
Abstract
The mechanism and predictive biomarkers of sinonasal inverted papilloma (IP) transformation into squamous cell carcinoma (SCC) are still unclear. We investigated the genetic mutations involved and the predictive biomarkers. Fourteen patients with SCC arising from IP and six patients with IPs without malignant transformation (sIP) were included. DNA was extracted separately from areas of normal tissue, IP, dysplasia, and SCC. Whole exome sequencing and immunohistochemistry was performed. Major oncogenic mutations were observed in the progression from IP to SCC. The most frequently mutated genes were TP53 (39%) and CDKN2A (27%). Mutations in TP53 and/or CDKN2A were observed in three of six IPs with malignant transformation (cIP); none were observed in sIPs. Tumor mutational burden (TMB) increased from IP to SCC (0.64/Mb, 1.11/Mb, and 1.25 for IP, dysplasia, and SCC, respectively). TMB was higher in the cIPs than in the sIPs (0.64/Mb vs 0.3/Mb). Three cIPs showed a diffuse strong or null pattern in p53, and one showed a total loss of p16, a distinct pattern from sIPs. Our result suggests that TP53 and CDKN2A status can be predictive markers of malignant transformation of IP. Furthermore, immunohistochemistry of p53 and p16 expression can be surrogate markers for TP53 and CDKN2A status.
Similar content being viewed by others

Introduction
Sinonasal inverted papillomas (IP) are common benign mucosal neoplasms that occur in the sinonasal tract and are characterized by their inverted growth pattern1. Although IPs are classified as benign, they have the potential to progress into squamous cell carcinoma (SCC), with reported rates of malignant transformation ranging from 1.9 to 27%2. The development of IP is a complex process that involves various genetic and environmental factors; however, their progression to SCC is poorly understood.
Several studies investigated the molecular mechanisms underlying the tumorigenesis and malignant transformation of IP. Various genetic mutations, including EGFR, TP53, CDKN2A and KRAS mutations, as well as human papillomavirus (HPV) infection have been reported as potential mechanisms of malignant transformation of IP3,4,5,6,7,8,9. Although one study used whole exome sequencing to find the genetic alterations related to malignant transformation9, most other studies used targeted gene panels for next-generation sequencing3,6,7,8, which limited the detection of genetic variants to only the genes included in the panel10. Furthermore, targeted sequencing is often performed using only tumor tissue, making it difficult to distinguish between germline and somatic mutations. In addition, there is currently no reliable diagnostic method for predicting malignant transformation. A better understanding of the genetic alterations that contribute to malignant transformation may enable the development of more accurate diagnostic methods and more effective treatment strategies.
In this study, we aimed to investigate the genetic mutations involved in the stepwise progression of IP to SCC and explore potential biomarkers that could predict malignant transformation using whole exome sequencing with matched normal tissue. This approach has the potential to provide a more comprehensive understanding of the genetic alterations that contribute to malignant transformation and to identify new targets for early detection and prevention of IP progression to SCC.
Materials and methods
Sample selection and DNA extraction
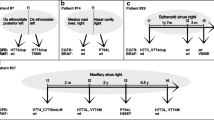
We included 14 patients who were diagnosed with and treated for SCC arising from IP (SCC-IP) at Seoul National University Bundang Hospital between 2004 and 2020. In addition, six patients who were diagnosed with IP without malignant transformation ("sIP") were included as a comparison group. The hematoxylin and eosin stained slides for each case were reviewed by two pathologists (S.K. and H.K.) to select the areas for sequencing and immunohistochemistry (IHC). In each case, we distinguished each component of normal mucosae, IP, IP with dysplasia, and invasive SCC for macro-dissection. DNA was extracted separately from each component. The list of patients and samples used for sequencing and IHC is shown in Fig. 1. The study protocol was approved by the Institutional Review Board of Seoul National University Bundang Hospital (IRB No. B-2008-630-307), and the study was performed in accordance with the Declaration of Helsinki. Informed consent was obtained from each patient, except for those who died.

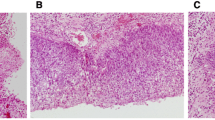
Representative histologic images of normal tissue (a), inverted papilloma (b), dysplasia (c), and squamous cell carcinoma (d) with the list of samples included in the sequencing and immunohistochemistry in each patient (× 20 magnification). IP inverted papilloma, SCC squamous cell carcinoma, SCC-IP patients with squamous cell carcinoma arising from inverted papilloma, sIP patients with inverted papilloma without malignant transformation.
