
Abstract
Glioblastoma (GBM) is a highly aggressive, infiltrative malignancy that cannot be completely cured by current treatment modalities, and therefore requires more precise molecular subtype signatures to predict treatment response for personalized precision therapy. Expression subtypes of GBM samples from the Cancer Genome Atlas (TCGA) were identified using BayesNM and compared with existing molecular subtypes of GBM. Biological features of the subtypes were determined by single-sample gene set enrichment analysis. Genomic and proteomic data from GBM samples were combined and Genomic Identification of Significant Targets in Cancer analysis was used to screen genes with recurrent somatic copy-number alterations phenomenon. The immune environment among subtypes was compared by assessing the expression of immune molecules and the infiltration of immune cells. Molecular subtypes adapted to immunotherapy were identified based on Tumor Immune Dysfunction and Exclusion (TIDE) score. Finally, least absolute shrinkage and selection operator (LASSO) logistic regression was performed on the expression profiles of S2, S3 and S4 in TCGA-GBM and RPPA to determine the respective corresponding best predictive model. Four novel molecular subtypes were classified. Specifically, S1 exhibited a low proliferative profile; S2 exhibited the profile of high proliferation, IDH1 mutation, TP53 mutation and deletion; S3 was characterized by high immune scores, innate immunity and adaptive immune infiltration scores, with the lowest TIDE score and was most likely to benefit from immunotherapy; S4 was characterized by high proliferation, EGFR amplification, and high protein abundance, and was the most suitable subtype for bevacizumab. LASSO analysis constructed the best prediction model composed of 13 genes in S2 with an accuracy of 96.7%, and the prediction model consisting of 17 genes in S3 with an accuracy of 86.7%, and screened 14 genes as components of the best prediction model in S4 with an accuracy of 93%. To conclude, our study classified reproducible and robust molecular subtypes of GBM, and these findings might contribute to the identification of patients responding to immunotherapy, thereby improving GBM prognosis.
Similar content being viewed by others

Introduction
Gliomas is a highly aggressive and the most common intracranial primary malignant tumor1,2. A new version of tumor taxonomy based on clinical and molecular pathological features for the GBM classification has been updated published by the World Health Organization (WHO) (2021 version). Low-grade gliomas (LGG) of grades 1 and 2 are defined as slow-growing tumors with low infiltration of the brain parenchyma, and high-grade gliomas (HGG) of grades 3 and 4 are highly infiltrative hypodifferentiated or undifferentiated tumors with high infiltration of the brain parenchyma. Grade 4 tumors, which also known as glioblastoma (GBM), are the hairiest and most aggressive CNS tumor1,3,4. Currently, surgical resection, targeted agents and chemotherapy are widely applied treatment options, however, the prognosis of GBM remains unfavorable, with a median survival time of only 14–16 months5. Immunotherapy as an emerging second-line treatment option has been proven to be successful in other cancers6,7. High infiltration results in extensive heterogeneity in the formation of immune components in tumor microenvironment, which explains the inability of immunotherapy to achieve breakthroughs in GBM8,9.
Numerous investigations have portrayed novel molecular expression tumor classification based on the Cancer Genome Atlas (TCGA) with different markers, focusing on the expression of signature genes to classify cancer subtypes10,11,12,13. These studies contributed to the development of anti-tumor targeted agents14,15. Molecular subtype-related studies have also classified GBM subtypes in terms of different features10,16. Liu et al. reported the identification of two subtypes in TCGA's low-grade glioma dataset based on cytomorpologic biomarkers of GBM10. In addition, Munquad et al. based on high-throughput data from different transcriptomes and methylomes in order to develop a deep learning model based on convolutional neural networks will be and used to recognize subtypes of GBM17. The portrayal of GBM molecular subtypes could improve personalized treatment selection for patients18. Verhaak's earliest study characterized four GBM subtypes, Proneural, Neural, Classical, and Mesenchymal, which exhibited abnormal expression of PDGFRA, IDH1, EGFR, and NF1 or copy number variation (CNV), respectively19. However, we still lack a systematic and in-depth insights into the current GBM subtyping. A deeper delineation of GBM molecular subtypes could offer a robust basis for scientific therapeutic instruction and immunotherapy development.
In this research, we presented the first classification of molecular subtypes of GBM in TCGA using Bayesian non-negative matrix factorization (BayesNMF) algorithm and consensus clustering. This clustering method has also previously helped to identify six expression subtypes for head and neck squamous cell carcinoma20. The genomic and proteomic data from the Clinical Proteomic Tumor Analysis Consortium (CPTAC), cellular data from Dependency Map, and proteomic data from the Cancer Proteome Atlas (TCPA) demonstrated the consistency and reproducibility of the discovered molecular subtypes. Based on the requirement of cytolytic immune responses for effective natural antitumor immunity, simple expression metrics of effector molecules (PRF1, GZMA) that mediate cytolytic activity could quantify the cytolytic activity of GBM, thereby allowing us to actively identify molecular subtypes that are sensitive to spontaneous cytolytic activity21. The molecular and pathological features of each subtype were explored based on expression profiles, and response to immunotherapy was assessed by TIDE. Our analysis developed novel molecular subtypes in GBM, and LASSO logistic regression analysis was also performed on the expression profiles of specific subtype to identify subtype-specific model, laying the foundation for a deeper characterization of cancer pathogenesis.
