
Abstract
Clematis nannophylla is a perennial shrub of Clematis with ecological, ornamental, and medicinal value, distributed in the arid and semi-arid areas of northwest China. This study successfully determined the chloroplast (cp) genome of C. nannophylla, reconstructing a phylogenetic tree of Clematis. This cp genome is 159,801 bp in length and has a typical tetrad structure, including a large single-copy, a small single-copy, and a pair of reverse repeats (IRa and IRb). It contains 133 unique genes, including 89 protein-coding, 36 tRNA, and 8 rRNA genes. Additionally, 66 simple repeat sequences, 50 dispersed repeats, and 24 tandem repeats were found; many of the dispersed and tandem repeats were between 20–30 bp and 10–20 bp, respectively, and the abundant repeats were located in the large single copy region. The cp genome was relatively conserved, especially in the IR region, where no inversion or rearrangement was observed, further revealing that the coding regions were more conserved than the noncoding regions. Phylogenetic analysis showed that C. nannophylla is more closely related to C. fruticosa and C. songorica. Our analysis provides reference data for molecular marker development, phylogenetic analysis, population studies, and cp genome processes to better utilise C. nannophylla.
Similar content being viewed by others


Introduction
Clematis are herbaceous or woody vines of the family Ranunculaceae, with several erect shrubs or perennial herbs1. Clematis has high ornamental and medicinal value and belongs to the Ranunculaceae family2,3,4. Clematis is widely distributed worldwide, with approximately 300 species. It is mainly distributed in China, which has the richest plant resources4, with more than 100 species1,5. C. nannophylla is mainly distributed in arid and semi-arid mountain slope environments in Northwest China6 and has good stress tolerance. In addition, they possess important pharmaceutical, economic, and ecological properties. However, the systematic classification of Clematis poses challenges owing to their intricate nature and extensive morphological variability4. Currently, most studies have focused on morphological, physiological, ecological, and pharmacological activities7,8,9, whereas there are few basic molecular studies on germplasm resource identification, genetic breeding, resource conservation, and phylogeny. Furthermore, the chloroplast (cp) genome data of Clematis previously tested were submitted directly without detailed analysis, thus limiting our overall understanding of their phylogeny and genome evolution. However, studies on the endemic plant C. nannophylla are even fewer in northwestern arid and semi-arid areas of China, limiting the protection and development of this plant species. Therefore, it is imperative to better understand the taxonomic status and predict the future populations of C. nannophylla to guide more efficient germplasm resource utilisation, conservation, and breeding strategies.
As the organelles are involved in angiosperm photosynthesis, cps provide energy for plant metabolism. Cps are semi-autonomous genetic organelles that contain a unique genome and gene expression system10. Maternal inheritance of cp genomes is prevalent in most angiosperms. Nevertheless, in a minority of instances, it is inherited paternally or through a biparental mode11. Compared to the mitochondrial genome, cp genes exhibit greater stability in their genome structure and a heightened rate of evolution. Angiosperm cp genomes are known for their structural and sequence conservation12. The cp genome has a distinctive quadripartite structure consisting of a large single-copy (LSC) region and small single-copy (SSC) regions separated by a pair of long inverted repeat (IRa and IRb) regions10,13. Although the cp genome is conserved, recent studies have identified many genetic mutations in the cp genome, such as loss of gene or intron fragments, insertion and deletion of bases, changes in the length of reverse repeat regions, insertion/deletion of partial fragments, expansion or deletion of entire reverse repeat regions, and gene rearrangement14,15,16,17, which may lead to variations in plant structure and adaptation and contribute to plant species identification and future selective breeding18.
Furthermore, plant cp genomes encompass a substantial wealth of molecular data, serving as a valuable asset for plant systematics, population genomics, and phylogenetic investigations19; for instance, they can be employed in DNA barcoding, research on transplants, and the examination of population-level evolutionary patterns. Moreover, they offer valuable genetic indicators for establishing phylogenetic connections20,21,22. However, the cp genome of C. nannophylla has not yet been determined, and a comprehensive analysis of the cp genome structure of Clematis genus persists. There are numerous reports on the cp genome of Clematis23,24,25,26,27. However, few comparative analyses of cp genome structure are available28. The Clematis cp genomes were used as related species for studying other species29. Recent phylogenetic analyses using complete cp genome sequences have provided important insights into two small genera closely related to Clematis, Archiclematis and Naravelia, and have suggested that they should be included in Clematis30.
Therefore, to establish the taxonomic boundaries and phylogenetic relationships between C. nannophylla and other groups, we determined the cp genome characteristics of C. nannophylla. This study aimed to (1) obtain the complete sequence of the cp genome of C. nannophylla and (2) analyse the phylogenetic positions of 78 coding genes in C. nannophylla. (3) The coding and non-coding regions of the cp genome were compared between C. nannophylla and three other Clematis species, and the effective regions of the cp genome of C. nannophylla were determined. (4) Phylogenetic studies of the Clematis genus based on the complete cp genome and protein sequences have clarified the phylogenetic relationship and evolution of C. nannophylla.
Materials and methods
Plant material, DNA extraction, and genome sequencing
Healthy and mature leaves of C. nannophylla were sampled from Guide County (36° 7′ 19.92″ N, 101° 35′ 10.68″ E, Altitude: 2192.80 m), Qinghai Province, China, and preserved in liquid nitrogen for further study. Haifeng and Ying used macroscopic botanical identification methods to classify plant materials. Plant specimens were obtained from the College of Animal Science and Veterinary Science, Qinghai University, China, under voucher number QXYTXL220715. The leaves of C. nannophylla were conserved in Drikold, delivered to Genesky Biotechnologies Inc. for cp genome extraction and sequencing, and then assembled and further analysed by Genesky Biotechnologies Inc.
DNA extraction, sequencing, and assembly
Sample quality control
Firstly, Nanodrop was used to detect the concentration and purity of the sample, and the concentration was ≥ 20 ng/µL, the total amount was ≥ 100 ng, and OD260/OD280 = 1.8–2.2. The integrity of the DNA samples was tested by agarose gel electrophoresis, which required the main band of genomic DNA to be visible without evident degradation or dispersion.
Random DNA library construction
A random sequencing library was constructed using a transposable enzyme library-building kit. The library was constructed quickly and efficiently using transposition enzymes to randomly interrupt DNA and attach splices to both ends of the fragment.
PCR amplification of DNA libraries
A high-fidelity polymerase was used to amplify the original library to ensure a sufficient library volume in the sequencer. PCR was used to introduce a specific index and sequencing connectors at both ends of the library. The number of PCR amplification cycles was maintained between 12 and 15. The bias introduced by excessive amplification cycles was reduced to ensure sufficient product yield.
Size selection of library fragments
For enlarged libraries, fragment size screening was performed using the Agencourt SPRIselect fragment screening kit while purifying the libraries. A double-sized selection screening method was used in this study. First, the SPRI magnetic beads were used to remove the left side of the target area. The large fragment on the right-side size selection was removed, and a sequencing library with a fragment peak value of 300 bp was screened.