Whole exome sequencing
DNA was extracted using the GeneRead DNA FFPE kit (Qiagen) following the manufacturer's protocol. The quality and quantity of purified DNA were assessed by fluorometry (Qubit, Invitrogen) and gel electrophoresis. Briefly, 200 ng of each sample was ligated to Illumina’s adapters and PCR-amplified. The samples were concentrated to < 1000 ng in 12 μL DW using a SpeedVac machine and hybridized with RNA probes, SureSelectXT Human All Exon V5 at 65 °C 1 min–37 °C 3 s, 60 cycles. After hybridization, the captured targets were pulled down by biotinylated probe/target hybrids using streptavidin-coated magnetic beads (Dynabeads My One Streptavidine T1; Life Technologies Ltd.) and buffers. The selected regions were then PCR-amplified using Illumina PCR primers. Libraries were quantified using the Agilent 4200 Bioanalyzer (Agilent) and KAPA Library Quantification Kit (Kapa Biosystems).
The high quality-libraries were pooled and sequenced on the Illumina NovaSeq6000 platform (Illumina) with 150 bp paired-end by following the manufacturer’s protocols. Image analysis were performed using the NovaSeq6000 control Software version 1.3.1 and the output base calling data was de-multiplexed with bcl2fastq version v2.20.0.422 generating fastQC files. Sequencing reads were aligned to the human reference genome hg19 using Burrows Wheeler Aligner (BWA) (v.0.7.17)11. After the alignment of the reads to reference genome, the duplicated reads were further removed using MarkDuplicates in Picard (v.2.20.7). Next, base quality score recalibration (BQSR) process was conducted to adjust the quality score using BaseRecalibrator in Genome Analysis Toolkit (GATK) (v.4.1.3)12. For germline and somatic variants calling, GATK12 HaplotypeCaller and Mutect2 were utilized, respectively. Further, the LearnReadOrientationModel and FilterMutectCalls of GATK12 were employed to filter orientation bias, technical artifacts and sequencing error. In addition to matched normal samples, gnomAD database was utilized to further exclude germline variants. Only variants with a minimum of 10 supporting reads were included. All variants were then annotated using Ensembl VEP v10013 considering the effects on transcripts, proteins, and regulatory regions. For known or overlapping variants, allele frequencies and disease or phenotype information were included. For downstream analysis, the variants call format (VCF) files were converted to mutation annotation format (MAF) files using vcf2maf. The variants annotated as PASS were summarized and visualized using R packages maftools14.
p53 and p16 immunohistochemistry
Immunostaining for p53 and p16 were performed using monoclonal mouse anti-human p53 (clone DO-7, 1:1000, Dako, Carpinteria, CA, USA) primary antibody and monoclonal mouse p16 (clone E6H4, CINtec®, Ventana Medical Systems, Inc., Tucson, AZ, USA) primary antibody on an automated platform (Benchmark Ultra; Ventana Medical Systems) according to the manufacturer’s instructions. The results were independently interpreted by two pathologists (S.K. and H.K.). P53 expression was classified as diffuse strong positive if there was a diffuse strong nuclear staining in > 80% of tumor cell nuclei, total loss if there was complete absence of staining, and patchy positive if there was variable nuclear staining in 1–80% of tumor cell nuclei15. P16 expression was classified as diffuse strong positive if there was a diffuse strong nuclear and cytoplasmic staining in > 90% of tumor cells, total loss if there was complete absence of staining, and patchy positive if there was variable nuclear and/or cytoplasmic staining16.
Human papillomavirus genotyping
HPV status was determined by HPV genotyping. HPV genotyping was performed using peptide nucleic acid probe-based fluorescence melting curve analysis in a real-time PCR system (PANA RealTyper™ HPV Kit, PANAGENE, Daejeon, Republic of Korea) according to the manufacturer’s instructions. It provides a qualitative detection of 40 HPV genotypes, including genotyping information of 20 high-risk types (16, 18, 26, 31, 33, 35, 39, 45, 51, 52, 53, 56, 58, 59, 66, 68, 69, 70, 73, 82) and 2 low-risk types (6, 11), or the presence of 18 low-risk types (30, 32, 34, 40, 42, 43, 44, 54, 55, 61, 62, 67, 74, 81, 83, 84, 87, 90) without genotyping.