Materials and methods
TCGA GBM data
For this study, we collected GBM data from multiple databases. RNA sequencing data (RNA-seq) for the GBM sequencing project (TCGA-GBM) were obtained from the TCGA (https://portal.gdc.cancer.gov/) database, and clinical data and somatic mutation data (maf file format) of the samples in TCGA-GBM were also procured. The SangerBox database (http://sangerbox.com/login.html)22 was used to screen sample statistics and those with missing status were eliminated. For RNA-seq data, fragments Per Kilobase of exon model per Million mapped fragments (FPKM) were first converted into transcripts Per Kilobase of exon model per Million mapped reads (TPM), followed by log2(TPM + 1) processing. After processing, RNA-seq data from 153 samples were included in this study.
Genomic, proteomic and copy number variation data for GBM
Genomics and proteomics data for 99 GBM samples were sourced from the Clinical Proteomic Tumor Analysis Consortium (CPTAC, https://cptac-data-portal.georgetown.edu/datasets) database. Copy number variant data were extracted from the cBioPortal for Cancer Genomics (https://www.cbioportal.org/) database. Reverse phase protein array (RPPA) data were sourced from The Cancer Proteome Atlas (TCPA, https://www.tcpaportal.org/tcpa/download.html). In addition, the omics data and CRISPR knockout data for Cancer Cell Line Encyclopedia (CCLE) GBM cell line samples were obtained from the DepMap (https://depmap.org/portal/; DepMap Public 21Q3 dataset). Out of 1377 cell lines, we used 47 GBM cell lines.
Vulnerability analysis of different subtype-specific cancers
The CERES obtained from the Cancer Dependency Map (DepMap) is used as a measure of cell line cancer susceptibility. The CERES is a calculation used to estimate the dependency of each cell line tested on a given gene knockout. Typically, the CERES is interpreted as follows: a score of 0 indicates that the gene is not essential in a given cell line, while -1 indicates a high dependence23.
Identification of molecular subtypes in GBM by Bayesian non-negative matrix factorization (BayesNMF)
For the molecular subtypes of novel features in GBM, the expression features of 153 samples in TCGA-GBM were discriminated by consensus clustering based on BayesNMF with reference to the method of Tan et al.24. The top 25% of highly variable genes were considered to feature across samples, and the expression matrix of these genes was retained defined as R. The expression matrix R* was then transformed based on the median expression as the centered multiplicity. The distance matrix 1-C (Cij expressed the Spearman correlation between genes in sample i and sample j) and the consistency matrix Mk were determined. BayesNMF was run to select the optimal number of clusters K to achieve sample clustering. The differences between Proneural, Neural, Classical, and Mesenchymal subtypes found with Verhaak et al.19 were compared.
Single-sample gene set enrichment analysis (ssGSEA) in molecular subtypes
To discuss the biological differences in molecular subtypes, the h.all.v7.5.1.symbols.gmt gene set was obtained from the Molecular Signatures Database (MSigDB, https://www.gsea-msigdb.org/gsea/msigdb). The GSVA package25 was installed in R. The ssGSEA was implemented26,27. Features in molecular subtypes were defined based on pathway activity. Proliferative and immune features in subtypes and expression levels of T cells effector molecular proteins (PRF1, GZMA) were assessed based on markers in pan-cancer from previous studies28.
CPTAC cohort validation
The molecular subtypes in the GBM samples from the CPTAC cohort were clustered according to the BayesNMF implementation consensus clustering, which based on a normal likelihood and exponential priors, and derive an efficient Gibbs sampler to approximate the posterior density of the NMF factors29. Based on the h.all.v7.5.1.symbols.gmt gene set, ssGSEA was implemented on the clustered subtypes to explore the biological pathway differences and we further discussed the reproducibility and robustness of the novel subtypes of GBM.
Copy number variation analysis in TCGA and CPTAC cohorts
To uncover the mutational heterogeneity of cancer and to select new cancer driver genes, the somatic mutation data obtained from the TCGA cohort were analyzed. We determined representative mutated genes in GBM and genes with remarkable somatic copy-number alterations (SCNA) in novel subtypes of GBM using MutSig2CV30,31 and Genomic Identification of Significant Targets in Cancer (GISTIC) 2.032.
Identification of subtypes for immunotherapy
To identify molecular subtypes of GBM that could benefit from immunotherapy, we calculated the Tumor Immune Dysfunction and Exclusion (TIDE) score. Initially, a cohort (registration number GSE84010) treated with Temozolomide was retrieved in the Gene Expression Omnibus (GEO, https://www.ncbi.nlm.nih.gov/geo/). The marker genes based on the subtypes were clustered according to BayesNMF implementation consensus clustering to classify the samples in GSE84010 as GBM molecular subtypes. ssGSEA was implemented again to verify the reproducibility of the subtypes. The TIDE scores of the TCGA cohort samples were then obtained from the Tumor Immune Dysfunction and Exclusion website (http://tide.dfci.harvard.edu/) to compare the differences in TIDE scores among subtypes, with lower TIDE scores representing greater benefit from immunotherapy33. CIBERSORT is a tool for deconvoluting the expression matrix of 22 human immune cell subtypes based on the principle of linear support vector regression34. The proportion of immune cells in the TCGA-GBM cohort was computed by CIBERSORT assessment.
Construction of subtype prediction model
The protein data from TCGA-GBM cohort and TCPA were used to divide samples into training set data and test set data according to 8:2. The model was constructed by the LASSO logistic regression model. The glmnet package35 was installed in R. The cv.glmnet() function was called to construct the LASSO logistic regression model with marker genes, and the model with the minimum prediction error rate was selected by choosing the optimal lambda value.