Library quality check
The sequencing library was then inspected and quantified. Qubit was used to accurately quantify the library concentration for the accurate mixing of samples to ensure the proper and balanced data volume of each sample. An Agilent 2100 Bioanalyzer was used to determine the size distribution of the library fragments and to evaluate their suitability for computer use.
Library pooling and sequencing
Qualified samples were diluted with an equal molar number of samples mixed in the machine. The library was sequenced using an Illumina HiSeq platform (Illumina, USA) with a 2 × 150 double-ended sequencing strategy.
Data quality assessment and assembly
Quality assessment of the original sequencing data was performed using FastQC software and R. To ensure high-quality sequencing data and enhance the accuracy of subsequent biological information analysis. The initial data underwent quality control and filtering based on specific criteria: (1) Removal of sequences containing more than 3 N bases; (2) Elimination of sequences with less than 60% high-quality bases (Phred score ≥ 20); (3) Trimming of low-quality bases at the 3′ end; (4) Exclusion of sequences shorter than 60 bp. After quality control, clean reads for C. nannophylla totalled 85,520,496 reads. As the sequences may include non-target sequences, they were assembled into contigs using metaSPAdes V3.13.0 software, resulting in 138 Contigs for C. nannophylla. Subsequent assembly analysis was conducted against the reference genome C. florida (NC058885) to assess contig formation, correct contig orientation, and to determine the starting base position.
Annotation and analysis of the cp genome sequences
According to the reference species (Clematis florida:NC_058885, Clematis fruticosa:NC_065273, Clematis tomentella:NC_065291, Clematis songorica:NC_065290), cps were annotated with CPGAVAS2 software, GenBank files were mapped with CPGView software (http://www.1kmpg.cn/cpgview/), the collinearity between the sample and the corresponding reference genome was analysed using BLAST V 2.9.0 software (https://blast.ncbi.nlm.nih.gov/Blast.cgi), and the collinearity results were analysed using Circos V 0.69-6 software.
SSRs (Simple Sequence Repeats) were analysed using the Perl script MISA V1.0 (https://webblast.ipk-gatersleben.de/misa/index.php), and the minimum number of repeats of mononucleotides, dinucleotides, trinucleotides, tetranucleotides, pentanucleotides, and hexanucleotides was set to 10, 5, 4, 3, 3, and 3, respectively31,32. Tandem repeats were identified using the Tandem Repeats Finder v. 4.09 (https://tandem.bu.edu/trf/submit_options)33. REPuter software (https://bibiserv.cebitec.uni-bielefeld.de/reputer) identified dispersed repeats, including forward (F), reverse (R), complement (C), and palindromic (P) match repeats, with a minimal length of 8 bp and a Hamming distance of 331,34.
Nucleotides A, T, C, and G were acquired using the CodonW program (version 1.4.2, available at https://sourceforge.net/projects/codonw/)33. To assess bias in nucleotide usage within the coding genes of C. nannophylla, we employed parity rule 2 (PR2) analysis. Mapping was performed using Origin 2021 Ink34.
Phylogenetic analysis
Combined with 32 previously reported Clematis plastomes, we constructed a phylogenetic tree using the newly sequenced C. nannophylla complete cp genome and 32 other cp genomes, including one family and two outgroups, downloaded from the NCBI for Biotechnology Information database. MAFFT (v7.313) was used for multiple sequence alignment35. Aligned complete cp genome sequence data were utilised to determine the optimal sequence model (ML) using MEGA 11 software, with the GTR + I + G model identified as the best model. Phylogenetic relationships were analysed using MEGA 11 and the Maximum Likelihood (ML) method was used to construct a phylogenetic tree with 1000 bootstraps33,34.
Genome structure comparison
Based on the above results of the phylogenetic analysis, the MVISTA format files of the four Clematis species were submitted to an online analysis tool for comparative cp genomes (mVISTA software, http://genome.lbl.gov/vista/mvista/submit.shtml) with the shuffle-LAGAN mode using the annotation of C. fruticosa as a reference34,36. The analysis of the IR boundaries in four Clematis cp genomes involved examining the expansion and contraction using the IRscope tool (https://irscope.shinyapps.io/IRapp/).
Adaptive evolution and phylogenetic analyses
Based on the cp genomes of C. nannophylla and the four other Clematis plants in this study, the Ka/Ks values for each functional protein-coding gene and the nucleotide diversity (Pi) values of the four Clematis cp genomes were calculated using DnaSP v6.0 software at default settings37. The Origin 2021 software was used to plot the data.
Ethics approval and consent to participate
The sampling of three newly sequenced C. nannophylla species was approved by Qinghai province of China and met local policy requirements. Our experimental research, including the collection of plant materials, are complies with institutional, national or international guidelines.
Results
Features of the C. nannophylla cp genome
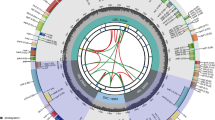
In total, 23,142,846 paired-end reads were obtained from the Illumina NovaSeq platform, with Q20 and Q30 values of 95.0% and 88.3%, respectively. The complete cp genome sequence of C. nannophylla was assembled de novo and uploaded to the NCBI for Biotechnology Information database (GenBank accession number: OQ581857). The circular cp genome of C. nannophylla is 158,091 bp in size (Fig. 1) and comprises an LSC (79,526 bp) region, two inverted repeat (IR, 31,045 bp) regions, and a small single-copy (SSC, 18,185 bp) region. The highest GC content was observed in the IR region (42.1%), whereas the lowest GC content was observed in the SSC region (31.3%); the average GC content of the whole genome was 38%.

Gene map of the C. nannophylla chloroplast genomes. From the center outward, the first track shows the dispersed repeats. The dispersed repeats consist of direct (D) and Palindromic (P) repeats. The second track shows the long tandem repeats as short blue bars. The third track shows the short tandem repeats or microsatellite sequences as short bars with different colors. The GC content along the genome is plotted on the fifth track. The base frequency at each site along the genome will be shown between the fourth and fifth tracks. The genes are shown on the sixth track.
There were 133 predicted functional genes in the C. nannophylla cp genome, including 89 protein-coding genes, 36 tRNA, and eight rRNA genes (Tables 1, 2). Protein-coding, tRNA, and rRNA genes accounted for 66.92%, 27.07%, and 6.02% of all annotated genes, respectively. Most genes and protein-coding genes were located in the LSC region, and only 9.02% were located in the SSC region.
Subsequently, we annotated all the assembled genes and their functions. These genes belong to four types: photosynthesis-related, self-replication-related, genes with unknown functions, maturases (matK), and proteases (clpP). A total of 22 annotated genes were double-copy genes, including 11 protein-coding genes, seven tRNAs, and four rRNAs. Sixteen genes (atpF, ndhA, ndhB, petB, petD, rpl16, rpl2, rpoC1, rps12, rps16, trnA-UGC, trnG-UCC, trnI-GAU, trnK-UUU, trnL-UAA and trnV-UAC) each contained one intron, whereas the protein-coding genes ycf3 and clpP contained two introns (Table 2). The longest intron (2,554 bp) was found in tunK-UUU, which completely encompassed matK, and the smallest intron (492 bp) was found in trnL-UAA.
