, Ye L, Lv X, Xu H, Yu H. Sequential control of biosynthetic pathways for balanced utilization of metabolic intermediates in Saccharomyces cerevisiae. Metab Eng. 2014;28C:8–18.
Gardner RG, Hampton RY. A ‘distributed degron’ allows regulated entry into the ER degradation pathway. EMBO J. 1999;18(21):5994–6004.
Ignea C, Trikka FA, Kourtzelis I, Argiriou A, Kanellis AK, Kampranis SC, Makris AM. Positive genetic interactors of HMG2 identify a new set of genetic perturbations for improving sesquiterpene production in Saccharomyces cerevisiae. Microb Cell Fact. 2012;11:162.
Hohmann S. PDC6, a weakly expressed pyruvate decarboxylase gene from yeast, is activated when fused spontaneously under the control of the PDC1 promoter. Curr Genet. 1991;20(5):373–8.
Solis-Escalante D, Kuijpers NG, Barrajon-Simancas N, van den Broek M, Pronk JT, Daran JM, Daran-Lapujade P. A minimal set of glycolytic genes reveals strong redundancies in Saccharomyces cerevisiae central metabolism. Eukaryot Cell. 2015;14(8):804–16.
Lee JW, Choi S, Park JH, Vickers CE, Nielsen LK, Lee SY. Development of sucrose-utilizing Escherichia coli K-12 strain by cloning beta-fructofuranosidases and its application for l-threonine production. Appl Microbiol Biotechnol. 2010;88(4):905–13.
Bruschi M, Boyes SJ, Sugiarto H, Nielsen LK, Vickers CE. A transferable sucrose utilization approach for non-sucrose-utilizing Escherichia coli strains. Biotechnol Adv. 2012;30(5):1001–10.
Archer CT, Kim JF, Jeong H, Park JH, Vickers CE, Lee SY, Nielsen LK. The genome sequence of E. coli W (ATCC 9637): comparative genome analysis and an improved genome-scale reconstruction of E. coli. BMC Genom. 2011;12:9.
Carlson M, Botstein D. Two differentially regulated mRNAs with different 5′ ends encode secreted with intracellular forms of yeast invertase. Cell. 1982;28(1):145–54.
Meijer MM, Boonstra J, Verkleij AJ, Verrips CT. Glucose repression in Saccharomyces cerevisiae is related to the glucose concentration rather than the glucose flux. J Biol Chem. 1998;273(37):24102–7.
Paddon CJ, Westfall PJ, Pitera DJ, Benjamin K, Fisher K, McPhee D, Leavell MD, Tai A, Main A, Eng D, et al. High-level semi-synthetic production of the potent antimalarial artemisinin. Nature. 2013;496(7446):528–32.
Brennan TC, Turner CD, Kromer JO, Nielsen LK. Alleviating monoterpene toxicity using a two-phase extractive fermentation for the bioproduction of jet fuel mixtures in Saccharomyces cerevisiae. Biotechnol Bioeng. 2012;109(10):2513–22.
Beekwilder J, van Rossum HM, Koopman F, Sonntag F, Buchhaupt M, Schrader J, Hall RD, Bosch D, Pronk JT, van Maris AJ, et al. Polycistronic expression of a beta-carotene biosynthetic pathway in Saccharomyces cerevisiae coupled to beta-ionone production. J Biotechnol. 2014;192:383–92.
Bzymek M, Lovett ST. Instability of repetitive DNA sequences: the role of replication in multiple mechanisms. Proc Natl Acad Sci USA. 2001;98(15):8319–25.
Lv X, Wang F, Zhou P, Ye L, Xie W, Xu H, Yu H. Dual regulation of cytoplasmic and mitochondrial acetyl-CoA utilization for improved isoprene production in Saccharomyces cerevisiae. Nat Commun. 2016;7:12851.
Yocum RR, Hanley S, West R Jr, Ptashne M. Use of lacZ fusions to delimit regulatory elements of the inducible divergent GAL1-GAL10 promoter in Saccharomyces cerevisiae. Mol Cell Biol. 1984;4(10):1985–98.
Paczia N, Nilgen A, Lehmann T, Gatgens J, Wiechert W, Noack S. Extensive exometabolome analysis reveals extended overflow metabolism in various microorganisms. Microb Cell Fact. 2012;11:122.
Vemuri GN, Eiteman MA, McEwen JE, Olsson L, Nielsen J. Increasing NADH oxidation reduces overflow metabolism in Saccharomyces cerevisiae. Proc Natl Acad Sci USA. 2007;104(7):2402–7.
Hou J, Lages NF, Oldiges M, Vemuri GN. Metabolic impact of redox cofactor perturbations in Saccharomyces cerevisiae. Metab Eng. 2009;11(4–5):253–61.
Boer VM, Crutchfield CA, Bradley PH, Botstein D, Rabinowitz JD. Growth-limiting intracellular metabolites in yeast growing under diverse nutrient limitations. Mol Biol Cell. 2010;21(1):198–211.
Seker T, Moller K, Nielsen J. Analysis of acyl CoA ester intermediates of the mevalonate pathway in Saccharomyces cerevisiae. Appl Microbiol Biotechnol. 2005;67(1):119–24.
Pronk JT, Yde Steensma H, VanDijken JP. Pyruvate metabolism in Saccharomyces cerevisiae. Yeast (Chichester, England). 1996;12(16):1607–33.
Chubukov V, Gerosa L, Kochanowski K, Sauer U. Coordination of microbial metabolism. Nat Rev Microbiol. 2014;12(5):327–40.
Heyland J, Fu J, Blank LM. Correlation between TCA cycle flux and glucose uptake rate during respiro-fermentative growth of Saccharomyces cerevisiae. Microbiology. 2009;155(12):3827–37.
