
Abstract
Fagopyrum dibotrys, belongs to Polygonaceae family, is one of national key conserved wild plants of China with important medicinal and economic values. Here, the complete chloroplast (cp) genome sequence of F. dibotrys is reported. The cp genome size is 159,919 bp with a typical quadripartite structure and consisting of a pair of inverted repeat regions (30,738 bp) separated by large single copy region (85,134 bp) and small single copy region (13,309 bp). Sequencing analyses indicated that the cp genome encodes 131 genes, including 80 protein-coding genes, 28 tRNA genes and 4 rRNA genes. The genome structure, gene order and codon usage are typical of angiosperm cp genomes. We also identified 48 simple sequence repeats (SSR) loci, fewer of them are distributed in the protein-coding sequences compared to the noncoding regions. Comparison of F. dibotrys cp genome to other Polygonaceae cp genomes indicated the inverted repeats (IRs) and coding regions were more conserved than single copy and noncoding regions, and several variation hotspots were detected. Coding gene sequence divergence analyses indicated that five genes (ndhK, petL rpoC2, ycf1, ycf2) were subject to positive selection. Phylogenetic analysis among 42 species based on cp genomes and 50 protein-coding genes indicated a close relationship between F. dibotrys and F. tataricum. In summary, the complete cp genome sequence of F. dibotrys reported in this study will provide useful plastid genomic resources for population genetics and pave the way for resolving phylogenetic relationships of order Caryophyllales.
Similar content being viewed by others


Introduction
The angiosperm chloroplast (cp) genome is more conserved than the nuclear and mitochondrial genome; typically its structure is quadripartite, containing a pair of inverted repeats (IRs), a large single-copy (LSC) region, and a small single-copy (SSC) region1. The cp genomes of plants are highly conserved in gene structure, organization, and content2. Because of its conserved and non-recombinant nature, cp genomes are used as a robust tool in genomics and evolutionary studies3. And some evolutionary hotspots of plant plastid genome such as single nucleotide polymorphisms and insertion/deletions can provide useful information to elucidate the phylogenetic relationships of taxonomically unresolved plant taxa4,5.
Traditionally, chloroplasts were firstly isolated by means of sucrose gradient centrifugation. And then pure cpDNA extracted from chloroplasts was used for cp genome sequencing. This approach often resulted in high quality cpDNA, but requires enough fresh leaf materials (20~100 g) and special high-speed refrigerated centrifuge6. Combined with high costs of traditional Sanger sequencing, only a small portion of the cp genomes were obtained, which are insufficient for determining evolutionary relationships and applying on plant phylogenetic and genomic studies. Recently, with the advent of next generation sequencing (NGS), the cost of DNA sequencing was dramatically decreased and numbers of genome sequences were generated. Therefore, it is comparatively simple to obtain chloroplast genome sequences for plant species by using NGS than by traditional Sanger sequencing. Nowadays, hundreds of flowering plant cp genomes were sequenced by NGS technology and were applied to phylogenetic analyses at different taxonomical levels7,8,9.
The 27 species in the genus Fagopyrum (Polygonaceae) are commonly called ‘buckwheat’10. Fagopyrum is primarily distributed in Eurasia, especially in southwest of China. Fagopyrum dibotrys (D. Don) Hara. is a perennial herb with important medicinal and economic values. Fagopyrum cymosum (Trev.) Meisn. was once commonly treated as the synonym of F. dibotrys, as there is no description in Latin when F. cymosum was firstly published11,12. The dried rhizomes of F. dibotrys (jin qiao mai) is one of the famous traditional Chinese medicines for the treatment of lung disease, dysentery, rheumatism, throat inflammation, and the grains of F. dibotrys have high nutritional value and health benefits13,14,15,16. Fagopyrum dibotrys was once widely distributed in China and was an important ecological and genetic resource17. The wild resource of F. dibotrys has declined dramatically, however, due to overexploitation, few natural populations remain. So far, F. dibotrys has been designated as a national key conserved wild plant of China by the State Council of Traditional Chinese Medicine and listed in the National Important Wild Conservation Plants in China13.
Because of the nutritional and medicinal value of F. dibotrys, research has mainly focused on its pharmaceutically active components. There is little data concerning its genetic diversity based on genetic markers (e.g. allozyme)18, and the phylogenetic position of F. dibotrys was inferred using few genetic markers (e.g. RAPD, ITS, rbcL and accD) only19,20,21. F. dibotrys was once considered as the wild ancestor of common and Tartary buckwheat. But molecular studies indicated that F. dibotrys is closer to Tartary buckwheat than to common buckwheat and F. dibotrys is not the ancestor of cultivated buckwheat20,21,22. Therefore, more genetic markers are needed to clarify its still debatable phylogenetic positon17. Although complete cp genome sequences of some Fagopyrum species are now available23,24,25, a comprehensive phylogenetic analysis based on whole cp genomes has not been published. Thus, the availability of complete cp genomes that include new variable and informative sites should help to elucidate a more accurate phylogeny.
In this study, we obtained the complete cp genome of F. dibotrys based on Illumina paired-end sequencing followed by a de novo and reference guided assembly. We analyzed the genome features of F. dibotrys and compared them with cp genomes from Polygonaceae species. We performed a phylogenomic analysis using cp genomes and 50 shared cp genes to reconstruct the phylogeny of order Caryophyllales and infer the preliminary phylogenetic position of F. dibotrys.
Results
Genome assembly and genome features of F. dibotrys
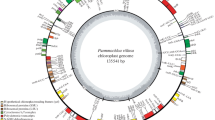
After Illumina paired-end sequencing, 24,970,664 reads were recovered with a sequence length of 125 bp. The total length of the reads was approximately 7.38 Gb and 24,959,432 clean reads were collected to assemble the F. dibotrys cp genome. Based on a combination of de novo and reference guided assembly, the cp genome of F. dibotrys was obtained. The complete cp genome of F. dibotrys was 159,919 bp in length and contained a pair of IRs (30,738 bp) which were separated by a small single copy (SSC) region (13,309 bp) and a large single copy (LSC) region (85,134 bp) (Fig. 1). All paired-end reads were mapped to the assembled cp genome with the mean coverage of 1,290.7. Coding regions (94,848, 59.31%) occupied over half of the cp genome, with the CDS (82,905 bp, 51.84%) regions forming the largest group, followed by rRNA genes (9,058 bp; 5.66%) and tRNA genes (2,885 bp; 1.80%). The remaining 40.71% is covered by intergenic regions, introns or pseudogenes (Table 1). The sequence of the chloroplast genome was deposited in GenBank (accession number: MF491390).