PR2 plot mapping analysis was performed using the protein-coding gene sequences of C. nannophylla (Fig. 2), which were constructed to show the relationship between A3/(A3 + T3) and G3/(G3 + C3), and the data were distributed into four quadrants in a scatter diagram. Most genes were located in the second quadrant, with ribosomal protein SSU genes in the first quadrant (G > C, A > T) and photosystem II genes in the third quadrant (C > G, T > A).

PR2 analysis for genes in the cp genome of C. nannophylla. A3, T3, C3, and G3 represent nucleotide A, T, C, and G content at the third position of synonymous codons.
Codon usage bias
As each amino acid corresponds to at least one or up to six codons, codon use varies widely among organisms and species38, and this difference in synonymous codon usage is referred to as codon preference. Natural selection, species mutations, and genetic drift may cause bias in codon use. We selected a codon bias unique to the CDS genome, and the results showed that 26,795 amino acids were detected in the cp genome of C. nannophylla (Fig. 3), of which leucine was the most abundant with 2744 codons (10.2%), followed by isoleucine with 2350 codons (8.8%), serine and glycine with 2070 and 1851 codons (7.7% and 6.9%, respectively), and cysteine was the least abundant, with 214 codons (1.2%) and 30 (49.18%) preferred codons (RSCU > 1). Methionine and tryptophan had RSCU values equal to 1, but the preferred codon was TTA, which encodes leucine (Leu) with an RSCU value of 1.806.

Counting of relative synonymous codon usage (RSCU) of amino acids in the cp genome of C. nannophylla. The colors of the histogram correspond to the colors of the codons.
Detection of cp genome repeat sequences and SSRs
The abscissa represents SSR repetition units, and the ordinate represents the number of SSRs of each type. To learn the repeat sequence of the Clematis cp genome, four categories of repeat sequences were detected and analysed. There were no complementary repeats in Clematis (Fig. 4), and the number of repeats was highest in C. songorica (75) and lowest in C. florida (71). The number of discrete replicates of C. nannophylla was 74, second only to C. songorica. Forward, palindromic, and tandem repeats were most common. A total of 50 dispersed repeats were found in the C. nannophylla cp genome, including 22 forward, 21 palindromic, and seven reverse repeats, which were more than 20 bp in length in C. nannophylla, which is different from other Clematis species. The most dispersed and tandem repeats were 20–30 bp and 10–20 bp, respectively.

Repeated sequences of the C. nannophylla cp genome. The abscissa is the length of the repeat sequence, and the ordinate is the number of repeat sequences. F stands for forward repetition, P for palindromic repetition, R for reverse repetition, and C for complementary repetition.
We detected 66 SSRs in the C. nannophylla cp genome using the MISA Perl script (Fig. 5). The SSRs were mainly distributed in the LSC region (45, 68.18%), followed by the IR region (15, 22.73%). Additionally, 49 SSRs were located in intergenic spaces, and 17 SSRs were located in genes, such as matK, psbC, rpoB, rpoC2, clpP, petB, rps3, ndhA, trnV-UAC, rpl16, and ycf1. The SSRs consisted of 39 mononucleotides, eight dinucleotides, three trinucleotides, nine tetranucleotides, one hexanucleotide, and six complex nucleotide repeats. Moreover, oligo A and T repetitions accounted for 21.21% and 36.36% of the total SSRs, respectively, whereas oligo C and G repetitions were uncommon, and only one mononucleotide (G10) was detected in C. nannophylla.

Counting of the types of SSRs.
Comparison of complete Cp genomes
The cp genome sequences of C. nannophylla were analysed using the BLAST program on the NCBI Biotechnology Information website (http://www.ncbi.nlm.nih.gov/blast). The C. fruticosa plant, which is most similar to C. nannophylla, was selected for this study (Fig. 6). Therefore, the complete cp genomes of the five Clematis species were represented using the mVISTA program with C. fruticosa as the reference.

Sequence alignment of C. nannophylla chloroplast genomes. With C. florida as a reference. The y-axis indicates the percent identity between 50 and 100%. Genome regions colored represent protein coding regions, rRNA coding regions, tRNA coding regions or conserved noncoding sequences (CNS). White peaks indicate the regions with sequence variation among Clematis species.
The results showed that the cp genome of Clematis was highly conserved and that the LSC and SSC regions were more divergent than the IR regions. Furthermore, the coding regions were more conserved than the non-coding regions in our alignment, and the differences between C. nannophylla and C. fruticosa were not statistically significant. There was only one evident difference between trnE-UUC-trnT-GGU. However, there were many divergent regions in C. florida. These divergent regions mainly included psbA-atpA, atpI-rpoC2, rpoB-psbD, psbE-petG, clpP, and rpoC2, most of which were found in the intergenic regions. The most divergent coding regions were clpP and rpoC2, known as hotspot regions, because they contain variations such as single-nucleotide polymorphisms and indels, which can be used as molecular markers in DNA barcoding and phylogenetic analysis of C. nannophylla.
IR expansion and contraction
As a highly conserved region of the cp genome, the expansion and contraction characteristics of the IR region are mainly responsible for changes in cp genome size and rearrangement. Therefore, to compare IR expansion and contraction in the cp genome of C. nannophylla with those of the four Clematis plants, we analysed the border structure of C. nannophylla and the four reference Clematis cp genomes (Fig. 7).

Comparisons of LSC, SSC, and IR border regions in the chloroplast genome of five Clematis species.
The genes located in the binding regions of LSC/IRb, IRb/SSC, SSC/IRa, and IRa/LSC were rpl36, infA, ycf1, trnN, ndhF, ycf1, trnN, rps8, and rps16. The rpl36 and infA genes were located at the junctions of the LSC/IRb border. The rpl36 gene was located in the LSC region, and the infA gene of C. florida was located exclusively in the IR region, 20 bp away from the LSC/IRb border, whereas those of other Clematis species extended into the LSC regions.
trnN was completely located in the IRb region of C.nannophylla and C. florida and 72 bp from the IRb/SSC boundary. However, the ycf1 gene was found at the IRb/SSC boundary of the other three Clematis species (C. fruticose, C. tomentella, and C. songorica), which extended into the SSC regions; IRb/SSC extended into ndhF genes in all Clematis species, except for C. florida.
The distribution of ycf1 and trnN at the SSC/IRa boundary was the same in all five Clematis species. All ycf1 genes were embedded at the SSC/IRa border, with 3943 and 1697 bp located in the SSC and IRa regions, respectively. The trnN genes are all located in the IRa region, 72 bp away from the SSC/IRa boundary.
Except for C. florida, rps8 genes completely located in the IRa region were 311 bp away from the IRa/LSC boundary, whereas infA genes were found in C. florida completely located in the IRa region, 20 bp away from the IRa/LSC boundary. The distance between rps16 and the IRa/LSC boundary in the five Clematis species ranged from 1,193 to 1,200 bp. Based on these results, The IR, LSC, and SSC regions of C. nannophylla were found to be slightly different from those of the other four Clematis species at the boundary, and the numbers and sequences of the genes in these regions were conserved.