Hanscho M, Ruckerbauer DE, Chauhan N, Hofbauer HF, Krahulec S, Nidetzky B, Kohlwein SD, Zanghellini J, Natter K. Nutritional requirements of the BY series of Saccharomyces cerevisiae strains for optimum growth. FEMS Yeast Res. 2012;12(7):796–808.
Vickers CE, Bongers M, Bydder SF, Chrysanthopoulos P, Hodson MP. Protocols for the production and analysis of isoprenoids in bacteria and yeast. Berlin: Springer; 2015. p. 1–30.
Verduyn C, Postma E, Scheffers WA, van Dijken JP. Effect of benzoic acid on metabolic fluxes in yeast: a continuous-culture study on the regulation of respiration and alcoholic fermentation. Yeast (Chichester, England). 1992;8(7):501–17.
van Hoek P, de Hulster E, van Dijken JP, Pronk JT. Fermentative capacity in high-cell-density fed-batch cultures of baker’s yeast. Biotechnol Bioeng. 2000;68(5):517–23.
Dietmair S, Timmins NE, Gray PP, Nielsen LK, Kromer JO. Towards quantitative metabolomics of mammalian cells: development of a metabolite extraction protocol. Anal Biochem. 2010;404(2):155–64.
Peng B, Shen Y, Li X, Chen X, Hou J, Bao X. Improvement of xylose fermentation in respiratory-deficient xylose-fermenting Saccharomyces cerevisiae. Metab Eng. 2012;14(1):9–18.
Authors’ contributions
BP, LKN and CEV designed the experiments. BP carried out the experiments. MP developed HPLC method and performed the HPLC analysis. AC carried out partial experiments. BP, LKN and CEV drafted and revised the manuscript. All authors contributed to the result analysis and the discussion of the research. All authors read and approved the final manuscript.
Acknowledgements
We thank Zhenyu Shi for assistance with feeding script programming and Ricardo A. Gonzalez Garcia for assistance with fed-batch fermentation. Metabolite analysis was performed in the Metabolomics Australia Queensland Node.
Competing interests
The authors declare that they have no competing interests.
Availability of supporting data
All data generated or analysed during this study are included in this published article (and its supplementary information files).
Funding
BP was supported by a University of Queensland International Postgraduate Research Scholarship. CEV was supported by Queensland Government Smart Futures and Accelerate Fellowships.
Author information
Authors and Affiliations
Corresponding author
Additional file
13068_2017_728_MOESM1_ESM.docx
Additional file 1: Table S1. Primers and PCR fragments amplified/used in this work. PXXX, promoter of gene XXX; T XXX , terminator of gene XXX; Y-GDNA, CEN.PK113-7D genomic DNA; sequence annealing to template in primers is shown in red and italics; over-lap sequence for over-lap extension PCR and Gibson Assembly is underlined; restriction sites used in cloning are shown in bold. Table S2. Molecular construction of plasmids used in this work. Figure S1. Plasmid rearrangement in the strain NC1D. Plasmid pPMVAd36 and [TRP1] from NC1D were digested by restriction enzymes NotI, SalI, SphI, BamHI and SbfI and gel figure was shown as the right-bottom figure. Figure S2. The growth profile of strain GH4 [CEN.PK113-5D derivative; ura3(1, 704 )::KlURA3] (1) on sucrose. The cells were pre-cultured on 40 g L-1 glucose. Mean values from duplicate experiments are shown. Figure S3. Logic charts for fed-batch feeding scripts: (a) carbon-source-restricted/DO-triggered fed-batch cultivation; (b) carbon-source-overflowed/carbon-source-pulsing fed-batch cultivation. Fs, feeding flow storage value; DOt, dissolved oxygen on-line value at time t; DOL, lowest dissolved oxygen storage value; T1, storage time; t, on-line time; μ, specific rate of feeding flow increasement; N, agitatation speed; Nmax, the maxium agitation speed; FVs, feeding volume storage value; FVt, feeding volume on-line value; Vt, culture volume on-line value. Figure S4. Growth (OD600) and process values (Dissovled oxygen, DO; oxygen transfer rate, OTR; carbon transfer rate; CTR; respiration quotient, RQ) in fed-batch cultivation for strain N391DA, with feeding logics in Fig. S1a employed. (a&b), 600 g L-1 glucose feeding; (c&d), 600 g L-1 sucrose feeding; (e&f), 400 g L-1 glucose and 158 g L-1 ethanol feeding. Figure S5. Growth (OD600) and process values (Dissovled oxygen, DO; oxygen transfer rate, OTR; carbon transfer rate; CTR; respiration quotient, RQ) in fed-batch cultivation for strain N391DA, with feeding logics in Fig. S1b employed. (a&b), 600 g L-1 glucose feeding with 10 g L-1 glucose pulse; (c&d), 600 g L-1 glucose feeding with 20 g L-1 glucose pulse; (e&f), 600 g L-1 sucrose feeding with 20 g L-1 sucrose pulse. Figure S6. The influence of nerolidol on yeast growth. Synthetic minimal medium was used, which contained 6.7 g L-1 yeast nitrogen base (Sigma-Aldrich #Y0626; pH 6.0) and 20 g L-1 glucose. Isomer-mixed nerolidol (Sigma-Aldrich #H59605) was used. Tween 80 was added to homogenize nerolidol into liquid medium. Mean values ± standard deviations are shown (N = 3).
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Peng, B., Plan, M.R., Carpenter, A. et al. Coupling gene regulatory patterns to bioprocess conditions to optimize synthetic metabolic modules for improved sesquiterpene production in yeast. Biotechnol Biofuels 10, 43 (2017). https://doi.org/10.1186/s13068-017-0728-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13068-017-0728-x