Results
Clinicopathologic characteristics
The clinicopathologic characteristics of the patients are summarized in Table 1. There was no significant difference in age (63.2 ± 12.1 vs. 62.9 ± 6.9 years), sex, and mean tumor size (4.1 cm vs. 3.4 cm) between the two groups (p = 0.935, 0.573, and 0.191, respectively). The five-year survival rate was 71.4% in the SCC-IP group and 100% in the sIP group without statistically significant difference (p = 0.763).
Genomic alteration related with malignant transformation of inverted papilloma
Various single nucleotide variants (SNVs) were identified in SCC-IP group. Top 50 genes that were frequently mutated are shown in Fig. 2. The most common mutated gene was TP53 (39%), followed by CDKN2A (27%), TTN (27%), PIK3CA (21%), and ARID1A (15%). When limited to SCC, the most frequently mutated genes were TP53 (43%), CDKN2A (36%), TTN (36%), ARID1A (21%), FAT1 (21%), KEAP1 (21%), and PIK3CA (21%). In contrast, rare mutations were identified in sIP group. The frequencies of commonly mutated genes in each tumor type subgroup are shown in Table 2. The entire list of the mutations can be found in Supplementary Table S1.

Genetic alterations and tumor mutational burden (TMB) in the benign inverted papilloma and squamous cell carcinoma arising from inverted papilloma (SCC-IP). sIP inverted papilloma without malignant transformation, cIP inverted papilloma with malignant transformation.
The tumor mutational burden (TMB) was calculated as a number of non-synonymous SNVs and indels per mega base (Mb) (Fig. 2). Mean TMB was higher in IP with malignant transformation (cIP) (0.64/Mb) than in sIP (0.3/Mb), and showed a tendency to gradually increase as cancer progressed within the SCC-IP group (0.64/Mb, 1.11/Mb, and 1.25 for IP, dysplasia, and SCC, respectively) (Fig. 2).
Multistep analysis of squamous cell carcinoma arising from inverted papilloma focusing on TP53 and CDKN2A
There were six cases which had matched IP and SCC component available for sequencing (SCC-IP-4, 7, 14, 8, 10, and 12). In the case of TP53 mutations, there were 2/6 (33.3%) cases in which mutations identical to those observed in SCC were already present in the IP, 2/6 (33.3%) cases in which no TP53 mutation was observed in the IP while SCC had one, and 2/6 (33.3%) cases in which TP53 mutation was not observed in neither IP nor SCC. For CDKN2A mutations, 2/6 (33%) cases showed the same mutations in both IP and SCC, 1/6 (17%) case showed mutations in SCC but not in the IP, and 3/6 (50%) cases showed no mutation in neither IP nor SCC. Taken together, 3/6 (50%) of cIP had the same TP53 and/or CDKN2A mutation as SCC.
In contrast, most of the observed mutations in dysplasia and in SCC were identical. There were seven cases which had matched dysplasia and SCC component available for sequencing (SCC-IP-3, 6, 11, 13, 8, 10, and 12). In all but one case, the mutational status of TP53 and CDKN2A in dysplasia and in SCC was the same. The exceptional case had nonsense TP53 mutation in dysplasia, but the SCC had no mutation (SCC-IP-10).
p53 and p16 immunohistochemistry and their correlation with mutational status
We first correlated the sequencing results with the IHC results in all samples to confirm the relationship between the presence or absence of TP53 and CDKN2A gene mutations and p53 and p16 protein expression (Table 3 and Fig. 3). Of the 17 samples which showed patchy positivity of p53 protein in IHC, all samples had wild-type TP53. When p53 expression was diffuse strong positive in IHC, 10/11 (91%) had missense/indel mutation of TP53 and 1/11 (9%) had wild type TP53. In the samples in which p53 expression showed total loss, nonsense mutation of TP53 was observed in 3/5 (60%) and wild type in 2/5 (40%).

Correlation between TP53 and CDKN2A genetic status and p53 and p16 protein expression.
For CDKN2A and p16 expression, 12/14 (86%) samples had wild type forms and 2/14 (14%) samples had missense/indel mutation of CDKN2A in p16 patchy positive tumors. Among five samples which showed diffuse strong positive expression in p16 IHC, all had wild type form of CDKN2A, while three had RB1 frameshift insertion mutation and the other two had high-risk HPV (type 16) infection. The three samples with the RB1 mutation belong to one case (SCC-IP-12), and the two samples with HPV infection belong to another case (SCC-IP-11). When there was total loss p16 expression, 6/14 (43%) samples had missense/indel mutation, 1/14 (7%) samples had nonsense mutation, and 7/14 (50%) samples had wild type form of CDKN2A.