Adaptive evolution analysis
Using C. nannophylla as a reference, the selection patterns of protein-coding genes were determined by examining synonymous and non-synonymous substitutions in the cp genomes of five Clematis species. The Ka/Ks ratios of 78 protein-encoding genes were compared among the five cp genomes (Fig. 8). The Ka/Ks values of most coding genes were less than 1 or 0, which could not be calculated, indicating that they were relatively conserved. In particular, all genes of Clematis species had Ka/Ks values less than 1, except C. florida. However, the Ka/Ks values of ycf1 in C. nannophylla and C. florida were greater than one. The Ka/Ks ratios of ndhB, rpoCl, and ycf1 in C. nannophylla were similar to those of C. fruticosa, C. songorica, and C. tomentella.

The 79 protein-coding genes of the C. nannophylla cp genome and four clematis species were used for Ka/Ks analysis.
The nucleotide diversity (Pi) values of the cp genomes of C. nannophylla and four other Clematis plants (C. fruticose, C. tomentella, C. songorica, and C. florida) were calculated to determine the divergent hotspots (Fig. 9). The Pi values within 600 bp of the five Clematis cp genomes were calculated. The minimum and maximum values for the entire genome sequence ranged from 0 to 0.014, with an average value of 0.001416.

Comparative analysis nucleotide diversity based on the five clematis. x-axis: gene name, y-axis: Pi value.
However, some highly variable loci, including trnF-ndhJ, ndhE-ndhG, ndhF-rpl32, ccsA-ndhD, ccsA, ndhD, trnS-trnL, ndhF-rpl32, rps15, and ndhE, were located more precisely. All these regions had much higher values than the other regions (Pi > 0.007), and most of these higher-value regions were located in the SSC region. In the LSC region, there were a few loci with Pi values greater than 0.007, whereas the IR region had the lowest Pi value. All Pi values were less than 0.003, indicating that the IR regions are substantially more conserved. Based on these results, we believe that rpl32, ccsA, ndhD, rps15, and ndhE, which have relatively high sequence deviations, are good candidates for interspecies phylogenetic analysis.
Phylogenetic analysis
Cp genomes play a significant role in the phylogenetic relationships and evolutionary histories of plants. To determine the phylogenetic position of C. nannophylla within Ranunculaceae, a phylogenetic tree was constructed using the best-fit model, GTR + G + I (Fig. 10). The analysis included the complete cp genomes of 17 Clematis species and six outgroup genera (three Aconitum species, two Nymphaea species, two Ranunculus species, one Naravelia species, one Nuphar species, and one Magnolia species). The resulting phylogenetic tree consisted of 30 nodes, with 29 nodes having a bootstrap support value of ≥ 80% and 26 nodes having a support value of 90%, indicating the high reliability of the clustering results. The 21 plant species were divided into two large and seven small groups. Magnoliaceae and Nymphaeaceae formed one large group, whereas 28 Ranunculaceae species formed another group, with subgroups including Clematis, Ranunculus, and Aconitum. The analysis revealed that C. nannophylla shares high homology with C. fruticose and C. songorica but shows a distant relationship with other Clematis species. Additionally, within the family Ranunculaceae, Clematis and Aconitum were identified as highly credible sister groups.

Molecular phylogenetic tree of 32 plants including 22 Clematis species based on complete chloroplast genomes. The tree was constructed by maximum likelihood analysis using MEGA11 with a bootstrap test of 1000 replications.
Discussion
Cp genome structure and size of C. nannophylla
Cps are important organelles for photosynthesis and energy production and are essential for plant growth and development10. Cps have a unique genome and gene expression system that play a crucial role in metabolism as a source of energy that supports plant life39. The complete C. nannophylla cp genome showed great similarities to most angiosperms in terms of GC content and quadripartite architecture, including two inverted repeats (IRs), an LSC region and a small SSC, which is common in plants23,28,39.
Furthermore, the cp genome of C. nannophylla contained 133 genes (including 89 protein-coding genes, 36 tRNAs, and eight rRNAs), and the GC% content of the genome was 38%. High GC content often correlates with earlier phylogenetic differentiation (such as Nymphaeaceae and Magnoliaceae)40. Generally, the complete cp genome of C. nannophylla is very similar to other reported cp genomes of Clematis plants in terms of length, structure, and gene composition23,28,41. There was no evidence of rearrangement, and good collinearity was observed. Aligning the entire cp genome revealed that C. nannophylla cp genomes were relatively well conserved; therefore, we concluded that C. nannophylla differentiated earlier among Ranunculaceae.
Cp genome repeat sequence of C. nannophylla
Plants contain numerous replicates in their genomes. However, the number, size, type, and location of repeats among different plants34 and repeats in the cp genome have been widely used to identify mutation hotspots and determine plant evolutionary relationships42. Fifty dispersed repeats were found in C. nannophylla, including 22 forward, seven reverse, and 21 palindromic repeats. The number of dispersed repeats was the same as that in other species of Clematis, and most of these dispersed repeats were in the LSC region. Most dispersed repeats were 20–30 bp long, indicating that short repeats occurred more frequently than long repeats among the dispersed repeats of C. nannophylla. Tandem repeats are generally considered the primary cause of genomic rearrangement and expansions43. Tandem repeats of C. nannophylla range from 10 to 20 bp, with most tandem repeats located in intergenic spaces or intron regions and a few in the same gene region, ycf244.
Simple sequence repeats (SSR) usually consist of 1–6 nucleotide repeating units and have been recognised as important molecular markers for studying population variation45,46. As genetic information in the cp genome is inherited only from maternal progenitors, SSR in the cp genome are sensitive to population genetic effects47 and have been widely used in the study of population evolution and polymorphism48. SSR varied in number and type according to species; 66 SSR repeats were screened in C. nannophylla, and their distribution was mainly found in the LSC and SSC regions. The number of variation sites in the IR region is reduced, mainly in the single-copy region49. Among the mononucleotide SSR repeats, the number of A/T mononucleotide repeats was significantly higher than that of G/C mononucleotide repeats. This pattern also exists in other angiosperms44,50. The dispersed tandem and SSR repeats identified above are responsible for cp genome rearrangement, gene replication, and gene expression; play a vital role in genomic rearrangement and sequence variation in cp genomes; and are helpful for phylogenetic studies. Rearrangements or sequence variations in these repeat units may also lead to substitutions, insertions, and deletions in the cp genome17,51,52. These repeat sequences have also been shown to be a source of information for the development of markers that play an important role in population and phylogenetic studies44 and can be used for future genetic structure, differentiation, and species identification of C. nannophylla. Therefore, they are a source of information for the development of markers and play an important role in population and phylogenetic studies44 for future genetic structure, differentiation, and species identification of C. nannophylla.
Codon usage bias in the cp genome of C. nannophylla
Codon usage bias is an important feature of genomic evolution and is of great significance in the study of molecular and exogenous gene expression53. PR2 further confirmed that most genes in C. nannophylla favour T and G in the coding chain rather than A and C and that the direct cause of this base asymmetry is the replication mechanism. However, asymmetry between coding and non-coding strands is an important cause of nucleotide skew54. However, the influence of replication mechanisms on base bias differed between the AT and CG asymmetries. Replication is generally strong for GC skew, whereas AT skew is caused by coding sequence-related mechanisms54,55.