Chloroplast genome map of F. dibotrys. The genes drawn outside of the circle are transcribed counterclockwise, while those inside are clockwise. Small single copy (SSC), large single copy (LSC), and inverted repeats (IRa, IRb) are indicated. GC content is shown. Gene function or identifiers are displayed using colors indicated by the inner legend.
The F. dibotrys cp genome was predicted to contain 131 genes, including 80 protein-coding genes, 28 tRNA genes and 4 rRNA genes (Table 1). Among these genes, five protein-coding genes (rpl2, ycf2, ndhB, rps7, ycf1), seven tRNA genes and four rRNA genes (rrn16, rrn23, rrn4.5, rrn5) were duplicated in IR regions. rpl23, which was repeated in the IR regions, was inferred to be a pseudogene. In the F. dibotrys cp genome, 18 genes contained introns, and 15 of them (9 peptide-coding genes and 6 tRNA genes) harbored one intron, whereas three genes (rps12, clpP, ycf3) harbored two introns. Of the 18 intron-containing genes, rpl2, ndhB, rps12, trnI-GAU, and trnA-UGG were located in the IR regions (Table 2). The rps12 gene is a trans-spliced gene with its N-terminal exon located in the LSC region and the two remaining exons located in the IR regions. The trnK-UUU has the largest intron (2,484 bp) and includes the additional gene matK. The overall AT content of F. dibotrys cp genome is 62.1% and the corresponding values in LSC, SSC and IR regions are 63.8%, 67.2% and 58.7%, respectively. The frequency of codon usage was calculated for the cp genome based on the sequences of protein-coding genes and tRNA genes, which was summarized in Table 3. Similar to the phenomenon detected in other angiosperms cp genes, codon usage was biased toward a high representation of U and A at the third codon position4,26.
Repeat analysis
We identified 11 forward repeats, 26 palindromic repeats, and 16 tandem repeats in the F. dibotrys cp genome (Table S1). Most of the repeats (77.78%) were between 20 and 50 bp and 63.90% of repeats were located in intergenic spacer regions and introns. Within the CDS region, ycf1 contained 4 tandem repeats, 5 palindromic repeats and 4 forward repeats, respectively (Table S1). Cp microsatellites (cpSSRs) are potentially useful markers for detection of polymorphisms in evolutionary studies of plants27. In the present study, a total of 48 SSR loci were detected for F. dibotrys cp genome, more than half of them (60.41%) were A and T mononucleotide repeats, followed by dinucleotide (22.91%), trinucleotide (8.33%) and tetranucleotide repeats (8.33%) (Table 4). Most SSRs were located in intergenic regions, but some of them were found in CDS regions such as ycf1, matK, rpoB, rpoA, ycf2, rpoC2, ndhC, ndhD, cemA, rpl22, atpA (Table 4).
Comparison of F. dibotrys to other Polygonaceae cp genomes
To understand the structural characteristics in cp genome of F. dibotrys, overall sequence alignment among seven Polygonaceae cp genomes were conducted using the annotation of F. dibotrys as a reference. The aligned chloroplast genome sequences were relatively conserved in seven Polygonaceae species, although some highly divergent regions were found. Similar to most angiosperm cp genomes, gene coding regions were more conserved than those of their noncoding counterparts (Fig. 2). Based on the alignment results, the most divergent non-coding regions among the eight cp genomes were trnH(GUG)-psbA, rps16-trnQ(UUG), psbI-trnS(GCU), trnS(GCU)-trnG(UCC), petN-psbM, psbM-trnD(GUC), trnE(UUC)-trnT(GGU), atpB-rbcL, psaA-ycf3, ycf3-trnS(GCA), rps4-trnT(UGU), psbE-petL, ycf2-trnL(CAA), ndhF-rpl32. Slightly sequence variation was observed among eight cp genomes in the atpF, rpoC2, rps19 and ycf1 gene. Most of these hotspot regions located in the LSC regions and only few regions located in the SSC or IR region (Fig. 3). Fagopyrum dibotrys cp genome of the present study was divergent in some intergenic regions (including the above non-coding regions) compared with the previous study25. F. dibotrys and other five Polygonaceae species were used to validate the discriminatory powers of these highly variable regions. The results indicated that almost all primer pairs amplified PCR products with the expected fragment size (Fig. S1, Supplementary Dataset 1), and these loci were able to discriminate more than two species. Our results indicated that these variable regions could be used as new genetic markers for authentication and phylogeny in Polygonaceae species.

mVISTA percent identity plot comparing the eight Polygonaceae plastid genomes with F. dibotrys as a reference. The top line shows genes in order (transcriptional direction indicated by arrows). The y-axis represents the percent identity within 50–100%. The x-axis represents the coordinate in the chloroplast genome. Genome regions are color coded as protein-coding (exon), tRNA or rRNA, and conserved noncoding sequences (CNS). The asterisk indicated the cp genome of F. dibotrys obtained in the present study.

Nucleotide diversity (Pi) by sliding window analysis in the aligned whole cp genomes of seven Polygonaceae species. Window length: 600 bp, step size: 200 bp.
Although genomic structure and size were relatively conserved in seven Polygonaceae cp genomes, the IR/SC boundary regions still varied slightly (Fig. 4). Five genes, including rps19, ndhF, rps15, ycf1, rpl2 and trnH, were found in the junctions of LSC/IR and SSC/IR regions of eight cp genomes. Inconsistent with other cp genomes, only ndhF gene was detected across the IRb/SSC border in these seven species. Rps15 was found to be 9 bp, 64 bp, 2 bp and 3 bp away from the SSC/IRa border in three Rumiceae species (R. palmatum, O. sinensis and R. wittrockii), F. tataricum and F. dibotrys (KY275181); but its 5′ end was extended 2 bp, 3 bp and 23 bp to the SSC/IRa border in F. esculentum, F. dibotrys and F. luojishanense, respectively (Fig. 4).