Cell culture
The GBM cell line U87 and the normal cell line HEB were obtained from ATCC. Cells should be cultured at 37 °C and 5% CO2 using Dulbecco's Modified Eagle Medium (DMEM) medium (from Gibco) with 10% fetal bovine serum, 1% glutamine, and 1% antibiotic/antifungal solution.
Quantitative real-time polymerase chain reaction (qRT-PCR)
Total RNA was extracted using Trizol reagent (Invitrogen). Total RNA was reverse transcribed to cDNA by Takara PrimeScript™ RT Reagent Kit and following its instructions. Subsequently, Takara TB Green™ Premix Ex Taq™ II instructions were used in order to perform a qRT-PCR reaction with the following reaction conditions: pre-denaturation at 95 °C for 30 s and 40 cycles (95 °C for 5 s and 60 °C for 30 s). Finally, the 2-ΔΔCt method was used to calculate the relative expression of the target genes with GAPDH as the internal reference gene. The primer sequences of the target gene were as follows: CDAN1: GCAGGATCAACCCAACTCCG (F), and CTCGCTCCTCTTGCAGACTTC (R); UBE2M: ATGAGGGCTTCTACAAGAGTGG (F), and ATTGTCTCACACTTCACCTTGG (R).
Statistical analysis
Statistical analyses in this study were conducted by R software (version: 4.1.0). The bilateral Wilcoxon rank sum test and chi-square test were chosen. All analyses were considered statistically significant at P < 0.05.
Results
Four novel molecular subtypes in GBM classified at the genomic level
To make it easier to understand the flow of this study, we have produced Supplementary Fig. 1. In this study, we aimed to classify novel molecular subtypes in GBM at the genomic level using a consensus clustering approach based on BayesNMF (Supplementary Table 1). Our findings indicated the existence of four novel molecular subtypes (S1, S2, S3, and S4) (Fig. 1A). To further explore the properties of the molecular subtypes, we compared the differences between the subtypes (Proneural, Neural, Mesenchymal, Classical) with that of Verhaak et al.19. The classification by Verhaak et al. provided the basis and perspective for molecular classification of GBM, but the study by wan et al. demonstrated that the original Verhaak 840 gene set was unable to robustly cluster the larger cohorts now available on different platforms36. It was observed that 46.2% of the samples in S1 were the Neural subtype, 82.8% of the samples in S2 were the Proneural subtype, 74% of the samples in S3 were the Mesenchymal subtype, and 57.9% of the samples in S4 were the Classical subtype. Furthermore, S2 was closely related to Proneural and S3 was closely related to Mesenchymal subtypes, with 70.6% of Proneural mapping to S2 and 66.1% of Mesenchymal subtypes mapping to S3 (Fig. 1B). These results indicate that our classification of GBM is highly consistent with previous classifications, with each subtype associated with specific phenotype.

Four novel GBM molecular subtypes. (A): Heat map of marker genes expression in GBM subtypes. (B): Comparison of four novel GBM molecular subtypes with Proneural, Neural, Classical, and Mesenchymal subtypes. And the number in each box represents the sample number, and the percentage in the middle refers to the proportion of the sample in all samples; The percentage on the right is the proportion of samples of the corresponding subtype in the whole row; The lower percentage is the proportion of samples of the corresponding subtype in the whole column.
Next, we discussed the biological meaning between the four molecular subtypes and performed ssGSEA. The results indicated that S1 exhibited a low proliferative profile, S2 and S4 exhibited a high proliferative profile, and S3 exhibited a high immune profile. In terms of survival trend, the prognosis of S2 and S4 was worse than that of S1 and S3 (Supplementary Fig. 2). Moreover, we also found that S2 exhibited low immune and inflammatory features (Fig. 2A). Genomic level features in the four molecular subtypes were identified by the pan-oncogenomic features in TCGA, the four molecular subtypes showed distinct mutational profiles, and S3 exhibited overall higher Aneuploidy Score, Stromal Fraction (Fig. 2B, C). Furthermore, to explore the association between the four subtypes and driver events (point mutations, insertional deletions and copy number variants), we analyzed cancer driver genes and copy number variants. Our results indicated that S2 was enriched to IDH1 mutation, TP53 mutation and loss, S3 was enriched to NF1 deletion, and S4 was enriched to EGFR amplification/mutation (Fig. 2D).

Biological highway analysis and copy number variation analysis in GBM subtypes. (A): ssGSEA results showing the biological pathway differences in GBM subtypes. (B): Number of Segments, Aneuploidy Score, Stromal Fraction in GBM subtypes. (C): Waterfall diagram demonstrating the variation differences of 17 genes in GBM subtypes. (D): Comparison of CNV differences in GBM subtypes.
The immune landscape in S1-4
The immune landscape in S1-4 was also discussed. S2 showed lower Macrophage Regulation, Lymphocyte Infiltration Signature Score, IFN-gamma Response, and TGF-beta Response. In contrast, S3 exhibited higher Macrophage Regulation, Lymphocyte Infiltration Signature Score, IFN-gamma Response, and TGF-beta Response (Fig. 3A). Important molecules in the immune system, including immune checkpoint molecules and human leukocyte antigen (HLA) encoding MHC proteins, showed higher expression levels in S3 than the other three molecular subtypes (Supplementary Fig. 3A, B). According to the immune characteristics in pan-cancer, the highest T Cells Follicular Helper, Th2 Cells were found in S2, and the highest Dendritic Cells, Monocytes, Neutrophils were displayed in S3 (Fig. 3B). We also evaluated the performance of immune score, stromal score37 and the overall score of immune cell types related to adaptive immunity and the overall score of immune cell types related to innate immunity38, respectively, among the four molecular subtypes and found that all four scores were the highest in S3 (Supplementary Fig. 4A, B). These results indicated that the S3 had a higher immunogenicity and was more easily recognized by the immune system and elicits an immune response.