Focusing on the six cases which had paired IP and SCC component available for sequencing, 4/6 (67%) cases showed aberrant expression (diffuse strong positive or total loss) of p53 and/or p16 in both IP and SCC. The other 2/6 (33%) cases, which showed patchy positive p53 and p16 expression in the IP, exhibited diffuse strong positive expression of p53 in the SCC, which acquired TP53 mutation during malignant transformation. In contrast, all sIP showed patchy positive p53 and p16 staining. The results of IHC and the mutational status of sIP and cIP are shown in Fig. 4.

Result of p53, p16 immunohistochemistry, and TP53 and CDKN2A mutations in inverted papilloma with and without malignant transformation (× 20 magnification). sIP inverted papilloma without malignant transformation, cIP inverted papilloma with malignant transformation.
Human papillomavirus infection in squamous cell carcinoma arising from inverted papilloma
High-risk HPV (type 16) was detected in two samples that belonged to one case (SCC-IP-11). Both dysplasia and SCC had HPV infection. As mentioned above, these samples showed diffuse strong positive p16 expression.
Discussion
In this study, we found that TP53 and CDKN2A could be involved in the early stage of the stepwise progression of IP to SCC and that the assessment of TP53 and CDKN2A status could be a predictive marker of malignant transformation of IP. Moreover, using IHC, we found that p53 and p16 expression could be used as surrogate marker for TP53 and CDKN2A mutational status, respectively, and aberrant expression of p53 and/or p16 could be a predictive marker of malignant transformation of IP.
Both TP53 and CDKN2A are tumor suppressor genes and are observed with high frequency in many tumors17,18. Recent studies have shown some conflicting results of TP53 mutation in cIP. Brown et al. reported that TP53 mutations and CDKN2A mutations/deletions were related to malignant transformation, based on the result that they were observed only in the carcinoma but not in the matched IP8. In contrast, Yasukawa et al. reported that most of the TP53 mutations observed in dysplasia and SCC were already present in IP and there was little difference in mutations observed between IP and SCC6. In this study, TP53 and CDKN2A mutations, which were identical to those present in SCC, were observed in 50% (3/6) of cIP, and dysplasia and SCC showed nearly identical mutations. Furthermore, TP53 and CDKN2A mutations were not observed in sIP. This suggests that TP53 and CDKN2A mutations are involved in the early stage of malignant transformation and can be used as biomarkers of early detection of cIP.
There was a strong correlation between TP53 mutation and the aberrant expression of p53. It was concordant with previous studies on gastric19 and ovarian cancers15. However, p16 expression did not show a correlation as strong as p53 expression did. When p16 was patchy positive, it was likely that CDKN2A was wild type. However, when p16 showed diffuse strong positive staining, there was no CDKN2A mutation, while when there was total loss of p16 expression, half of the cases had CDKN2A mutation. Traditionally, p16 IHC was used to differentiate high-grade squamous intraepithelial lesion, an HPV-associated squamous lesion of the lower anogenital tract20, and as a surrogate marker for HPV testing in HPV-mediated oropharyngeal squamous cell carcinoma21. In this context, it was meaningful if p16 expression was manifested as diffuse strong positive, and total loss of p16 would not play a role in the data interpretation. However, recently there has been some reports that the total loss of p16 expression is related to CDKN2A mutation, and it is argued that not only the diffuse strong positive expression but also the total loss of p16 expression should be regarded as an abnormal phenotype16,22. Moreover, it has been reported that total loss of p16 expression is more frequently seen in SCC-IP than in sIP and is a risk factor for the recurrence of sIP, although the mutational status of CDKN2A was not evaluated23,24. In this study, 7/9 (78%) samples with CDKN2A SNV showed total loss of p16. Conversely, when there was a total loss of p16, CDKN2A mutation was found in 7/14 (50%). Additionally, 3/6 (50%) of cIP showed total loss of p16 expression whereas none of the sIP did. Therefore, it is reasonable to consider the total loss of p16 as an aberrant expression and a predictive marker of malignant transformation. In addition, five samples that showed diffuse strong p16 expression had either high-risk HPV infection or RB1 mutation, which may explain the aberrant p16 expression without CDKN2A mutation.