Codon usage patterns are evolutionary features of the genome. In plants, codon usage bias is related to gene expression and is mainly affected by natural selection and mutation pressure, with differences between species56. In the cp genome of C. nannophylla, there are 30 high-frequency codons (RSCU > 1); leucine is the most important amino acid, and cysteine is the least important, consistent with the codons observed in other higher plants53,57,58. The use of synonymous codons is not random, and the analysis of codon preferences can provide valuable information for understanding species adaptation and molecular evolution.
Comparative genomic analysis of the cp genome of C. nannophylla
The IR regions of angiosperm cp genomes are highly conserved. The expansion and contraction of the IR region boundaries are common evolutionary events in most angiosperms that may lead to variations in cp genome length, gene replication or reduction, and the origin of pseudogenes59,60. This study found that the IR expansion and contraction of C. nannophylla showed great similarity with those other plants of Clematis, and these regional genotypes and distribution locations are similar38. However, only minor differences are observed near the IRb/SSC boundaries. trnN was not ycf1 at the IRb/SSC boundary of C. nannophylla and C. florida, and infA was not observed near the IRa/LSC boundary, possibly the result of contraction and expansion of the IR region; this is also an important reason for the differences in cp genome length33. The infA gene is transcribed as a polycistronic mRNA, a component of the ribosomal protein (rpl23) operon, while the ycf1 gene is a functional gene that encodes essential products for cell survival61. Therefore, the loss (or pseudogenisation) of infA and ycf1 may have resulted from gene transfer to the nucleus. However, there is no evidence suggesting that infA and ycf1 are transferred from the cp genome to the nuclear genome of Clematis. Further studies on the transcriptomes of these two genes are required to elucidate the effects of length variation on Clematis.
Owing to the highly conserved structure and nucleotide content of cp genomes, mutation hotspots of cp genomes can be quickly and accurately identified by comparative analysis. Therefore, mutation hotspots are often used as a basis for highly variable markers (DNA barcodes) in population genetics and phylogenetic studies62,63. In this study, we compared the cp genome structures of five Clematis species using mVISTA (with C. fruticosa as a reference) and found that the non-coding region was more prone to mutations than the coding region. Furthermore, variation in the SC region was higher than that in the IR region, similar to the results of previous plant studies28,62,64. psbA-atpA, atpI-rpoC2, rpoB-psbD, psbE-petG, clpP, and rpoC2 were the most highly variable regions detected in C. nannophylla. To determine the degree of variation in these highly variable regions in C. nannophylla, nucleotide variability in DNASP v6 was used to identify differences among the cp genomes of Clematis and mutation hotspots. Nucleotide diversity (Pi) indicates the degree of variation in the nucleic acid sequences of each species, and sites with high variability can be selected as molecular markers for population genetics33,65. In the present study, the nucleotide diversity analysis showed that the gene sequences in the LSC and SSC regions were more variable than those in the IR regions, which is consistent with the results found in Asteraceae and Fagaceae plants33,66.
By analysing the cp genome sequence variation of five Clematis species, we identified 13 hypervariable regions (Pi > 0.006) in the LSC and SSC regions, which is of great significance for the study of molecular barcodes. Highly variable regions, such as ndhF, ccsA, and ndhD, have also been found in two Korean endemic Clematis species28. Simultaneously, the same highly variable regions, ccsA and rpl32, were found in Fagus longipetiolata of Fagaceae. The ccsA gene is also considered the locus for understanding cp genome evolution in Fagus longipetiolata of Fagaceae33, Litsea65, Pterocarpus62, and Prosopis genera67. Furthermore, the Pi values of the 13 height-variable regions in this study were greater than 0.006, corresponding to the height-variable regions. Overall, these highly diverse regions provide a wealth of information for the development of molecular markers for the identification of Clematis species as well as for the analysis of the phylogenetic relationships and population genetics of C. nannophylla.
Adaptive evolution analysis of the cp genome of C. nannophylla
By comparing C. nannophylla with four other species of Clematis, we detected the protein-coding region genes in C. nannophylla under selection pressure. Ka/Ks is generally used to express the selection pressure on protein-coding genes68. When Ka/Ks was greater than 1, it indicated a positive selection effect, and when Ka/Ks was less than 1, it indicated a purification selection effect69. In this study, the Ka/Ks values of most genes in C. nannophylla were less than 1 compared to those of the other four plants, indicating that purification selection played an important role in the cp genomes of the five Clematis species. However, only the Ka/Ks of the ycf1 (C. nannophylla and C. florida) genes was greater than 1, indicating that the ycf1 gene was selected for adaptation to the living environment; ycf1 was also positively selected in previous studies45. The ycf1 gene, the largest gene in the cp and the most potent cp DNA barcode, encodes the ATP-binding (ABC) protein in the cp. ycf1 is characterised by species specificity61,70, rapid mutation rate, and rapid evolution69, and has been verified to have classification potential at the subgenus level. In C. nannophylla, regions with high purification selectivity were mainly distributed in self-replication (proteins of large ribosomal subunits and subunits of RNA polymerase), photosystem genes (subunits of the photosystem and NADH dehydrogenase), other genes, and unknown genes (ycf), similar to the evolution of cp genes in Pterocarpus, Artemisia maritima, and Artemisia absinthium62,66, suggesting that strong purification selection preserves specific gene residues and gene functions in these species. Compared to the other four Clematis species, the Ka/Ks of ndhB of C. nannophylla was approximately 0.9, which was significantly higher than that of the other genes. ndh is thought to be positively selected for species at relatively high altitudes71, which may be due to the higher elevation of the distribution area of C. nannophylla compared to the remaining four species of clematis.
Phylogenetic analysis of the cp genome of C. nannophylla
Cp genomes contain a large amount of genetic information that is useful for inferring evolutionary and phylogenetic relationships72. Many researchers have used complete cp genome sequences to resolve phylogenetic relationships at various taxonomic levels, and a strong phylogenetic tree can intuitively represent the relatedness of species and evolutionary relationships at various scales. In the present study, we reconstructed a phylogenetic tree with the complete cp genomes of 32 species using the ML method, with six outgroups. The results showed that C. nannophylla was more closely related to C. fruticosa and C. songorica but less closely related to C. florida, which is consistent with the results of the classification based on morphological characteristics. C. nannophylla, C. fruticosa, C. tomentella, and C. songarica belonged to sect. Fruticella, whereas C. florida belongs to sect. Viticella belongs to the Clematis group6. The present study also shows that Clematis is monophyletic and divides into two large subclades, and Clematis forms sister relationship with Aconitum41.
