
Abstract
Aeluropus littoralis, a halophyte grass, is widely distributed from the Mediterranean to the Indian subcontinent through the Mongolian Gobi. This model halophyte has garnered increasing attention owing to its use as forage and its high tolerance to environmental stressors. The chloroplast genomes of many plants have been extensively examined for molecular, phylogenetic and transplastomic applications. However, no published research on the A. littoralis chloroplast (cp) genome was discovered. Here, the entire chloroplast genome of A. littoralis was assembled implementing accurate long-read sequences. The entire chloroplast genome, with an estimated length of 135,532 bp (GC content: 38.2%), has a quadripartite architecture and includes a pair of inverted repeat (IR) regions, IRa and IRb (21,012 bp each), separated by a large and a small single-copy regions (80,823 and 12,685 bp, respectively). The features of A. littoralis consist of 133 genes that synthesize 87 peptides, 38 transfer RNAs, and 8 ribosomal RNAs. Of these genes, 86 were unique, whereas 19 were duplicated in IR regions. Additionally, a total of forty-six simple sequence repeats, categorized into 32-mono, four-di, two-tri, and eight-tetranucleotides, were discovered. Furthermore, ten sets of repeats greater than 20 bp were located primarily in the LSC region. Evolutionary analysis based on chloroplast sequence data revealed that A. littoralis with A. lagopoides and A. sinensis belong to the Aeluropodinae subtribe, which is a sister to the Eleusininae in the tribe Cynodonteae and the subfamily Chloridoideae. This subfamily belongs to the PACMAD clade, which contains the majority of the C4 photosynthetic plants in the Poaceae. The newly constructed A. littoralis cp genome offers valuable knowledge for DNA barcoding, phylogenetic, transplastomic research, and other biological studies.
Similar content being viewed by others


Introduction
Chloroplasts, tiny organelles found only in photosynthetic eukaryotic cells1,2, are unique because they have their own DNA and ribosomes3. Aside from their photosynthetic function, chloroplasts play an essential role in the biosynthesis of fatty acids, starch, and several amino acids4,5. The first complete chloroplast (cp) genome sequence was reported by Ohyama6 for the common liverwort species Marchantia polymorpha, followed by that for the tobacco plant Nicotiana tabacum7. To date, large numbers of chloroplast genomes have been sequenced, examined, and deposited in the NCBI organelle genome database (https://www.ncbi.nlm.nih.gov/genome/browse#!/organelles/); an expected rise in number as researchers exploit cutting-edge NGS technologies. In general, the chloroplast genome is circular and contains several genes vital for the maintenance of organelle and its functions, as well as those encoding ribosomal and transfer RNA1,3,8. The circular chloroplast genome of terrestrial plants is approximately 120–180 bp long9, with quadripartite features consisting of two inverted repeat regions (IR) separated by large (LSC) and small single copy (SSC) region1,10,11.
Poaceae is a large family of monocotyledons that are commonly known as grasses and are of particular interest to humans and animals. In recent decades, the picture of the evolutionary history of the grass family has developed using different techniques: restriction site maps of the chloroplast genome; sequences of the chloroplast genes (such as ndhF, rpoC2, rbcL, matK, and rps4); and sequences of several nuclear genes (such as phytochrome B and granule-bound starch synthase), sequences of nuclear ribosomal DNA (ITS), and ribosomal RNA sequences (18S rDNA)12. Molecular phylogenetic analyses have facilitated the division of the Poaceae family into 12 subfamilies, including three early-divergent small subfamilies, Anomochlooideae, Puelioideae, and Pharoideae, which include 4, 11 and 12 species, respectively13. The remaining nine subfamilies form two large sister clades: the PACMAD clade, which contains six subfamilies (Panicoideae, Arundinoideae, Chloridoideae, Micrairoideae, Aristidoideae, and Danthonioideae), and the BEP clade (synonym: BOP), which contains three subfamilies (Bambusoideae, Oryzoideae (synonym: Ehrhartoideae) and Pooideae)12,13,14,15,16,17. It has been reported that the C4 photosynthesis pathway has evolved 22 to 24 times in grasses, and it exists only in the PACMAD clade, whereas the BEP (BOP) clade contains only C3 taxa18.