Comparison of chloroplast genome borders of LSC, SSC, and IRs among seven Polygonaceae species. The asterisk indicated the cp genome of F. dibotrys obtained in the present study.
Divergence of coding gene sequence
To detect the selective pressure on the 78 cp genes of four Fagopyrum species. We calculated the rates of synonymous (dS) and nonsynonymous (dN) substitutions and the dN/dS ratio (Fig. 5). The average dS values between paired Fagopyrum species (F. dibotrys vs F. tataricum/F. dibotrys vs F. esculentum subsp. ancestrale/F. dibotrys vs F. luojishanense/F. tataricum vs F. esculentum subsp. ancestrale/F. esculentum subsp. ancestrale vs F. luojishanense/F. tataricum vs F. luojishanense) were 0.0038/0.0236/0.0840/0.0241/0.0873/0.0873, 0.0085/0.0511/0.1571/0.0489/0.1724/0.1547 and 0.0002/0.0089/0.0215/0.0091/0.0190/0.0217 in the LSC, SSC, and IR regions respectively, with a total average value of 0.0042/0.0266/0.0903/0.0266/0.0949/0.0926 across all regions (Table S2). The dN values ranged from 0 to 0.0640, with a total average value of 0.0010/0.0053/0.0148/0.0055/0.0159/0.0148 across all whole cp genomes. Most dN/dS ratios were less than 1, possibly indicating that cpDNA genes were under purifying selection. Only five genes (ndhK, petL, rpoC2, ycf1, ycf2) had dN/dS values >1, indicating that these genes had undergone positive selection (Table S2).

The dS and dN/dS values of 78 protein-coding genes from four Fagopyrum cp genomes (Fd: F. dibotrys; Fe: F. esculentum subsp. ancestrale; Ft: F. tataricum; Fl: F. luojishanense).
Phylogenetic analysis
In the present study, complete cp genomes and 50 shared cp genes shared among order Caryophyllales were utilized to depict the phylogenetic relationships. Phylogenetic analyses were performed using maximum parsimony (MP), maximum likelihood (ML) and Bayesian inference (BI) methods. Two Santalales species, Osyris alba and Champereia manillana were set as outgroup. The dataset comprised of 382,668/39,085 (cp genomes/50 cp genes) nucleotide positions with 73,706/8,726 informative sites. The results of ML analyses based on two different datasets (i.e. complete cp genomes and 50 shared genes) were showed in Fig. 6, which shared identical topology of phylogenetic tree inferred from the MP and BI analysis. The Pentastar in the phylogenetic tree indicated that the support rate of branch was 100/100/1.0. The results showed same phylogenetic signals for the complete cp genomes and 50 shared genes of species in order Caryophyllales, and only a few species showed inconsistent interspecific relationships based on these two datasets (Fig. 6A,B). Our phylogenetic trees supported the monophyly of order Caryophyllales and three families including Droseraceae, Polygonaceae and Caryophyllaceae also formed a monophyletic clade with high bootstrap values (MP and ML analyses) and posterior probability value (BI analysis). Interestingly, two Amaranthaceae species clustered in the same clade were embedded in the family Chenopodiaceae, which corroborated the close relationship between these two families28. We found all the Fagopyrum species formed a monophyletic clade with high resolution, and F. dibotrys was placed along with F. tataricum.

Phylogenetic tree reconstruction of 42 taxa using maximum likelihood, maximum parsimony and Bayesian inference based on datasets of the 50 shared genes and entire genome sequence. (A) The dataset of 50 shared genes. (B) The entire genome sequence dataset. ML topology was shown with ML bootstrap value/MP bootstrap value /Bayesian posterior probability given at each node. The Pentastar in the phylogenetic tree indicated that the support rate of branch is 100/100/1.0.
Discussion
In this study, the complete cp genome of Fagopyrum dibotrys was assembled by using Illumina sequencing reads derived from the whole genome. This strategy without prior isolation of the cpDNA, provided a new way to obtain the cp genome and had been successful in many studies4,29,30,31. The cp genome will provide a series of resources for evolutionary and genetic studies about this endangered medicinal plant.
The cp genome of F. dibotrys possess the typical angiosperm quadripartite structure with two short inverted repeat regions separated by two single copy regions (Fig. 1) and the gene content with a size in range with other Polygonaceae species3,32,33. Notably, we found the newly sequenced cp genome of F. dibotrys was almost identical with the previously published one25, and the sequence divergences of these two genomes were mainly distributed in non-coding regions (trnS-trnG, rpoB-trnC, psbM-trnD, ndhC-trnV, atpB-rbcL, trnP-psaJ). Although cp genome is remarkably conserved relative to gene content, some variable regions that include insertions/deletions could be detected34. Therefore, some variable regions were found in the two cp genomes of F. dibotrys. According to the alignment result, no significant structural rearrangements, such as inversions or gene relocations were detected in these eight cp genomes. The eight plastomes of Polygonaceae were relatively well conserved, and most variations were detected in intergenic regions (Fig. 2). DNA barcodes are defined as the DNA sequences with a sufficiently high mutation rate to identify a species within a given taxonomic group and are confirmed as reliable tools for the identification of medicinal plants35,36. Here, highly variable in regions such as atpF, rpoC2, rps19 and ycf1, trnH(GUG)-psbA, rps16-trnQ(UUG), psbI-trnS(GCU), trnS(GCU)-trnG(UCC) petN-psbM, psbM-trnD(GUC), trnE(UUC)-trnT(GGU), atpB-rbcL, pasA-ycf3, ycf3-trnS(GCA), rps4-trnT(UGU), psbE-petL, ycf2-trnL(CAA), ndhF-rpl32 were detected. As our results showed, most of them located in LSC region and these regions can discriminate some Polygonaceae species successfully (Fig. S1). Therefore, the above highly variable regions could be used as specific DNA barcodes for authentication of the source plant in family Polygonaceae, and these regions also provide sufficient genetic markers for resolving the phylogeny of family Polygonaceae.