Immune-related scores and immune cell scores in GBM subtypes. (A): Scores of Macrophage Regulation, Lymphocyte Infiltration Signature Score, IFN-gamma Response, TGF-beta Response in GBM subtypes calculated by ssGSEA. (B): Immune cell score in GBM computed by CIBERSORT. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
Subtype-specific cancer vulnerability and validation of specific gene expression levels
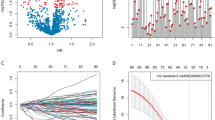
Glioma cell lines in CCLE were first classified into specific subtypes using subtype-specific marker genes (Supplementary Tables 1 and 2). Due to the small number of high-grade glioma cell lines, only three subtypes, S2(n = 4), S3(n = 36), and S4(n = 6), were eventually mapped (Supplementary Fig. 5A). Here, we selected the GBM somatic copy number variant genes identified in this study (Supplementary Table 3). Among them, genes with subtype-specific cancer susceptibility need to fulfill two conditions: first, to ensure that the CERES median for each driver gene is less than -0.5 in the subtype to reflect the fact that the subtype cell line is dependent on the gene; and, second, that the CERES median for the gene in the specific subtype is lower than that in the rest of the cell line by 0.3.Therefore, we found that only 2 genes (CDAN1 and UBE2M) fulfill the above conditions. However, the differences in these genes were non-significant due to the small sample size used (Supplementary Fig. 5B).
To validate CDAN1 and UBE2M, we examined the mRNA expression levels of these two genes in the GBM cell line U87 based on qRT-PCR. As shown in Supplementary Fig. 5C, we observed that the mRNA expression levels of CDAN1 and UBE2M were significantly upregulated in GBM cells compared to normal control cells. These results indirectly confirm the validity of our identified GBM subtypes.
Proteomics revealed specific protein regulation in S2-4
To further explore the features on protein expression levels in the four GBM subtypes, GBM samples in CPTAC was clustered as S1 (N = 15), S2 (N = 18), S3 (N = 35), and S4 (N = 21) based on marker genes in S1-4. Consistent with the ssGSEA results in TCGA, the S2 and S4 subtypes in CPTAC exhibited a high proliferative profile and the S3 subtype exhibited a high immune profile, this also deepened our conjecture that S3 may be immune infiltrating type (Fig. 4A). Because there was no significantly activated pathway in S1, subsequent studies were focused on protein regulation in S2, S3, and S4. GISTIC results showed the SCNA phenomenon of EGFR and CDKN2A in S2, S3, and S4. Specifically, CDKN2A had more CNV deletion, whereas there was more amplification in EGFR (Fig. 4B). Then we compared the protein abundance of EGFR and CDKN2A, and found that the protein abundance of CDKN2A was high in S2 and S3 but low in S4. The protein abundance of EGFR was low in S2 and S3 but high in S4 (Fig. 4C). Based on these results, we hypothesized that CNV loss of CDKN2A and CNV gain of EGFR in S4 may be one of the main reasons for the decreased expression of CDKN2A and the increased expression of EGFR.

Proteomics data analysis in CPTAC GBM. (A): Molecular subtypes of GBM in CPTAC GBM cohort based on marker genes. (B): Histogram of CNV for CDKN2A and EGFR information in S2-4. (C): Protein abundance of CDKN2 and EGFR in GBM subtypes. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
Our initial GSVA results indicated that S2 and S4 subtypes possessed a high proliferative profile and S3 possessed a high immune profile. To further investigate the potential biological differences, we performed a more in-depth characterization at the proteomic level. In the CPTAC cohort, CNV deletion of PD-L1 was more common in S3 and S4, but CNV amplification of PD-L1 was more common in S2. We also noticed the highest level of PD-L1 expression and phosphorylation in S3 (Fig. 5A). Among S2-S4, EGFR had the highest CNV gain in S4, EGFR had the highest CNV loss in S2, and EGFR had an intermediate CNV gain and CNV loss in S3, and, the highest levels of EGFR expression, protein abundance and phosphorylation were observed in S4 (Fig. 5B). In the TCGA cohort, the CNV phenomenon of PD-L1 in S2-S4 was very different from that of EGFR. The CNV rate of PD-L1 in S2 was low, and the CNV pattern in S4 was almost all CNV loss. EGFR had a higher CNV rate in S4 than in S2 and S3, and the CNV pattern in each subtype was CNV gain. S2 had the lowest expression levels of both PD-L1 and EGFR, the highest expression level of PD-L1 in S3, and the highest expression level of EGFR in S4. (Fig. 5C). Based on previous studies, we compared the expression patterns of Proliferation scores and effectors of T cells (IFN-gamma Response) in S1-4. High levels of Proliferation scores were observed in S2 and S4, and the lowest IFN-gamma Response scores were observed in S2 (Fig. 5D). The highest PRF1, GZMA protein abundance was also observed in S3 (Fig. 5E). Thus, there was heterogeneity among S2, S3 and S4 in the genomic features of immune molecules, which may lead to different responses to immunotherapy.