A recent meta-analysis demonstrated that high-risk HPV subtypes 16 and 18 infection was associated with increased risk of malignant transformation of IP25. However, the prevalence of high-risk HPV in SCC-IP seems to be low, ranging from 0 to 25%4,26,27,28,29. In this study, 1 out of 14 SCC-IP patients had high-risk HPV (type 16) and showed diffuse strong positive p16 expression, which implicated the role of high-risk HPV in the pathogenesis of SCC-IP. However, most other SCC-IP specimens did not express high-risk HPV infection. This result was similar to those shown in previous studies in Korea, which did not find HPV infection in any cIP specimen30,31. Further studies are needed to clarify the association between HPV infection and malignant transformation of IP.
TMB is defined as the number of mutations per megabase32, and whole exome sequencing is generally regarded as the gold standard for TMB measurement33. The threshold for high tumor mutational burden (TMB-H) was 10/Mb in KEYNOTE-158 study, based on which the FDA has approved a PD-1 inhibitor, pembrolizumab, for all solid tumors with TMB greater than 10/Mb. Although it is still controversial whether the cut-off value of 10/Mb can be applied universally across all solid tumors34, 1.25/Mb, the mean value of TMB of SCC in this study, is much lower than 10/Mb, the cut-off value. Even the highest TMB in this study was 2.66/Mb, which is still considerably low. Therefore, SCC-IP can be regarded as tumors with low TMB. As low-TMB tumors are not suitable candidates for immunotherapy, it is important to identify cIP before it transforms into SCC.
Previous studies have reported frequent EGFR mutations in IP, especially exon 20 insertions3,4,5,26,29,35. However, in this study, EGFR mutation was not found in any of the cases. This discrepancy can be explained from two points of view: the association with SCC, and geographical distribution. Sahnane et al. reported that EGFR mutation was less frequent in SCC-IP (30%) than in sIP (72), and EGFR-wild-type IP had higher tendency of malignant transformation than EGFR-mutated IP at 5-year follow-up26. In this study, 27/33 (82%) samples are from SCC-IP, which might partially explain why all the samples were EGFR wild-type. In the aspect of geographical distribution, Yasukawa et al. reported the frequency of EGFR mutations to be 20%, 38%, and 0% in IP, dysplasia, and SCC-IP, respectively, in the samples from Hokkaido University Hospital, Japan6, whereas Udager et. al reported the frequency of EGFR mutations to be 88% in the samples from University of Michigan, USA3. As the EGFR mutation frequency differs significantly between Japan and USA, it can be assumed that there are difference in geographical distribution. However, Wang et al. reported a high frequency of EGFR mutations (78%) in Chinese patients, although the study included sIP only35. Furthermore, Sasaki et al. reported that 90% of sIP and 88% of SCC-IP in Japanese patients harbored EGFR mutations5, while Cabal et al. found EGFR exon 20 mutations in 38% of sIP and 50% of SCC-IP in Spanish patients29. Therefore, the difference of the frequency of EGFR mutations cannot be explained by geographical distribution alone and further studies are needed.
Nevertheless, this study has a few limitations. This was a retrospective study that included patients from a single institute; therefore, the number of patients was relatively small and we were unable to obtain peripheral blood lymphocytes. The normal mucosae that were used for sequencing were adjacent to IP or SCC and, therefore, may have already harbored some of the mutations of IP or SCC, potentially leading to false negative results. Moreover, the samples for separate sequencing of each component in SCC-IP were obtained synchronously, which may not directly reflect the time course of malignant transformation. However, we sought to compare the differences in genetic mutations between the regions of IP, dysplasia, and SCC tissue in the same patient. In malignant transformation, we suggest that there may be genetic evidence of the same spectrum. In addition, the synchronousness of each component may have some advantages in preoperative biopsy because performing p53 and p16 staining on the preoperative biopsy specimen can help determine the presence of coexisting SCC component and can be clinically helpful during surgical resection in deciding the extent of resection and the necessity of intraoperative frozen examination, etc.
As mentioned above, there was no clinicopathologic difference between sIP and cIP; therefore, additional tests to differentiate between the two are of high importance.
In conclusion, aberrant expression of p53 and/or p16 is indicative of genetic alterations of TP53 and CDKN2A, which could be used as a predictive marker of malignant transformation of IP to SCC.