The contraction and expansion at the borders of the IR regions are the main reasons for the size variation of cp genomes37. Despite the similar lengths of the IR regions of F. dibotrys and the other Polygonaceae species, some expansion and contraction were observed, with the IR regions ranging from 30,651 bp in R. wittrockii to 30,956 bp in R. palmatum. In this study, only ndhF gene was detected across the IRb/SSC border in seven Polygonaceae species, which was caused by a duplication of the normally single-copy gene ycf1. In general, ycf1, which was located in IRb, is considered a pseudogene in several angiosperm cp genomes. However, no stop codons were detected in the coding sequence of ycf1, thus the long length of ycf1 affected the differences of gene distribution at the SC/IR borders. We deduced that the expansion of the IR caused a duplication of ycf1. Gene duplications caused an expansion of the IR in Eucommia ulmoides as well38.
Repeat elements are correlated with plastome rearrangement and recombination39,40. In this study, a low number of repeats was detected in the F. dibotrys cp genome, and most repeats were located in intergenic regions or in ycf1. Repeats in the ycf1 gene are commonly observed41. Most of the repeated regions identified in the present study showed similar characteristics to the congeneric species3. Cp microsatellites (cpSSRs) usually showed high variation within the same species and which are potentially useful markers for population genetics27. In this study, some SSRs were identified that could be used to infer the population genetic structure and help to develop more conservation strategies for F. dibotrys. These SSR markers also be useful for genetic diversity studies of other Polygonaceae species.
Sequence divergence of protein coding genes was evaluated by calculating the synonymous (dS) substitution rates; all of the genes showed a low sequence divergence (dS < 0.1). Our analyses indicated that most cp genes were under purifying selection (dN/dS < 1); similar results were reported for other cp genomes30,42,43. Only five genes (ndhK, petL, rpoC2, ycf1, ycf2) had dN/dS ratio >1 as expected of genes under positive selection. Eleven genes in plant cp genome (ndhA-ndhK) encode NAD(P)H dehydrogenase (NDH) complex which plays important role in photosystem I cyclic electron transport and chlororespiration44,45. Because the NDH monomer is sensitive to high light intensity, we deduced that the genes encoded NAD(P)H dehydrogenase might have changed drastically to develop new functions for stress resistance45,46. Previous research reported that genes belong to subunits of cytochrome were under positive selection in some species47,48, we therefore inferred that petL for cytochrome b6/f complex subunit proteins may have a high evolution rate in the cp genome of F. luojishanense. The gene rpoC2 was associated with PPR7 protein, we thus speculated it may have coevolved with nuclear genes49. The ycf1 and ycf2 are two of the largest genes encoding for a putative membrane protein50,51 and in two Fagopyrum cp genomes these two genes may have rapidly evolved3.
Cp genomes with sufficient informative sites have been proven to be effective in resolving difficult phylogenetic relationships7,8. Until now, the phylogeny of Caryophyllales was analyzed using only a few genetic markers, and the phylogenetic positon of F. dibotrys is still needed to be clarified. Here, the phylogeny of the Caryophyllales was rebuilt using MP, ML, Bayesian methods based on complete cp genomes and 50 shared PCGs. Phylogenetic trees inferred from different methods showed an identical topology with high resolution values at most clades. And trees rebuilt based on complete cp genomes and 50 shared genes also showed identical topology except some Droseraceae species, which was mainly caused by the unusual structure, plastome-wide rearrangements and gene losses in Droseraceae cp genomes. We thus presumed that shared genes may provide more reliable phylogenetic signals for the species with unusual structure of cp genome. In our study, species of the Polygonaceae formed a monophyletic clade and showed a paraphyletic relationship with species in the Droseraceae, which was consistent with the previous phylogenetic study based on rbcL and matK52. Two species from Cactaceae and Aizoaceae species showed a paraphyletic relationship, which was in accordance with the phylogeny inferred from cpDNA53. Our phylogenetic analyses provided robust support for the monophyly of species in the Amaranthaceae, Chenopodiaceae and Caryophyllaceae; previous studies of the phylogeny of the Caryophyllales resulted in similar findings, but with relatively low support values53. Unexpectedly, two species of Amaranthaceae were clustered with the Chenopodiaceae species, indicating a close relationship between these two taxa. Previous phylogenetic and morphological research showed that Amaranthaceae and Chenopodiaceae were closely related families and had long been considered a single evolutionary lineage28. Therefore, our study further confirmed the close relationships of these two families. We found that all Fagopyrum species formed one monophyletic clade along with three Rumiceae species, and F. dibotrys was related to F. tataricum, as in the phylogeny reported by Zhou et al.54 using ITS and matK. Our phylogeny based on cp genomes further confirmed that F. dibotrys is not the ancestor of cultivated buckwheat and F. dibotrys is closer to Tartary buckwheat than to common buckwheat20,21,22. Although our results clarified the phylogenetic relationships of some Caryophyllales species based on the available cp genomes, more complete cp genome sequences are need to resolve the comprehensive phylogenies of this order, especially since limited taxon sampling may produce discrepancies in tree topologies4,55.
Conclusions
Our study reported the complete chloroplast genome of Fagopyrum dibotrys, which provided valuable plastid genomic resources for this endangered medicinal plant. The cp genome organization and gene content are similar to that of congeneric species. We also identified SSRs that could be used for population genetics studies within Fagopyrum. The comparative analysis of the genome structure of seven Polygonaceae plants showed several variation hotspots, which could be used to develop more specific DNA barcodes for the authentication of Polygonaceae speies. And these highly variable regions also presented a solid resource for phylogenetic studies in the family Polygonaceae. Coding gene sequence divergence analyses indicated that only a few genes were subject to positive selection. We depicted the phylogenetic relationships of some species belong to order Caryophyllales and confirmed the phylogenetic relationship between F. dibotrys and common buckwheat.
Materials and Methods
Plant material
Young leaves of F. dibotrys were collected from Pingli, Shaanxi, China (32°23′33″N, 109°21′61″E). Voucher specimen of F. dibotrys was deposited at Xi’an Botanical Garden Herbarium (XBGH).