Conclusion
In summary, the complete cp genome of C. nannophylla was sequenced and compared with those of other related species, providing an important reference for the phylogeny of C. nannophylla. Although the cp genomes of C. nannophylla were identical to those of other Clematis species in terms of genome structure, gene content, and GC content, there were some differences in the boundaries of the IR region. Nucleotide diversity analysis indicated hotspots in the LSC and SSC regions of the cp genes in C. nannophylla, which could provide informative markers for the phylogenetic analysis of C. nannophylla. Purification selection played an important role in the cp genomes of five Clematis species, whereas ycf1 was positively selective (C. nannophylla and C. florida). Phylogenetic analysis showed that C. nannophylla is closely related to C. fruticosa, C. tomentella, and C. songarica, and the well-resolved phylogenetic tree showed the monophyletic origin of the genera Clematis and Aconitum as sister genera. The cp genome information obtained in this study provides reference data for molecular marker development, phylogenetic analysis, population studies, and cp genome processing, as well as for better exploitation and utilisation of C. nannophylla. These results can guide more efficient germplasm resource utilisation, conservation, and breeding strategies.
Data availability
All annotated chloroplast genomes have been deposited in GenBank (https://www.ncbi.nlm.nih.gov/genbank/), accession number OQ581857.
Abbreviations
- Cp:
-
Chloroplast
- LSC:
-
Large single copy
- SSC:
-
Small single copy
- IR:
-
Inverted repeat
- IRa:
-
Inverted repeat a
- IRb:
-
Inverted repeat b
- SSR:
-
Simple Sequence Repeat
- ML:
-
Maximum likelihood
- Ka:
-
Non-synonymous substitutions
- Ks:
-
Synonymous substitutions
- Pi:
-
Nucleotide diversity
References
Hu, Q. et al. Physiological and gene expression changes of Clematis crassifolia and Clematis cadmia in response to heat stress. Front. Plant Sci. 12, 624875. https://doi.org/10.3389/fpls.2021.624875 (2021).
Hao, D.C. et al. Chemical and biological research of clematis medicinal resources. Chin. Sci. Bull. 58, 1120–1129. https://doi.org/10.1007/s11434-012-5628-7 (2013).
Li, R. et al. Dose-response characteristics of Clematis triterpenoid saponins and clematichinenoside AR in rheumatoid arthritis rats by liquid chromatography/mass spectrometry-based serum and urine metabolomics. J. Pharm. Biomed. Anal. 136, 81–91. https://doi.org/10.1016/j.jpba.2016.12.037 (2017).
Liu, D. et al. Complete sequence and comparative analysis of the mitochondrial genome of the rare and endangered Clematis acerifolia, the first clematis mitogenome to provide new insights into the phylogenetic evolutionary status of the genus. Front. Genet. 13, 1050040. https://doi.org/10.3389/fgene.2022.1050040 (2023).
Qian, R. et al. Metabolomic and Transcriptomic Analyses Reveal New Insights into the Role of Metabolites and Genes in Modulating Flower Colour of Clematis tientaiensis. Horticulturae 9, 14. https://doi.org/10.3390/horticulturae9010014 (2023).
Committee of the Flora of China,1980. Chinese Academy of Sciences. Flora of China. Vol. 28. Ranunculaceae (2), Dicotyledonous Plant Class, Angiosperma Phylum. (Science Press, 1980).
Lyu, R. et al. Phylogeny and Historical Biogeography of the East Asian Clematis Group, Sect. Tubulosae, Inferred from Phylogenomic Data. Journal of Molecular Science 24, 3056. https://doi.org/10.3390/ijms24033056 (2023).
Zhao, X. et al. Variation of the floral traits and sexual allocation patterns of Clematis tangutica to the altitudinal gradient of the eastern Qinghai-Tibet Plateau. Biologia 78, 55–65. https://doi.org/10.1007/s11756-022-01178-5 (2023).
Teshome, N. et al. Evaluation of Wound Healing and Anti-Inflammatory Activity of Hydroalcoholic Leaf Extract of Clematis simensis Fresen (Ranunculaceae). Clinical, Cosmetic and Investigational Dermatology 15, 1883–1897. https://doi.org/10.2147/CCID.S384419 (2022).
Daniell, H., Lin, C.S., Yu, M. & chang, W.J. Chloroplast genomes: Diversity, evolution, and applications in genetic engineering. Genome Biology 17, 134. https://doi.org/10.1186/s13059-016-1004-2 (2016).
Hu, Y.C. et al. Occurrence of plastids in the sperm cells of Caprifoliaceae: Biparental plastid inheritance in angiosperms is unilaterally derived from maternal inheritance. Plant Cell Physiol. 49, 958–968. https://doi.org/10.1093/pcp/pcn069 (2008).
Huang, Y., Wang, J., Yang, Y.P., Fan, C.Z. & Chen, J.H. Phylogenomic analysis and dynamic evolution of chloroplast genomes in Salicaceae. Front. Plant Sci. 8, 1050. https://doi.org/10.3389/fpls.2017.01050 (2017).
He, X.Y. et al. The complete chloroplast genome of Carpesium abrotanoides L. (Asteraceae): Structural organization, comparative analysis, mutational hotspots and phylogenetic implications within the tribe Inuleae. Biologia 77(7), 1861–1876. https://doi.org/10.1007/s11756-022-01038-2 (2022).
Blazier, J.C. et al. Divergence of RNA polymerase α subunits in angiosperm plastid genomes is mediated by genomic rearrangement. Sci. Rep. 6, 24595. https://doi.org/10.1038/srep24595 (2016).
Guisinger, M.M., Kuehl, J.V., Boore, J.L. & Jansen, R.K. Extreme reconfiguration of plastid genomes in the angiosperm family Geraniaceae: Rearrangements, repeats, and codon usage. Mol. Biol. Evol. 28, 583–600. https://doi.org/10.1093/molbev/msq229 (2011).
Chumley, T.W. et al. The complete chloroplast genome sequence of Pelargonium × hortorum: Organization and evolution of the largest and most highly rearranged chloroplast genome of land plants. Mol. Biol. Evol. 23, 2175–2190. https://doi.org/10.1093/molbev/msl089 (2006).
Abdullah, M.F. et al. Correlations among oligonucleotide repeats, nucleotide substitutions, and insertion-deletion mutations in chloroplast genomes of plant family Malvaceae. J. Syst. Evol. 2, 388–402. https://doi.org/10.1111/jse.12585 (2021).
Shen, X.F. et al. Complete chloroplast genome sequence and phylogenetic analysis of the medicinal plant Artemisia annua. Molecules 22, 1330. https://doi.org/10.3390/molecules22081330 (2017).
Yu, X.L. et al. Complete chloroplast genomes of Ampelopsis humulifolia and Ampelopsis japonica: Molecular structure, comparative analysis, and phylogenetic analysis. Plants 8(10), 410. https://doi.org/10.3390/plants8100410 (2019).
Choi, K.S., Chung, M.G. & Park, S. The complete chloroplast genome sequences of three Veroniceae Species (Plantaginaceae): Comparative analysis and highly divergent regions. Front. Plant Sci. 7, 355. https://doi.org/10.3389/fpls.2016.00355 (2016).
Li, B. et al. Development of chloroplast genomic resources for Akebia quinata (Lardizabalaceae). Conserv. Genet. Resour. 8, 447–449. https://doi.org/10.1007/s12686-016-0593-0 (2016).
Wang, L., Wuyun, T.N., Du, H.Y., Wang, D.P. & Cao, D.M. Complete chloroplast genome sequences of Eucommia ulmoides: Genome structure and evolution. Tree Genet. Genomes 12, 12. https://doi.org/10.1007/s11295-016-0970-6 (2016).