The C4 plant Aeluropus littoralis is a perennial plant belonging to the Poaceae family, the Chloridoideae subfamily, and the Cynodonteae tribe19. Aeluropus littoralis is a monocotyledonous halophyte grass that processes salt glands and performs C4-type photosynthesis. This long stoloniferous grass species often has rooting stems19,20 and leaves that are close, short, stiff, flat and pointed at the top. The plant can withstand salt (NaCl) concentrations of up to 600 mM19,21 and is also considered drought and heat tolerant. It undergoes vegetative reproduction via its rhizomes and can also produce seeds19,20,22. Owing to these characteristics, A. littoralis can serve as a natural forage grass, growing in salt marshes and arid soils19,23,24. The subfamily Chloridoideae, to which the Aeluropus genus belongs, is a monophyletic group within the PACMAD clade of grasses (chloridoid grasses), as shown by molecular phylogenetic studies13,25. This subfamily includes approximately 131–140 genera with 1400–1700 species, the majority of which can thrive in arid regions and marginal salty land12,15,17. The most recent classification based on chloroplast and ITS sequences revealed that the Chloridoideae subfamily is classified into five tribes: Centropodieae, Triraphideae, Eragrostideae, Zoysieae, and Cynodonteae12,14,15,26. This subfamily is an important group for studying the evolutionary transition from C3 to C4 photosynthesis in grasses since the majority of its species uses the C4 photosynthetic pathway13. The C4 grasses are known to be particularly tolerant to drought, salt, and high temperature. This tolerance allows them to colonize harsh habitats through a unique network of anatomical, physiological, and molecular adaptations related to water, temperature, salinity, and excess light stresses16. For this purpose, they are considered important reservoirs of genes and promoters to improve resilience to abiotic stresses in cereals27. With progress in sequencing techniques over the last decade, plastomes have been increasingly adopted in grass phylogenetic studies28. By analyzing 122 sequenced nuclear loci from 47 species and 56 housekeeping genes, it was shown that Aeluropus pungens and Odyssea paucinervis form an independent Aeluropus subclade26. The same results were reported using nuclear sequences for two species (Aeluropus pungens and Odyssea paucinervis), which were classified into an independent subtribe named Aluropodinae under the Cynodonteae tribe16. Additionally, a phylogenetic tree was generated from the combined plastid data (rps16-trnK spacer, rps16 intron, rpoC2, rpl32-trnL spacer, ndhF, ndhA intron, ccsA) and the nuclear region (ITS). The plastid data place the plant Odyssea paucinervis as a sister to Neobouteloua paucirracemosa in Dactylocteniinae26. However, when the nuclear ITS sequences were used, the same plant was placed as a sister to Aeluropus in Aeluropodinae26. Referring to results based on 111 complete plastomes, the genus Aeluropus belongs to the Chloridoideae subfamily, the Cynodonteae tribe and the subtribe Aeluropodinae17. These authors demonstrated that in Cynodonteae, Eleusininae and Aeluropodinae are the third diverged lineages17. In their work, the subtribe Aeluropodinae included only Aeluropus lagopoides and Aeluropus sinensis.
Several research teams have characterized the chloroplast genomes of various plants for molecular selection, DNA barcoding, phylogenetic determination, and transplastomic purposes29,30,31,32. However, no published data were found in the literature on the chloroplast genome of A. littoralis. In this work, for the first time, we reported the entire chloroplast genome of A. littoralis, which we assembled based on the sequences of HiFi reads generated by the PacBio sequencing platform. Additionally, we examined simple sequence repeats (SSRs) and provided an overview of its general characteristics, gene contents, and organization. Lastly, we assessed its phylogenetic linkage to other chloroplast genomes in Poaceae family members. Our research sheds valuable light on the structural diversity and evolutionary history of chloroplast genomes in this widely distributed family of grasses.
Results
Assembly of chloroplast genome
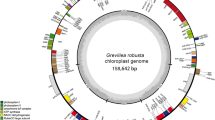
The A. littoralis cp genome was assembled using selected chloroplast-related HiFi sequences obtained from the mapping of raw HiFi reads against a selected group of related cp genomes. The filtered 6907 reads with a mean length of 17,935 bp and a maximum length of 37,947 bp, accounting for 31,327,386 bp and ~ X230 coverage, were employed as input data for the cp genome assembly. The resulting 135,532 bp in length of A. littoralis cp genome with 38.2% GC content displayed a regular quadripartite structure architecture (Fig. 1), including an LSC of 80,823 bp, an SSC of 12,685 bp, and a pair of IRs of 21,012 bp each (Table 1). In addition, mapping of the HiFi long reads revealed that the A. littoralis cp genome exhibited two haplotypes, which differed in the 5’-3’ orientation of the SSC region and had an abundance ratio closer to 1:1. Indeed, a total of 1268 and 1076 long-reads were mapped to haplotype A (with a frequency of 0.54) and to haplotype B (with a frequency of 0.46), respectively (Fig. S1). The references of all reads mapped to either haplotype A or B were reported in Supplementary Table S1.

A. littoralis chloroplast genome map. Genes shown inside the circle are transcribed clockwise, whereas genes outside are transcribed counterclockwise. The light gray inner circle shows the AT content, the dark gray corresponds to the GC content.