Genomic and proteomic analysis in the CPTAC GBM cohort. (A): PD-L1 expression, CNV, protein abundance, and phosphorylation levels in GBM subtypes. (B): EGFR expression, CNV, protein abundance, and phosphorylation levels in GBM subtypes. (C): PD-L1 and EGFR expression, CNV in TCGA-GBM cohort. (D): GBM subtype proliferation score and immune score in TCGA-GBM cohort. (E): Protein abundance of T cell effectors PRF1 and GZMA in the CPTAC GBM cohort.
S3 was a potential molecular subtype for immune checkpoint therapy response
A GBM sequencing cohort that received immunotherapy (Temozolomide treatment), GSE84010, was retrieved from the GEO database. Based on the expression matrix of marker genes, the samples could be classified as S1, S2, S3 and S4. The GSEA results were consistent with the trend of sample characteristics in the TCGA cohort, with S2 and S4 exhibiting high proliferative properties and S3 exhibiting high immune properties (Fig. 6A). In comparison with the subtypes identified by Verhaak et al.19, the majority of cases in S2 belonged to the Proneural subtype, the majority of cases in S3 belonged to the Mesenchymal subtype, and the majority of cases in S4 belonged to the Classical subtype (Fig. 6B). These results were highly consistent with in the TCGA cohort, further indicating that the novel subtypes of GBM in this study were robust. To identify the subtypes responding to immunotherapy, the TIDE score analysis was conducted. Due to the absence of significantly activated pathways in S1 and the absence of typical molecular typing features. We focused on the immunotherapy response in S2, S3, and S4. Samples in S4 had the highest TIDE scores, indicating the least sensitivity to immunotherapy (Fig. 6C). The lowest percentage of response to immunotherapy in S4 was also validated (Fig. 6D). In contrast, TIDE scores were lower in S3, indicating a greater benefit from taking immunotherapy (Fig. 6C, D). These results suggested that the S3 subtype was potentially a key molecular subtype for indicating response to immunotherapy in GBM. Since S4 had the lowest response rate to immunotherapy, we found the GBM cohort (GSE84010 dataset) treated with bevacizumab on the basis of radiotherapy/temozolomide, and analyzed the survival difference of the four subtypes after removing samples that could not be subclassified. We found that S4 had a significantly better prognosis in the bevacizumab group than in the other three subtypes, whereas in the placebo group, there was no significant difference in prognosis among the four subtypes, suggesting that S4 is the most suitable subtype for bevacizumab (Supplementary Fig. 6).

Immunotherapy response prediction. (A): Comparison of subtype classification and biological activity of samples in GSE84010. (B): Comparison of GBM subtypes with Proneural, Neural, Classical, and Mesenchymal subtypes in GSE84010. (C): TIDE scores of GBM subtypes in GSE84010. (D): Immunotherapy response rate of GBM subtypes in GSE84010.
Biomarkers for the identification of S2, S3 and S4 subtypes
Our results confirmed that the BayesNMF-based approach to construct a novel subtype classification in GBM was feasible. But as more than 400 marker genes were identified in all four subtypes by this analysis method, marker genes in the model required compression. Based on marker genes or marker proteins in S2, S3 and S4, we constructed LASSO logistic regression model in TCGA-GBM cohort and RPPA cohort to predict S2, S3 and S4 subtypes by selecting the model with the smallest prediction error rate depending on the smallest lambda value (Supplementary Table 4). In the TCGA-GBM cohort, the LASSO logistic regression model with 13 genes reached an accuracy of 96.7% in predicting the S2 subtype, the LASSO logistic regression model with 17 genes reached an accuracy of 86.7% in predicting the S3 subtype, and the LASSO logistic regression model with 14 genes reached an accuracy of 93.3% in predicting the S4 subtype. In the RPPA cohort, the LASSO logistic regression model with 17 marker proteins reached an accuracy of 84.6% in predicting the S2 subtype, and the LASSO logistic regression model with 18 marker proteins reached an accuracy of 84.6% in predicting the S4 subtype (Table 1). These results indicated that we could accurately predict S2, S3 and S4 subtypes using marker genes expression data or protein expression data.
Discussion
Previous GBM studies identified the presence of somatic mutations in the IDH gene and common deletions of 1p and 19q in chromosomes, which determined the relevant subtypes39. Verhaak et al.19 also identified four GBM subtypes (Proneural, Neural, Classical, Mesenchymal). In 2021, WHO updated the classification summary, refining the clinicopathological classification and molecular characterization of GBM1. However, GBM is an “immune-cold” tumor, and strong tumor heterogeneity and high tumor plasticity have been challenging to accurately identify molecular subtypes40,41,42,43,44. Unclear molecular mechanisms hinder effective treatment for GBM patients and prognosis prediction5.