Data availability
All analysed and derivative raw data are available from the corresponding author (J.H.W. and H.K.) upon reasonable request.
Change history
17 July 2024
A Correction to this paper has been published: https://doi.org/10.1038/s41598-024-67395-x
References
Lisan, Q., Laccourreye, O. & Bonfils, P. Sinonasal inverted papilloma: From diagnosis to treatment. Eur. Ann. Otorhinolaryngol. Head Neck Dis. 133, 337–341 (2016).
Nudell, J., Chiosea, S. & Thompson, L. D. Carcinoma ex-Schneiderian papilloma (malignant transformation): A clinicopathologic and immunophenotypic study of 20 cases combined with a comprehensive review of the literature. Head Neck Pathol. 8, 269–286 (2014).
Udager, A. M. et al. High-frequency targetable EGFR mutations in sinonasal squamous cell carcinomas arising from inverted sinonasal papilloma. Cancer Res. 75, 2600–2606 (2015).
Udager, A. M. et al. Human papillomavirus (HPV) and somatic EGFR mutations are essential, mutually exclusive oncogenic mechanisms for inverted sinonasal papillomas and associated sinonasal squamous cell carcinomas. Ann. Oncol. 29, 466–471 (2018).
Sasaki, E., Nishikawa, D., Hanai, N., Hasegawa, Y. & Yatabe, Y. Sinonasal squamous cell carcinoma and EGFR mutations: A molecular footprint of a benign lesion. Histopathology 73, 953–962 (2018).
Yasukawa, S. et al. Genetic mutation analysis of the malignant transformation of sinonasal inverted papilloma by targeted amplicon sequencing. Int. J. Clin. Oncol. 23, 835–843 (2018).
Uchi, R. et al. Genomic sequencing of cancer-related genes in sinonasal squamous cell carcinoma and coexisting inverted papilloma. Anticancer Res. 41, 71–79 (2021).
Brown, N. A. et al. TP53 mutations and CDKN2A mutations/deletions are highly recurrent molecular alterations in the malignant progression of sinonasal papillomas. Mod. Pathol. 34, 1133–1142 (2021).
Viitasalo, S. et al. Exome sequencing reveals candidate mutations implicated in sinonasal carcinoma and malignant transformation of sinonasal inverted papilloma. Oral. Oncol. 124, 105663 (2022).
Sun, Y. et al. Next-generation diagnostics: Gene panel, exome, or whole genome?. Hum. Mutat. 36, 648–655 (2015).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
McKenna, A. et al. The genome analysis toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303 (2010).
McLaren, W. et al. The Ensembl variant effect predictor. Genome Biol. 17, 122 (2016).
Mayakonda, A., Lin, D. C., Assenov, Y., Plass, C. & Koeffler, H. P. Maftools: Efficient and comprehensive analysis of somatic variants in cancer. Genome Res. 28, 1747–1756 (2018).
Kobel, M. et al. Optimized p53 immunohistochemistry is an accurate predictor of TP53 mutation in ovarian carcinoma. J. Pathol. Clin. Res. 2, 247–258 (2016).
Matson, D. R. et al. A “Null” pattern of p16 immunostaining in endometrial serous carcinoma: An under-recognized and important aberrant staining pattern. Int. J. Gynecol. Pathol. 41, 378–388 (2021).
Olivier, M., Hollstein, M. & Hainaut, P. TP53 mutations in human cancers: Origins, consequences, and clinical use. Cold Spring Harb. Perspect. Biol. 2, a001008 (2010).
Serra, S. & Chetty, R. p16. J. Clin. Pathol. 71, 853–858 (2018).
Hwang, H. J. et al. Prediction of TP53 mutations by p53 immunohistochemistry and their prognostic significance in gastric cancer. J. Pathol. Transl. Med. 54, 378–386 (2020).
Darragh, T. M. et al. The lower anogenital squamous terminology standardization project for HPV-associated lesions: Background and consensus recommendations from the College of American Pathologists and the American Society for Colposcopy and Cervical Pathology. Arch. Pathol. Lab. Med. 136, 1266–1297 (2012).
El-Naggar, A. K. & Westra, W. H. p16 expression as a surrogate marker for HPV-related oropharyngeal carcinoma: A guide for interpretative relevance and consistency. Head Neck 34, 459–461 (2012).