Chloroplast genome sequencing, assembly and annotation
Total genomic DNA was extracted from the fresh leaves of F. dibotrys using a CTAB-based protocol56. The DNA library was prepared according to the method of Zhou et al.30 and then a paired-end library was sequenced using Illumina hiseqTM 2500 platform with the average read length of 125 bp. The raw reads were trimmed using NGS QC Toolkit_v2.3.3 with default cut-off values57. After trimming of low quality reads and adapters, the clean reads were mapped to the cp genome of F. esculentum subsp. ancestrale (EU254477) using Bowtie 2–2.2.6 with default values58. The matched paired-end reads were assembled using SPAdes-3.6.059. After de novo assembly, some ambiguous regions were picked out to extend length with MITObim v1.860. Eventually, the complete chloroplast genome was annotated using DOGMA61 and the primary annotated results were manually verified according to the reference cp genome in Geneious R9 v 9.0.2 (Biomatters Ltd., Auckland, New Zealand). The circular plastid genome map was completed using the online program OrganellarGenome DRAW62
Genome analysis, codon usage, repeat structure and sequence divergence
Whole chloroplast gene distribution of all seven Polygonaceae species was performed and visualized using mVISTA software with the annotation of F. dibotrys as a reference63. The nucleotide diversity (Pi) and sequence polymorphisms of seven Polygonaceae species were analyzed using DNAsp 6.064. In order to validate the divergence hotspot regions and develop specific DNA barcodes for discriminating species in Polygonaceae. The primer pairs were designed based on the sequence of F. dibotrys cp genome (Table S3) and validated using the genomic DNA of F. dibotrys and other 5 Polygonaceae species including Rumex crispus, Rheum hotaoense, Reynoutria japonica, Rheum palmatum, and Fallopia multiflora. PCR amplification to validate these hotspot regions were performed in a reaction volume of 25 μL with 12.5 μL 2 × Taq PCR Master Mix, 0.4 μM of each primer, 2 μL template DNA and 10.1 μL ddH2O. All amplifications were carried out in SimpliAmp™ Thermal Cycler (Applied Biosystems, Carlsbad, CA,USA) as follow: denaturation at 94 °C for 5 min, followed by 35 cycles of 94 °C for 50 s, at specific annealing temperature (Tm) for 45 s, 72 °C for 90 s and 72 °C for 7 min as final extension. PCR products were visualized on 2% after staining with agarose gels ethidium bromide and then the DNA fragments were sequenced by Sangon Biotech (Shanghai, China).
The codon usage frequency was calculated by using MEGA665. Dispersed and palindromic repeats of F. dibotrys cp genome were identified using REPuter with a minimum repeat size of 30 bp and a sequence identity >90%66. Tandem repeat sequences were searched using the Tandem Repeats Finder program with the following parameters: 2 for alignment parameters match, 7 for mismatch and indel, respectively67. Simple sequence repeats (SSRs) were analyzed using MISA (http://pgrc.ipk-gatersleben.de/misa/) with the parameters of ten for mono, five for di-, four for tri-, and three for tetra-, penta, and hexa-nucleotide motifs. In order to detect whether plastid genes were under selection pressure, the nonsynonymous (dN), synonymous (dS), and dN/dS values of each protein coding gene in the three Fagopyrum cp genomes were analyzed using PAML packages 4.0 with YN algorithm68.
Phylogenetic analysis
In this study, 45 cp genomes available in GenBank were recovered to infer the phylogenetic relationships among 42 species belonging to the order Caryophyllales. Osyris alba and Champereia manillana were set as out-group (Table S4). First, multiple alignments were performed using complete cp genomes based on the conserved structure and gene order of the chloroplast genomes. All the nucleotide sequences were aligned using MAFFT v7.30869 with default parameters. Three methods were employed to construct phylogenetic trees, including maximum parsimony (MP), maximum likelihood (ML) and Bayesian inference (BI). Maximum parsimony (MP) analyses were performed using PAUP 4.0b1070 and addition-sequence was set as 1,000 replications for Heuristic search. The Maximum likelihood (ML) analyses were conducted using IQ-TREE71 with the best best-fit model selected by ModelFinder in the IQ-TREE package72 (Table S5) and the bootstrap replicates were 1,000. Bayesian inference (BI) was conducted using MrBayes v3.2.673 with the nucleotide substitution model inferred from Modeltest 3.774 (Table S5). The Markov chain Monte Carlo (MCMC) algorithm was run for 2 million generations and sampled every 100 generations. The first 25% of trees generated were discarded as burn-in and the remaining trees were used to build a majority-rule consensus tree with posterior probability (PP) values for each node. Due to gene loss, inversion and unusual structure were detected in the cp genomes of some species (e.g. Carnegiea gigantea, Dionaea muscipula and Drosera rotundifolia). The above three phylogenetic-inference methods were used to infer the phylogenetic tree from 50 shared cp genes using the same settings (Table S6).
Data availability
The complete chloroplast sequence generated and analyzed during the current study are available in GenBank, https://www.ncbi.nlm.nih.gov/genbank/ (accession numbers are described in the text).
References
Bendich, A. J. Circular chloroplast chromosomes: the grand illusion. The Plant Cell 16, 1661–1666, https://doi.org/10.1105/tpc.160771 (2004).
Asaf, S. et al. Comparative analysis of complete plastid genomes from wild soybean (Glycine soja) and nine other Glycine species. PLOS ONE 12, e0182281, https://doi.org/10.1371/journal.pone.0182281 (2017).
Cho, K.-S. et al. Complete chloroplast genome sequence of tartary buckwheat (Fagopyrum tataricum) and comparative analysis with common buckwheat (F. esculentum). PLOS ONE 10, e0125332, https://doi.org/10.1371/journal.pone.0125332 (2015).
Eguiluz, M., Rodrigues, N. F., Guzman, F., Yuyama, P. & Margis, R. The chloroplast genome sequence from Eugenia uniflora, a Myrtaceae from Neotropics. Plant Systematics and Evolution, https://doi.org/10.1007/s00606-017-1431-x (2017).