In this study we identified four novel molecular subtypes of GBM, S1, S2, S3, and S4 by consensus hierarchical clustering based on the BayesNMF method. The identity of the classical subtype is defined by the most common genomic aberration in GBM. The majority of cases in S2 belonged to the Proneural subtype, the majority of cases in S3 belonged to the Mesenchymal subtype, and the majority of cases in S4 belonged to the Classical subtype. S1 was dominated by neural and classical subtypes, and the high proportion of these two subtypes prevented us from clearly defining S1. However, in terms of pathways enriched in S1, its metabolism is active and its proliferative activity is very low. S2 was enriched to IDH1 mutations, TP53 mutations and deletions. The most typical features in Proneural were point mutations in IDH1 and TP53 mutations and loss of heterozygosity19. High abundance of Dendritic Cells, Monocytes, and Neutrophils was observed in S3. Dendritic Cells were specialized immune cells that presented antigens and were important regulators in the innate immune response45. In an inflammatory environment, Monocytes induce Monocytes-derived Dendritic Cells (MoDC) production46. In GBM, Neutrophils regulate T cells abundance and tumor-associated macrophage/ Monocytes abundance47. Reduced Dendritic Cells activity induces T Cells depletion48. It could be noted that the high abundance of Dendritic Cells, Monocytes, and Neutrophils in S3 formed a pro-immune active environment. High levels of EGFR expression and CNV amplification were observed in Classical subtypes19, and EGFR amplification and high protein abundance were detected in S4. Highly proliferative and aggressive GBM in which wtEGFR is typically expressed is an essential cause of relapse after treatment49. We observed that S4 exhibited a higher proliferative profile in which proliferation-associated signaling pathways are activated. The obtained four molecular subtypes were compared with the classification of Verhaak et al., and it was found that each subtype provided by Verhaak was also specifically reflected in the new molecular subtypes, which also increased the rationality of this classification of GBM.
S3 was the highly immunocompetent subtype among the four subtypes, showing a high intensity of macrophage regulation, lymphocyte infiltration signature score, IFN-gamma response. Macrophages are the major immune cells that express PD-L1 in tumors, and tumors rich in PD-L1 TAM exhibit an activated immune state with high levels of immune-related gene expression that may contribute to ICI therapy50,51,52. Macrophages are the major immune cells that express PD-L1 in tumors, and tumors rich in PD-L1 TAM exhibit an activated immune state with high levels of immune-related gene expression that may contribute to ICI therapy53. High levels of IFN-γ-related gene expression signatures are an important feature of tumors that respond to PD-1 checkpoint blockade54. Moreover, immune score, adaptive immunity score and innate immunity score HLA molecules encoding MHC proteins also showed the highest levels in S3, indicating that the S3 had a higher immunogenicity and was more easily recognized by the immune system and elicits an immune response. Down-regulation of MHC class I molecules on tumor cells is an important mechanism of immune escape and acquired ICI resistance. Increasing the expression of MHC class I proteins helps to improve or restore anti-tumor cell immunity, so as to obtain clinical benefits55. These pieces of evidence all support a favorable response of S3 to ICI therapy. Our results obtained by TIDE analysis also did indicate that S3 had the highest response rate to ICI treatment. To unblock immune checkpoints in tumors, immune cross-presentation in the organism is crucial. Hammerich et al.56 noted that adsorption and induction of cross-initiated Dendritic Cells in tumors could enable anti-tumor killing T cell responses and immune checkpoint blockade. Intra-tumor Dendritic Cells enhance T cell responses and glioma rejection57. DCs in the TME play a key role in mediating the response to ICI drugs58. Quantitative expansion and activation of DCs promote the effectiveness of ICI treatment response59. A high abundance of Dendritic Cells was found in S3. The TIDE score indicated that the S3 subtype was less prone to immune escape after receiving immunotherapy.
Interestingly, the lowest TIDE score and the highest immunotherapy response were observed in S1. In our study, S1 did not have significantly activated biological pathways and showed low proliferative properties, which might account for its high response to immunotherapy.
We recognized some limitations of our study. The cohort studied presented predominantly white data, and the results may not necessarily apply to other races, and larger validation in more races is needed. GBM also has intratumor heterogeneity, which needs to be dissected by single-cell sequencing technology, which will facilitate a more complete and accurate intratumor classification. Subsequently, animal experiments are also a necessary step.
Collectively, we reported a new classification of GBM, which divided GBM into four subtypes, each with its own specific molecular features and showing varying degrees of response rates to immunotherapy. In addition, a prediction model with high accuracy was tailored for particularly important subtypes, providing a reference for specific and potentially targetable markers for subtype.
Data availability
The datasets generated during and/or analyzed during the current study are available in the GSE repository [GSE84010] (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc = GSE84010).
Abbreviations
- GBM:
-
Glioblastoma
- TCGA:
-
The Cancer Genome Atlas
- GISTIC:
-
Genomic identification of significant targets in cancer
- TIDE:
-
Tumor immune dysfunction and exclusion
- LASSO:
-
Least absolute shrinkage and selection operator
- WHO:
-
World Health Organization
- LGG:
-
Low-grade gliomas
- HGG:
-
High-grade gliomas
- CNV:
-
Copy number variation
- BayesNMF:
-
Bayesian non-negative matrix factorization
- CPTAC:
-
Clinical Proteomic Tumor Analysis Consortium
- TCPA:
-
The Cancer Proteome Atlas
- RNA-seq:
-
RNA sequencing
- RPPA:
-
Reverse phase protein array
- MSigDB:
-
Molecular signatures database
- ssGSEA:
-
Single sample gene set enrichment analysis
- SCNA:
-
Somatic copy-number alterations
- GEO:
-
Gene Expression Omnibus
- MoDC:
-
Monocytes-derived dendritic cells
References
Louis, D. N. et al. The 2021 WHO classification of tumors of the central nervous system: A summary. Neuro Oncol. 23(8), 1231–1251 (2021).
Kurdi, M. et al. Glioblastoma with PRMT5 gene upregulation is a key target for tumor cell regression. Oncologie 26(2), 239–246 (2024).