Schaefer, I. M. et al. Abnormal p53 and p16 staining patterns distinguish uterine leiomyosarcoma from inflammatory myofibroblastic tumour. Histopathology 70, 1138–1146 (2017).
Lin, G. C. et al. Epidermal growth factor receptor, p16, cyclin D1, and p53 staining patterns for inverted papilloma. Int. Forum Allergy Rhinol. 3, 885–889 (2013).
Menendez, M. et al. Loss of p16 expression is a risk factor for recurrence in sinonasal inverted papilloma. Rhinology 60, 453–461 (2022).
McCormick, J. P., Suh, J. D., Lee, J. T., Wells, C. & Wang, M. B. Role of high-risk HPV detected by PCR in malignant sinonasal inverted papilloma: A meta-analysis. Laryngoscope 132, 926–932 (2022).
Sahnane, N. et al. Comprehensive analysis of HPV infection, EGFR exon 20 mutations and LINE1 hypomethylation as risk factors for malignant transformation of sinonasal-inverted papilloma to squamous cell carcinoma. Int. J. Cancer 144, 1313–1320 (2019).
Rooper, L. M., Bishop, J. A. & Westra, W. H. Transcriptionally active high-risk human papillomavirus is not a common etiologic agent in the malignant transformation of inverted schneiderian papillomas. Head Neck Pathol. 11, 346–353 (2017).
Mehrad, M. et al. Transcriptionally active HPV and targetable EGFR mutations in sinonasal inverted papilloma: An association between low-risk HPV, condylomatous morphology, and cancer risk?. Am. J. Surg. Pathol. 44, 340–346 (2020).
Cabal, V. N. et al. EGFR mutation and HPV infection in sinonasal inverted papilloma and squamous cell carcinoma. Rhinology 58, 368–376 (2020).
Kim, J. Y., Yoon, J. K., Citardi, M. J., Batra, P. S. & Roh, H. J. The prevalence of human papilloma virus infection in sinonasal inverted papilloma specimens classified by histological grade. Am. J. Rhinol. 21, 664–669 (2007).
Hwang, C. S., Yang, H. S. & Hong, M. K. Detection of human papillomavirus (HPV) in sinonasal inverted papillomas using polymerase chain reaction (PCR). Am. J. Rhinol. 12, 363–366 (1998).
Merino, D. M. et al. Establishing guidelines to harmonize tumor mutational burden (TMB): In silico assessment of variation in TMB quantification across diagnostic platforms: Phase I of the Friends of Cancer Research TMB Harmonization Project. J. Immunother. Cancer. 8, e000147 (2020).
Doig, K. D., Fellowes, A., Scott, P. & Fox, S. B. Tumour mutational burden: An overview for pathologists. Pathology 54, 249–253 (2022).
Strickler, J. H., Hanks, B. A. & Khasraw, M. Tumor mutational burden as a predictor of immunotherapy response: Is more always better?. Clin. Cancer Res. 27, 1236–1241 (2021).
Wang, H. et al. EGFR and KRAS mutations in Chinese patients with sinonasal inverted papilloma and oncocytic papilloma. Histopathology 75, 274–281 (2019).
Funding
This study was funded by the National Research Foundation of Korea (NRF-2020-R1G1A1005390 to JHW) and by Seoul National University Bundang Hospital (Grant number 14-2020-0029 to HK).
Author information
Authors and Affiliations
Contributions
H.K. and J.H.W. conceptualized the study, designed the methodology, and supervised the study. J-W.K., S-W.C., T-B.W., and C-S.R. acquired samples. S.K. and J-W.K. drafted the manuscript. H.K., J.H.W., S.K., J-W.K., E.S.K., J.H.P., J-H.C., S-W.C., T-B.W., and C-S.R. edited and reviewed the manuscript. All the authors have read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The original online version of this Article was revised: The original version of the Article contained an error in the Author Information. “These authors contributed equally: Soohyeon Kwon, Jeong-Whun Kim, Jee Hye Wee and Hyojin Kim.” now reads: “These authors contributed equally: Soohyeon Kwon and Jeong-Whun Kim. These authors jointly supervised this work: Jee Hye Wee and Hyojin Kim.”
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kwon, S., Kim, JW., Kim, E.S. et al. Assessment of TP53 and CDKN2A status as predictive markers of malignant transformation of sinonasal inverted papilloma. Sci Rep 14, 14286 (2024). https://doi.org/10.1038/s41598-024-64901-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-64901-z
- Springer Nature Limited