Ruhfel, B. R., Gitzendanner, M. A., Soltis, P. S., Soltis, D. E. & Burleigh, J. G. From algae to angiosperms–inferring the phylogeny of green plants (Viridiplantae) from 360 plastid genomes. BMC Evolutionary Biology 14, 23, https://doi.org/10.1186/1471-2148-14-23 (2014).
Du, F. K. et al. An improved method for chloroplast genome sequencing in non-model forest tree species. Tree Genetics & Genomes 11, 114, https://doi.org/10.1007/s11295-015-0942-2 (2015).
Ma, P.-F., Zhang, Y.-X., Zeng, C.-X., Guo, Z.-H. & Li, D.-Z. Chloroplast phylogenomic analyses resolve deep-level relationships of an intractable bamboo tribe Arundinarieae (Poaceae). Systematic biology 63, 933–950, https://doi.org/10.1093/sysbio/syu054 (2014).
Carbonell-Caballero, J. et al. A phylogenetic analysis of 34 chloroplast genomes elucidates the relationships between wild and domestic species within the genus Citrus. Molecular biology and evolution 32, 2015–2035, https://doi.org/10.1093/molbev/msv082 (2015).
Zhang, S. D. et al. Diversification of Rosaceae since the Late Cretaceous based on plastid phylogenomics. New Phytologist 214, 1355–1367, https://doi.org/10.1111/nph.14461 (2017).
Zhou, M., Kreft, I., Woo, S. H., Chrungoo, N. & Wieslander, G. Molecular breeding and nutritional aspects of buckwheat. (Oxford: Academic Press, 2016).
Peng, Y., Sun, Z. & Xiao, P. The research and development of Fagopyrum dibotrys. Chin. Med. Mat. 27, 629–631 (1999).
Liu, G., Li, M., Zhu, Q., Li, Y. & Shui, S. The research advance on resource plant Fagopyrum dibotrys. Chinese Agricultural Science Bulletin 22, 380–389 (2006).
Chen, C. & Li, A. Transcriptome analysis of differentially expressed genes Involved in proanthocyanidin accumulation in the rhizomes of Fagopyrum dibotrys and an Irradiation-Induced mutant. Frontiers in Physiology 7, https://doi.org/10.3389/fphys.2016.00100 (2016).
Wang, K.-J., Zhang, Y.-J. & Yang, C.-R. Antioxidant phenolic constituents from Fagopyrum dibotrys. Journal of ethnopharmacology 99, 259–264, https://doi.org/10.1016/j.jep.2005.02.029 (2005).
De Francischi, M., Salgado, J. & Leitao, R. Chemical, nutritional and technological characteristics of buck wheat and non-prolamine buckwheat flours in comparison of wheat flour. Plant Foods for Human Nutrition (Formerly Qualitas Plantarum) 46, 323–329, https://doi.org/10.1007/BF01088431 (1994).
Guo, Y.-Z., Chen, Q.-F., Yang, L.-Y. & Huang, Y.-H. Analyses of the seed protein contents on the cultivated and wild buckwheat Fagopyrum esculentum resources. Genetic resources and crop evolution 54, 1465–1472, https://doi.org/10.1007/s10722-006-9135-z (2007).
Ji, H. Studies on community ecology and genetic diversity of Fagopyrum cymosum (Trev.) Meisn. (Southwest University, 2007).
Yamane, K. & Ohnishi, O. Phylogenetic relationships among natural populations of perennial buckwheat, Fagopyrum cymosum Meisn., revealed by allozyme variation. Genetic Resources and Crop Evolution 48, 69–77, https://doi.org/10.1023/A:1011265212293 (2001).
Sharma, T. & Jana, S. Species relationships in Fagopyrum revealed by PCR-based DNA fingerprinting. TAG Theoretical and Applied Genetics 105, 306–312, https://doi.org/10.1007/s00122-002-0938-9 (2002).
Yasui, Y. & Ohnishi, O. Phylogenetic relationships among Fagopyrum species revealed by the nucleotide sequences of the ITS region of the nuclear rRNA gene. Genes & Genetic Systems 73, 201–210, https://doi.org/10.1266/ggs.73.201 (1998).
Yasui, Y. & Ohnishi, O. Interspecific relationships in Fagopyrum (Polygonaceae) revealed by the nucleotide sequences of the rbcl and accd genes and their intergenic region. American Journal of Botany 85, 1134–1142, https://doi.org/10.2307/2446346 (1998).
Ohnishi, O. Search for the wild ancestor of buckwheat III. The wild ancestor of cultivated common buckwheat, and of tatary buckwheat. Economic Botany 52, 123, https://doi.org/10.1007/bf02861199 (1998).
Logacheva, M. D., Samigullin, T. H., Dhingra, A. & Penin, A. A. Comparative chloroplast genomics and phylogenetics of Fagopyrum esculentum ssp. ancestrale– A wild ancestor of cultivated buckwheat. BMC Plant Biology 8, 59, https://doi.org/10.1186/1471-2229-8-59 (2008).
Yang, J. et al. The complete chloroplast genome sequence of Fagopyrum cymosum. Mitochondrial DNA Part A 27, 2410–2411, https://doi.org/10.3109/19401736.2015.1030619 (2016).
Wang, C.-L. et al. Comparative analysis of four buckwheat species based on morphology and complete chloroplast genome sequences. Scientific Reports 7, 6514, https://doi.org/10.1038/s41598-017-06638-6 (2017).
Ravi, V., Khurana, J. P., Tyagi, A. K. & Khurana, P. An update on chloroplast genomes. Plant Systematics & Evolution 271, 101–122, https://doi.org/10.1007/s00606-007-0608-0 (2008).
Provan, J., Powell, W. & Hollingsworth, P. M. Chloroplast microsatellites: new tools for studies in plant ecology and evolution. Trends Ecol Evol 16, 142–147, https://doi.org/10.1016/S0169-5347(00)02097-8 (2001).
Pratt, D. B. Phylogeny and morphological evolution of the Chenopodiaceae-Amaranthaceae alliance Doctor thesis, Iowa State University, (2003).
Zhou, T., Zhao, J., Chen, C., Meng, X. & Zhao, G. Characterization of the complete chloroplast genome sequence of Primula veris (Ericales: Primulaceae). Conservation Genetics Resources 8, 455–458, https://doi.org/10.1007/s12686-016-0595-y (2016).