Park, B.K., Ghimire, B., Ha, Y.H., Son, D.C. & Kim, D.K. Complete chloroplast genome of Clematis taeguensis (Ranunculaceae), an endemic species from South Korea. Mitochondrial DNA Part B 6(4), 1496–1497. https://doi.org/10.1080/23802359.2021.1910080 (2021).
Guo, S., Liu, Y., Li, Z., He, M. & Wu, W. The complete chloroplast genome sequence of Clematis chinensis Osbeck. Mitochondrial DNA Part B 7(11), 2015–2017. https://doi.org/10.1080/23802359.2022.2148823 (2022).
Zhang, R. et al. Characterization of the complete chloroplast genome of Clematis potaninii (Ranunculaceae), a medicinal and ornamental plant. Mitochondrial DNA Part B 7(7), 1273–1274. https://doi.org/10.1080/23802359.2022.2097023 (2022).
Yang, Y.C., Wang, N., Zhang, W. & Zhou, T. The complete chloroplast genome of Clematis fruticosa Turcz. (Ranunculaceae). Mitochondrial DNA Part B 5(2), 1908–1909. https://doi.org/10.1080/23802359.2020.1754951 (2020).
Dong, Y., Zhu, Q. & Yue, J. The complete chloroplast genome of Clematis florida Thunb. (Ranunculaceae), an ornamental and medicinal plant from Henan province, China. Mitochondrial DNA Part B 7(3), 471–473. https://doi.org/10.1080/23802359.2022.2049460 (2022).
Choi, K.S. et al. Two Korean endemic Clematis chloroplast genomes: Inversion, reposition, expansion of the inverted repeat region, phylogenetic analysis, and nucleotide substitution rates. Plants 10, 397. https://doi.org/10.3390/plants10020397 (2021).
Liu, H. et al. Comparative analysis of complete chloroplast genomes of Anemoclema, Anemone, Pulsatilla, and Hepatica revealing structural variations among genera in tribe Anemoneae (Ranunculaceae). Frontiers in plant science 9, 1097. https://doi.org/10.3389/fpls.2018.01097 (2018).
Liu, H.-J. et al. Complete chloroplast genomes of Archiclematis, Naravelia and Clematis (Ranunculaceae), and their phylogenetic implications. Phytotaxa 343, 214–226. https://doi.org/10.11646/phytotaxa.343.3.2 (2018).
Kurtz, S. et al. Versatile and open software for comparing large genomes. Genome Biol https://doi.org/10.1186/gb-2004-5-2-r12 (2004).
Sun, C.Q., Chen, F.D., Teng, N.J., Xu, Y.C. & Dai, Z.L. Comparative analysis of the complete chloroplast genome of seven Nymphaea species. Aquat. Bot. 170(1), 103353. https://doi.org/10.1016/j.aquabot.2021.103353 (2021).
Liang, D.Q., Wang, H.Y., Zhang, J. Zhao, Y.X. & Wu, F. Complete chloroplast genome sequence of Fagus longipetiolata Seemen (Fagaceae): Genome structure, adaptive evolution, and phylogenetic relationships. Life 12, 92. https://doi.org/10.3390/life12010092 (2022).
Cui, G.X. et al. Complete chloroplast genome of Hordeum brevisubulatum: Genome organization, synonymous codon usage, phylogenetic relationships, and comparative structure analysis. PLoS ONE 16(12), e0261196. https://doi.org/10.1371/journal.pone.0261196 (2021).
Katoh, K. & Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. https://doi.org/10.1093/molbev/mst010 (2013).
Yan, C., Du, J.C., Gao, L., Li, Y. & Hou, X.L. The complete chloroplast genome sequence of watercress (Nasturtium officinale R. Br.): Genome organization, adaptive evolution and phylogenetic relationships in Cardamineae. Gene 699, 24–36. https://doi.org/10.1016/j.gene.2019.02.075 (2019).
Asaf, S. et al. Complete chloroplast genome of Nicotiana otophora and its comparison with related species. Front. Plant Sci. 7, 843. https://doi.org/10.3389/fpls.2016.00843 (2016).
Kovalenko, S.P. On the origin of genetically coded protein synthesis. Russ. J. Bioorg. Chem. 47, 1201–1219. https://doi.org/10.1134/S1068162021060121 (2021).
Dobrogojski, J., Adamiec, M. & Luciński, R. The chloroplast genome: A review. Acta Physiologiae Plantarum 42, 98. https://doi.org/10.1007/s11738-020-03089-x (2020).
Cai, Z.Q. et al. Complete plastid genome sequences of drimys, liriodendron, and piper: Implications for the phylogenetic relationships of Magnoliids. BMC Evol. Biol. 6, 77. https://doi.org/10.1186/1471-2148-6-77 (2006).
Park, I. et al. The complete chloroplast genome sequence of Aconitum coreanum and Aconitum carmichaelii and comparative analysis with other Aconitum species. PLoS ONE 12(9), e0184257. https://doi.org/10.1371/journal.pone.0184257 (2017).
Powell, W., Morgante, M., McDevitt, R., Vendramin, G.G. & Rafalski, J.A. Polymorphic simple sequence repeat regions in chloroplast genomes: Applications to the population genetics of pines. Proc. Natl. Acad. Sci. 92(17), 7759–7763. https://doi.org/10.1073/pnas.92.17.7759 (1995).
Zhao, Y.M., Zhang, X., Zhou, T., Chen, X.D. & Bing, B. Complete chloroplast genome sequence of Gynostemma guangxiense: Genome structure, codon usage bias, and phylogenetic relationships in Gynostemma (Cucurbitaceae). Braz. J. Bot. 46, 351–365. https://doi.org/10.1007/s40415-023-00874-z (2023).
Zhai, Y.Y. et al. Phylogenomic, phylogeography and germplasms authentication of the Rheum palmatum complex based on complete chloroplast genomes. J. Plant Res. 136, 291–304. https://doi.org/10.1007/s10265-023-01440-0 (2023).
Niu, Y.F., Su, T., Wu, C.H., Deng, J. & yang, F.Z. Complete chloroplast genome sequences of the medicinal plant Aconitum transsectum (Ranunculaceae): Comparative analysis and phylogenetic relationships. BMC Genomics 24, 90. https://doi.org/10.1186/s12864-023-09180-0 (2023).
Huang, S.N., Ge, X.J., Cano, A., Salazar, B.G.M. & Deng, Y.F. Comparative analysis of chloroplast genomes for five Dicliptera species (Acanthaceae): Molecular structure, phylogenetic relationships, and adaptive evolution. Peer J. 8, e8450. https://doi.org/10.7717/peerj.8450 (2020).
Jiang, M. et al. Sequencing, characterization, and comparative analyses of the plastome of Caragana rosea var. rosea. Int. J. Mol. Sci. 19, 1419. https://doi.org/10.3390/ijms19051419 (2018).
Jeong, Y.M., Chung, W.H., Mun, J.H., Kim, N. & Yu, H.J. De novo assembly and characterization of the complete chloroplast genome of radish (Raphanus sativus L.). Gene 551, 39–48. https://doi.org/10.1016/j.gene.2014.08.038 (2014).