Ma, T. et al. Mesenchymal stem cell exosomes: A promising delivery system for glioma therapy. Oncologie 26(2), 179–193 (2024).
Gong, L. & Jia, M. ABCC8 is correlated with immune cell infiltration and overall survival in lower grade glioma. BIOCELL 47(1), 109–123 (2023).
McKinnon, C., Nandhabalan, M., Murray, S. A. & Plaha, P. Glioblastoma: Clinical presentation, diagnosis, and management. BMJ 374, n1560 (2021).
Kwapisz, D. Pembrolizumab and atezolizumab in triple-negative breast cancer. Cancer Immunol. Immunother. 70(3), 607–617 (2021).
Chaft, J. E. et al. Neoadjuvant atezolizumab for resectable non-small cell lung cancer: An open-label, single-arm phase II trial. Nat. Med. 28(10), 2155–2161 (2022).
DeCordova, S. et al. Molecular heterogeneity and immunosuppressive microenvironment in glioblastoma. Front. Immunol. 11, 1402 (2020).
Liu, Z. & Huang, X. A model based on eight iron metabolism-related genes accurately predicts acute myeloid leukemia prognosis. BIOCELL 47(3), 593–605 (2023).
Liu, X. P. et al. Clinical significance and molecular annotation of cellular morphometric subtypes in lower-grade gliomas discovered by machine learning. Neuro Oncol. 25(1), 68–81 (2023).
Collisson, E. A., Bailey, P., Chang, D. K. & Biankin, A. V. Molecular subtypes of pancreatic cancer. Nat. Rev. Gastroenterol. Hepatol. 16(4), 207–220 (2019).
Yin, L., Duan, J. J., Bian, X. W. & Yu, S. C. Triple-negative breast cancer molecular subtyping and treatment progress. Breast Cancer Res. 22(1), 61 (2020).
Yan, D. et al. Exploration of combinational therapeutic strategies for HCC based on TCGA HCC database. Oncologie 24(1), 101–111 (2022).
Jaaks, P. et al. Effective drug combinations in breast, colon and pancreatic cancer cells. Nature 603(7899), 166–173 (2022).
Wang, Z. et al. Construction and validation of a novel pyroptosis-related four-lncRNA prognostic signature related to gastric cancer and immune infiltration. Front. Immunol. 13, 854785 (2022).
Chehade, G. et al. Long-term survival in patients with IDH-wildtype glioblastoma: Clinical and molecular characteristics. Acta Neurochir. (Wien) 165(4), 1075–1085 (2023).
Munquad, S., Si, T., Mallik, S., Das, A. B. & Zhao, Z. A deep learning-based framework for supporting clinical diagnosis of glioblastoma subtypes. Front. Genet. 13, 855420 (2022).
Yang, K. et al. Glioma targeted therapy: Insight into future of molecular approaches. Mol. Cancer 21(1), 39 (2022).
Verhaak, R. G. et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 17(1), 98–110 (2010).
Zhu, Y. et al. High-resolution profiling of head and neck squamous cells carcinoma identifies specific biomarkers and expression subtypes of clinically relevant vulnerabilities. Curr. Med. Chem. https://doi.org/10.2174/0109298673276128231031112655 (2023).
Rooney, M. S., Shukla, S. A., Wu, C. J., Getz, G. & Hacohen, N. Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell 160(1–2), 48–61 (2015).
Shen, W. et al. Sangerbox: A comprehensive, interaction-friendly clinical bioinformatics analysis platform. iMeta 1(3), e36 (2022).
Meyers, R. M. et al. Computational correction of copy number effect improves specificity of CRISPR-Cas9 essentiality screens in cancer cells. Nat. Genet. 49(12), 1779–1784 (2017).
Tan, V. Y. & Fevotte, C. Automatic relevance determination in nonnegative matrix factorization with the beta-divergence. IEEE Trans. Pattern Anal. Mach. Intell. 35(7), 1592–1605 (2013).
Hanzelmann, S., Castelo, R. & Guinney, J. GSVA: Gene set variation analysis for microarray and RNA-seq data. BMC Bioinform. 14, 7 (2013).
Barbie, D. A. et al. Systematic RNA interference reveals that oncogenic KRAS-driven cancers require TBK1. Nature 462(7269), 108–112 (2009).
Song, Z. et al. CHDTEPDB: Transcriptome expression profile database and interactive analysis platform for congenital heart disease. Congenit. Heart Dis. 18(6), 693–701 (2023).
Thorsson, V. et al. The immune landscape of cancer. Immunity 48(4), 812–30 e14 (2018).
Schmidt, M. N. (ed.) Bayesian Non-Negative Matrix Factorization (Springer, Berlin Heidelberg, 2009).
Lawrence, M. S. et al. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature 505(7484), 495–501 (2014).
Lawrence, M. S. et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 499(7457), 214–218 (2013).
Mermel, C. H. et al. GISTIC2.0 facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancers. Genome Biol. 12(4), R41 (2011).
Jiang, P. et al. Signatures of T cell dysfunction and exclusion predict cancer immunotherapy response. Nat. Med. 24(10), 1550–1558 (2018).
Chen, B., Khodadoust, M. S., Liu, C. L., Newman, A. M. & Alizadeh, A. A. Profiling tumor infiltrating immune cells with CIBERSORT. Methods Mol. Biol. 1711, 243–259 (2018).