Zhou, T. et al. Comparative transcriptome and chloroplast genome analyses of two related Dipteronia Species. Frontiers in Plant Science 7, https://doi.org/10.3389/fpls.2016.01512 (2016).
Guo, X. et al. Plastome phylogeny and early diversification of Brassicaceae. BMC Genomics 18, 176, https://doi.org/10.1186/s12864-017-3555-3 (2017).
Fan, K., Sun, X.-J., Huang, M. & Wang, X.-M. The complete chloroplast genome sequence of the medicinal plant Rheum palmatum L.(Polygonaceae). Mitochondrial DNA Part A 27, 2935–2936, https://doi.org/10.3109/19401736.2015.1060448 (2016).
Dagarova, S. S., Sitpayeva, G. T., Pak, J.-H. & Kim, J. S. The complete plastid genome sequence of Rheum wittrockii (Polygonaceae), endangered species of Kazakhstan. Mitochondrial DNA Part B 2, 516–517, https://doi.org/10.1080/23802359.2017.1361359 (2017).
Aldrich, J. & Cherney, B. W. & Merlin, E. The role of insertions/deletions in the evolution of the intergenic region between psbA and trnH in the chloroplast genome. Current Genetics 14, 137–146, https://doi.org/10.1007/bf00569337 (1988).
Techen, N., Parveen, I., Pan, Z. & Khan, I. A. DNA barcoding of medicinal plant material for identification. Current Opinion in Biotechnology 25, 103–110, https://doi.org/10.1016/j.copbio.2013.09.010 (2014).
Li, X. et al. Plant DNA barcoding: from gene to genome. Biological Reviews 90, 157–166, https://doi.org/10.1111/brv.12104 (2015).
He, L. et al. Complete chloroplast genome of medicinal plant Lonicera japonica: genome rearrangement, intron gain and loss, and implications for phylogenetic studies. Molecules 22, 249, https://doi.org/10.3390/molecules22020249 (2017).
Wang, L., Wuyun, T. N., Du, H., Wang, D. & Cao, D. Complete chloroplast genome sequences of Eucommia ulmoides: genome structure and evolution. Tree Genetics & Genomes 12, 1–15, https://doi.org/10.1007/s1129 (2016).
Weng, M. L., Blazier, J. C., Govindu, M. & Jansen, R. K. Reconstruction of the ancestral plastid genome in Geraniaceae reveals a correlation between genome rearrangements, repeats and nucleotide substitution rates. Molecular biology and evolution 31, 645–659, https://doi.org/10.1093/molbev/mst257 (2013).
Lu, L. et al. Phylogenetic studies and comparative chloroplast genome analyses elucidate the basal position of halophyte Nitraria sibirica (Nitrariaceae) in the Sapindales. Mitochondrial DNA Part A, 1–11, https://doi.org/10.1080/24701394.2017.1350954 (2017).
Curci, P. L., De Paola, D., Danzi, D., Vendramin, G. G. & Sonnante, G. Complete chloroplast genome of the multifunctional crop globe artichoke and comparison with other asteraceae. PLOS ONE 10, e0120589, https://doi.org/10.1371/journal.pone.0120589 (2015).
Rousseau-Gueutin, M. et al. The chloroplast genome of the hexaploid Spartina maritima (Poaceae, Chloridoideae): Comparative analyses and molecular dating. Molecular phylogenetics and evolution 93, 5–16, https://doi.org/10.1016/j.ympev.2015.06.013 (2015).
Xu, J.-H. et al. Dynamics of chloroplast genomes in green plants. Genomics 106, 221–231, https://doi.org/10.1016/j.ygeno.2015.07.004 (2015).
Kofer, W., Koop, H. U., Wanner, G. & Steinmüller, K. Mutagenesis of the genes encoding subunits A, C, H, I, J and K of the plastid NAD(P)H-plastoquinone-oxidoreductase in tobacco by polyethylene glycol-mediated plastome transformation. Molecular General Genetics 258, 166–173, https://doi.org/10.1007/s004380050719 (1998).
Yang, Y. et al. Comparative analysis of the complete chloroplast genomes of five Quercus species. Frontiers in Plant Science 7, 959, https://doi.org/10.3389/fpls.2016.00959 (2016).
Peng, L., Yamamoto, H. & Shikanai, T. Structure and biogenesis of the chloroplast NAD(P)H dehydrogenase complex. Biochimica et biophysica acta 1807, 945–953, https://doi.org/10.1016/j.bbabio.2010.10.015 (2011).
de Santana Lopes, A. et al. The complete plastome of macaw palm [Acrocomia aculeata (Jacq.) Lodd. ex Mart.] and extensive molecular analyses of the evolution of plastid genes in Arecaceae. Planta 247, 1011–1030, https://doi.org/10.1007/s00425-018-2841-x (2018).
Dong, W. L. et al. Molecular evolution of chloroplast genomes of Orchid species: Insights into phylogenetic relationship and adaptive evolution. International Journal of Molecular Sciences 19, 716, https://doi.org/10.3390/ijms19030716 (2018).
Jalal, A. et al. A small multifunctional pentatricopeptide repeat protein in the chloroplast of Chlamydomonas reinhardtii. Molecular Plant 8, 412–426, https://doi.org/10.1016/j.molp.2014.11.019 (2015).
Drescher, A., Ruf, S., Calsa, T., Carrer, H. & Bock, R. The two largest chloroplast genome‐encoded open reading frames of higher plants are essential genes. The Plant Journal 22, 97–104, https://doi.org/10.1046/j.1365-313x.2000.00722.x (2000).
Kikuchi, S. et al. Uncovering the protein translocon at the chloroplast inner envelope membrane. Science 339, 571–574, https://doi.org/10.1126/science.1229262 (2013).
Cuenoud, P. et al. Molecular phylogenetics of Caryophyllales based on nuclear 18S rDNA and plastid rbcL, atpB, and matK DNA sequences. American Journal of Botany 89, 132–144, https://doi.org/10.3732/ajb.89.1.132 (2002).