Luo, Y.K. et al. Comparative analysis of complete chloroplast genomes of 13 species in Epilobium, Circaea, and Chamaenerion and insights into phylogenetic relationships of onagraceae. Front. Genet. 12, 730495. https://doi.org/10.3389/fgene.2021.730495 (2021).
Wu, M.L., Yan, R.R., Xu, X., Gou, G.Q. & Dai, Z.X. Characterization of the plastid genome of the vulnerable endemic Indosasa lipoensis and phylogenetic analysis. Diversity 15(2), 197. https://doi.org/10.3390/d15020197 (2023).
Do, H.D.K. & Kim, J.H. A dynamic tandem repeat in monocotyledons inferred from a comparative analysis of chloroplast genomes in Melanthiaceae. Front. Plant Sci. 8, 693. https://doi.org/10.3389/fpls.2017.00693 (2017).
Wang, M.L. et al. Phylogenomic and evolutionary dynamics of inverted repeats across Angelica plastomes. BMC Plant Biol. 21(1), 26. https://doi.org/10.1186/s12870-020-02801-w (2021).
Wang, Y.Z. et al. Comparative analysis of codon usage patterns in chloroplast genomes of ten Epimedium species. BMC Genomic Data 24, 3. https://doi.org/10.1186/s12863-023-01104-x (2023).
Mrázek, J. & Karlin, S. Strand compositional asymmetry in bacterial and large viral genomes. Proc. Natl. Acad. Sci. USA 95, 3720–3725. https://doi.org/10.1073/pnas.95.7.3720 (1998).
Romiguier, J. & Roux, C. Analytical biases associated with GC-content in molecular evolution. Front. Genet. 8, 16. https://doi.org/10.3389/fgene.2017.00016 (2017).
Sheng, J., She, X., Liu, X., Wang, J. & Hu, Z. Comparative analysis of codon usage patterns in chloroplast genomes of five Miscanthus species and related species. PeerJ 9, e12173. https://doi.org/10.7717/peerj.12173 (2021).
Wang, Z.J. et al. Comparative analysis of codon Bias in the chloroplast genomes of Theaceae species. Front. Genet. 13, 824610. https://doi.org/10.3389/fgene.2022.824610 (2022).
Li, G., Zhang, L. & Xue, P. Codon usage pattern and genetic diversity in chloroplast genomes of Panicum species. Gene. 802, 145866. https://doi.org/10.1016/j.gene.2021.145866 (2021).
Jiang, D.Z. et al. Complete chloroplast genomes provide insights into evolution and phylogeny of Zingiber (Zingiberaceae). BMC Genomics 24, 30. https://doi.org/10.1186/s12864-023-09115-9 (2023).
Bai, X.J., Wang, G., Ren, Y., Su, Y.Y. & Han, J.P. Insights into taxonomy and phylogenetic relationships of eleven Aristolochia species based on chloroplast genome. Front. Plant Sci. 14, 1119041. https://doi.org/10.3389/fpls.2023.1119041 (2023).
Drescher, A., Ruf, S., Calsa, T. Carrer, H. & Bock, R. The two largest chloroplast genome-encoded open reading frames of higher plants are essential genes. Plant J. 22(2), 97–104. https://doi.org/10.1046/j.1365-313x.2000.00722.x (2010).
Hong, Z. et al. Comparative analyses of five complete chloroplast genomes from the genus Pterocarpus (Fabacaeae). Int. J. Mol. Sci. 21, 3758. https://doi.org/10.3390/ijms21113758 (2020).
Abdullah, M.F. et al. Chloroplast genome of Hibiscus rosa-sinensis (Malvaceae): Comparative analyses and identification of mutational hotspots. Genomics 112, 581–591. https://doi.org/10.1016/j.ygeno.2019.04.010 (2020).
Liu, H.Y. et al. The chloroplast genome of Lilium henrici: Genome structure and comparative analysis. Molecules 23(6), 1276. https://doi.org/10.3390/molecules23061276 (2018).
Zhang, Y.Y. et al. Comparative chloroplast genomics of Litsea Lam. (Lauraceae) and its phylogenetic implications. Forests 12, 744. https://doi.org/10.3390/f12060744 (2021).
Shahzadi, I., Abdullah, M.F., Ali, Z., Ahmed, I. & Mirza, B. Chloroplast genome sequences of Artemisia maritima and Artemisia absinthium: Comparative analyses, mutational hotspots in genus artemisia and phylogeny in family Asteraceae. Genomics 112(2), 1454–1463. https://doi.org/10.1016/j.ygeno.2019.08.016 (2019).
Asaf, S., Khan, A. L., Khan, A. & Al-Harrasi, A. Unraveling the chloroplast genomes of two prosopis species to identify its genomic information, comparative analyses and phylogenetic relationship. Int. J. Mol. Sci. 21, 3280. https://doi.org/10.3390/ijms21093280 (2020).
Lohmueller, K.E. et al. Natural selection affects multiple aspects of genetic variation at putatively neutral sites across the human genome. PLoS Genet. 7(10), e1002326. https://doi.org/10.1371/journal.pgen.1002326 (2011).
Nekrutenko, A., Makova, K.D. & Li, W.H. The Ka/Ks ratio test for assessing the protein-coding potential of genomic regions: An empirical and simulation study. Genome Res. 12(1), 198–202. https://doi.org/10.1101/gr.200901 (2002).
Dong, W.P., Liu, J., Yu, J., Wang, L. & Zhou, S.L. Highly variable chloroplast markers for evaluating plant phylogeny at low taxonomic levels and for DNA barcoding. PLoS One 7(4), e35071. https://doi.org/10.1371/journal.pone.0035071 (2012).
Chen, Z. et al. Comparative analysis of chloroplast genomes within Saxifraga (Saxifragaceae) takes insights into their genomic evolution and adaption to the high-elevation environment. Genes (Basel). 13(9), 1673. https://doi.org/10.3390/genes13091673 (2022).
Firetti, F. et al. Complete chloroplast genome sequences contribute to plant species delimitation: A case study of the Anemopaegma species complex. Am. J. Bot. 104(10), 1493–509. https://doi.org/10.3732/ajb.1700302 (2017).
Acknowledgements
We sincerely thank Genesky Biotechnologies Inc., Shanghai for performing the high throughput sequencing.
Funding
This work was supported by the “Chief Scientist Program of Qinghai Province, 2024-018-SFC-0004”, in Qinghai Province.
Author information
Authors and Affiliations
Contributions
Q.J.P.: conceived and designed the study, performed the experiments, contributed materials and data analysis, and wrote the paper. L.Y.: Methodology, Investigation, Supervision, review, editing. W.Y.L.: Methodology, Investigation, review editing. M.Y.S.: Resources, Supervision, Writing, review, editing.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Qin, J., Ma, Y., Liu, Y. et al. Phylogenomic analysis and dynamic evolution of chloroplast genomes of Clematis nannophylla. Sci Rep 14, 15109 (2024). https://doi.org/10.1038/s41598-024-65154-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-65154-6
- Springer Nature Limited