Simon, N., Friedman, J., Hastie, T. & Tibshirani, R. Regularization paths for cox’s proportional hazards model via coordinate descent. J. Stat. Softw. 39(5), 1–13 (2011).
Teo, W. Y. et al. Relevance of a TCGA-derived glioblastoma subtype gene-classifier among patient populations. Sci. Rep. 9(1), 7442 (2019).
Yoshihara, K. et al. Inferring tumour purity and stromal and immune cell admixture from expression data. Nat. Commun. 4, 2612 (2013).
Charoentong, P. et al. Pan-cancer immunogenomic analyses reveal genotype-immunophenotype relationships and predictors of response to checkpoint blockade. Cell Rep. 18(1), 248–262 (2017).
Horbinski, C., Berger, T., Packer, R. J. & Wen, P. Y. Clinical implications of the 2021 edition of the WHO classification of central nervous system tumours. Nat. Rev. Neurol. 18(9), 515–29 (2022).
Wen, P. Y. et al. Glioblastoma in adults: A Society for Neuro-Oncology (SNO) and European Society of Neuro-Oncology (EANO) consensus review on current management and future directions. Neuro Oncol. 22(8), 1073–1113 (2020).
Galon, J. & Bruni, D. Approaches to treat immune hot, altered and cold tumours with combination immunotherapies. Nat. Rev. Drug Discov. 18(3), 197–218 (2019).
Nduom, E. K. et al. PD-L1 expression and prognostic impact in glioblastoma. Neuro Oncol. 18(2), 195–205 (2016).
Hambardzumyan, D., Gutmann, D. H. & Kettenmann, H. The role of microglia and macrophages in glioma maintenance and progression. Nat. Neurosci. 19(1), 20–27 (2016).
Quail, D. F. & Joyce, J. A. The microenvironmental landscape of brain tumors. Cancer Cell. 31(3), 326–341 (2017).
Veglia, F. & Gabrilovich, D. I. Dendritic cells in cancer: The role revisited. Curr. Opin. Immunol. 45, 43–51 (2017).
Segura, E. & Amigorena, S. Identification of human inflammatory dendritic cells. Oncoimmunology 2(5), e23851 (2013).
Friebel, E. et al. Single-cell mapping of human brain cancer reveals tumor-specific instruction of tissue-invading leukocytes. Cell 181(7), 1626–42 e20 (2020).
Wang, J., Liu, P., Xin, S., Wang, Z. & Li, J. Nrf2 suppresses the function of dendritic cells to facilitate the immune escape of glioma cells. Exp. Cell Res. 360(2), 66–73 (2017).
Eskilsson, E. et al. EGFR heterogeneity and implications for therapeutic intervention in glioblastoma. Neuro Oncol. 20(6), 743–752 (2018).
Xiang, X., Wang, J., Lu, D. & Xu, X. Targeting tumor-associated macrophages to synergize tumor immunotherapy. Signal Transduct. Target. Ther. 6(1), 75 (2021).
Wang, D., Chen, H. & Hu, Y. Polarized autologous macrophages (PAM) can be a tumor vaccine. Oncologie 24(3), 441–449 (2022).
Xiao, Y., Jiang, J., Chen, Y., Huang, Y. & Wei, C. PD-1 relevant genes predict the prognosis of breast cancer and their prediction effect in tumor response to immunotherapy. Oncologie 24(4), 729–742 (2022).
Paijens, S. T., Vledder, A., de Bruyn, M. & Nijman, H. W. Tumor-infiltrating lymphocytes in the immunotherapy era. Cell Mol. Immunol. 18(4), 842–859 (2021).
Ayers, M. et al. IFN-gamma-related mRNA profile predicts clinical response to PD-1 blockade. J. Clin. Invest. 127(8), 2930–2940 (2017).
Rana, P. S., Ignatz-Hoover, J. J. & Driscoll, J. J. Targeting proteasomes and the MHC class I antigen presentation machinery to treat cancer, infections and age-related diseases. Cancers (Basel) https://doi.org/10.3390/cancers15235632 (2023).
Hammerich, L. et al. Systemic clinical tumor regressions and potentiation of PD1 blockade with in situ vaccination. Nat. Med. 25(5), 814–824 (2019).
Flores, C. T. et al. Lin(-)CCR2(+) hematopoietic stem and progenitor cells overcome resistance to PD-1 blockade. Nat. Commun. 9(1), 4313 (2018).
Cohen, M. et al. The interaction of CD4(+) helper T cells with dendritic cells shapes the tumor microenvironment and immune checkpoint blockade response. Nat. Cancer 3(3), 303–317 (2022).
Prokopi, A. et al. Skin dendritic cells in melanoma are key for successful checkpoint blockade therapy. J. Immunother. Cancer https://doi.org/10.1136/jitc-2020-000832 (2021).
Funding
This study was supported by Guangdong Province Medical Science and Technology Research Fund (A2024087).
Author information
Authors and Affiliations
Contributions
All authors contributed to this present work: [D.D.L.] & [A.P.L.] designed the study, [S.H.] acquired the data. [G.W.Y.] & [D.L.] drafted the manuscript, [H.L.Z.] and [B.P.] revised the manuscript. I certify that all authors commented on previous versions of the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Luo, D., Luo, A., Hu, S. et al. Genomics and proteomics to determine novel molecular subtypes and predict the response to immunotherapy and the effect of bevacizumab in glioblastoma. Sci Rep 14, 17630 (2024). https://doi.org/10.1038/s41598-024-68648-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-68648-5
- Springer Nature Limited