Downie, S. R. & Palmer, J. D. A chloroplast DNA phylogeny of the Caryophyllales based on structural and inverted repeat restriction site variation. Systematic Botany 19, 236–252, https://doi.org/10.2307/2419599 (1994).
Zhou, M. L. et al. Phylogenetic relationship of four new species related to southwestern Sichuan Fagopyrum based on morphological and molecular characterization. Biochemical Systematics and Ecology 57, 403–409, https://doi.org/10.1016/j.bse.2014.09.024 (2014).
Leebens-Mack, J. et al. Identifying the basal angiosperm node in chloroplast genome phylogenies: sampling one’s way out of the Felsenstein zone. Mol Biol Evol 22, 1948–1963, https://doi.org/10.1093/molbev/msi191 (2005).
Doyle, J. J. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem bull 19, 11–15 (1987).
Patel, R. K. & Jain, M. NGS QC Toolkit: a toolkit for quality control of next generation sequencing data. PloS one 7, e30619, https://doi.org/10.1371/journal.pone.0030619 (2012).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nature methods 9, 357–359, https://doi.org/10.1038/nmeth.1923 (2012).
Bankevich, A. et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. Journal of Computational Biology 19, 455–477, https://doi.org/10.1089/cmb.2012.0021 (2012).
Hahn, C., Bachmann, L. & Chevreux, B. Reconstructing mitochondrial genomes directly from genomic next-generation sequencing reads—a baiting and iterative mapping approach. Nucleic acids research 41, e129–e129, https://doi.org/10.1093/nar/gkt371 (2013).
Wyman, S. K., Jansen, R. K. & Boore, J. L. Automatic annotation of organellar genomes with DOGMA. Bioinformatics 20, 3252–3255, https://doi.org/10.1093/bioinformatics/bth352 (2004).
Lohse, M., Drechsel, O., Kahlau, S. & Bock, R. OrganellarGenomeDRAW–a suite of tools for generating physical maps of plastid and mitochondrial genomes and visualizing expression data sets. Nucleic Acids Res 41, W575–W581, https://doi.org/10.1093/nar/gkt289 (2013).
Frazer, K. A., Pachter, L., Poliakov, A., Rubin, E. M. & Dubchak, I. VISTA: computational tools for comparative genomics. Nucleic acids research 32, W273–W279, https://doi.org/10.1093/nar/gkh458 (2004).
Rozas, J. et al. DnaSP 6: DNA sequence polymorphism analysis of large data sets. Molecular Biology and Evolution 34, 3299–3302, https://doi.org/10.1093/molbev/msx248 (2017).
Tamura, K., Stecher, G., Peterson, D., Filipski, A. & Kumar, S. MEGA6: molecular evolutionary genetics analysis version 6.0. Molecular biology and evolution 30, 2725–2729, https://doi.org/10.1093/molbev/mst197 (2013).
Kurtz, S. et al. REPuter: the manifold applications of repeat analysis on a genomic scale. Nucleic acids research 29, 4633–4642, https://doi.org/10.1093/nar/29.22.4633 (2001).
Benson, G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic acids research 27, 573, https://doi.org/10.1093/nar/27.2.573 (1999).
Yang, Z. PAML 4: phylogenetic analysis by maximum likelihood. Mol Biol Evol 24, 1586–1591, https://doi.org/10.1093/molbev/msm088 (2007).
Katoh, K. & Standley, D. M. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Molecular biology and evolution 30, 772–780, https://doi.org/10.1093/molbev/mst010 (2013).
Swofford, D. L. Commands used in the PAUP Block in PAUP 4.0: phylogenetic analysis using parsimony 132–135. (Smithsonian Institution, 1998).
Nguyen, L.-T., Schmidt, H. A., von Haeseler, A. & Minh, B. Q. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Molecular Biology and Evolution 32, 268–274, https://doi.org/10.1093/molbev/msu300 (2015).
Kalyaanamoorthy, S., Minh, B. Q., Wong, T. K. F., von Haeseler, A. & Jermiin, L. S. ModelFinder: fast model selection for accurate phylogenetic estimates. Nature Methods 14, 587, https://doi.org/10.1038/nmeth.4285 (2017).
Ronquist, F. et al. MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Systematic biology 61, 539–542, https://doi.org/10.1093/sysbio/sys029 (2012).
Posada, D. & Crandall, K. A. Modeltest: testing the model of DNA substitution. Bioinformatics 14, 817–818, https://doi.org/10.1093/bioinformatics/14.9.817 (1998).
Acknowledgements
This study was co-supported by the National Natural Science Foundation of China (Grand Nos 31770364, 31470401, 81001602) and Scientific Research Supporting Project for New Teacher of Xi’an Jiaotong University (1191319802).
Author information
Authors and Affiliations
Contributions
X.W. and T.Z. conceived and designed the experiment; G.B. and T.Z. collected samples and performed the experiment; T.Z. analyzed the data; T.Z. and X.W. wrote the manuscript; Y.Z. prepared figures and tables. All authors read and approved the final version.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wang, X., Zhou, T., Bai, G. et al. Complete chloroplast genome sequence of Fagopyrum dibotrys: genome features, comparative analysis and phylogenetic relationships. Sci Rep 8, 12379 (2018). https://doi.org/10.1038/s41598-018-30398-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-30398-6
- Springer Nature Limited
This article is cited by
-
Chloroplast genomes of Caragana tibetica and Caragana turkestanica: structures and comparative analysis
BMC Plant Biology (2024)
-
Phylogenetic relationships, selective pressure and molecular markers development of six species in subfamily Polygonoideae based on complete chloroplast genomes
Scientific Reports (2024)
-
The chloroplast genome of Camellia sinensis var. assamica cv. Duntsa (Theaceae) and comparative genome analysis: mutational hotspots and phylogenetic relationships
Genetic Resources and Crop Evolution (2024)
-
Chloroplast genome characteristics and phylogeny of the sinodielsia clade (apiaceae: apioideae)
BMC Plant Biology (2023)
-
Genomic exploration of Sesuvium sesuvioides: comparative study and phylogenetic analysis within the order Caryophyllales from Cholistan desert, Pakistan
BMC Plant Biology (2023)