Chloroplast genome annotation
A. littoralis cp genome annotation using the Chloe annotation package determined the presence of genes encoding for: 8 ribosomal RNA (rRNA), 38 transfer RNA (tRNA), and 87 different proteins (Table 2). An in-depth look at the 133 genes revealed that 46 of them are implicated in the photosynthesis process, including the rbcL gene encoding for the Rubisco large subunit and ndhA—K genes encoding NADPH dehydrogenase proteins. Additionally, the A. littoralis cp genome included 31 genes encoding for RNA polymerase subunits and ribosomal proteins and 46 genes (tRNA + rRNA genes) involved in transcription and translation processes. In addition, 10 genes were implicated in several functions, such as cytochrome synthesis, carbon metabolism, proteolysis, and RNA processing. Interestingly, a gene structure analysis indicated that 112 genes were intronless, while 21 annotated genes had introns; 19 of these genes harbored a single intron, and only 2, rps12 and PafI, contained 2 introns each (Supplementary Table S2).
Repeat sequence surveys
In A. littoralis cp genome, a full set of 46 simple sequence repeats (SSRs) were discovered. Among them, 69.56% (n = 32) were mononucleotide repeats composed of either A or T (Fig. 2). No penta- or hexa-nucleotide SSRs were detected in the A. littoralis cp genome. Interestingly, the identified SSRs were largely abundant in LSC, with a frequency of 78.26%, compared with those in SSC and IRs. Our repeat search identified 10 sets of repeats longer than 20 bp from the chloroplast genome of A. littoralis. The length of the repeats ranged between 20 and 67 bp. The majority of the repeats were in the LSC region, except for one in the SSC region. Seven of them were in intergenic spacers, two were in rpoC2, and one was in rps18 gene (Supplementary Table S3).

Simple sequence repeats (SSR) in the A. littoralis cp genome. (A) Frequency of identified SSR types. (B) Number of different identified SSR motifs.
Putative RNA editing site analysis
RNA editing is pivotal post-transcriptional regulatory process of cp-genes expression through nucleotide insertions, deletions, and substitutions1. By examining A. littoralis cp sequence, 78 RNA editing sites were predicted, involving 31 protein-coding genes. Remarkably, 31% of the predicted RNA editing sites were noticed within the ndh genes (ndhA [6], ndhB [7], ndhD [2], ndhF [5], ndhG [1], ndhH [2], and ndhK [1]; however, the rpoC2 gene had the largest number of predicted RNA editing sites [12], followed by matK [9], ndhB [7], ndhA [6], rpoB and ndhF [5 each], cemA [3], and atpA, ndhD, ndhH, rpl23, rps18, rps19, and ycf3 (2 each), whereas the other 17 genes had only one predicted editing site. All the predicted RNA editing sites involved the conversion of cytosine (C) to uracil (U), which may have caused amino acid changes. A major portion (76%) of the predicted RNA editing occurred in the second codon, and only 24% occurred in the first position of the codon (Supplementary Table S4).
Codon usage
The sequences of the 87 protein-coding genes were retrieved from the A. littoralis cp genome, and the codon number and codon usage frequency were evaluated. A total of 20,508 different codons were analyzed among the 87 protein-coding genes. The nucleotide triplet (AUU), which encodes the amino acid isoleucine, was the most abundant, with an average number of 847, while the UGC triplet, which encodes cysteine, was the least abundant (56), except for the stop codons (Fig. 3). Among the 20 amino acids, leucine, isoleucine, glycine, and serine were the most abundant, with 2221 (10.82%), 1686 (8.22%), 1544 (7.52%), and 1482 (7.23%) codons, respectively; in addition, the rarest one was cysteine, with 221 (1.07%) codons. To identify codon usage profiles in the A. littoralis cp genome, the average relative synonymous codon usage (RSCU) values were estimated (Fig. 3). The look at these RSCU values revealed that thirty codons were most frequently used (RSCU > 1), whereas thirty-two codons showed little usage (RSCU < 1). Contrary, the AUG (methionine) and UGG (tryptophan) codons showed a lack of bias (RSCU = 1). Interestingly, within the codons with RSCU > 1, twenty-four were enriched in A/U, 12 (40%) ended in A, and 18 (60%) ended in U, suggesting that A/T nucleotide bases are preferred at the third position of the codon in the A .littoralis cp genome.

Codon usage patterns analysis of the A. littoralis chloroplast genome. (A) Frequency analysis of amino-acids in A. littoralis cp protein-coding genes. (B) RSCU values of 20 amino acid and stop codons in all protein-coding genes of the A. littoralis cp genome.
Comparisons of Aeluropus cp genomes boundary regions
To gain insight into the evolutionary history of the genus Aeluropus, the expansion and contraction variation in junction regions were monitored via the comparison of border genes and regions across the cp genomes of the genus Aeluropus (Fig. 4 and Table S5). As illustrated in Fig. 4 and Table S4, the cp genomes of the genus Aeluropus showed high identity in terms of gene order, gene number, as well as at their IRa/LSC and IRb/SSC boundary regions. The fragment size of rpl22-rps19 positioned in the IRb region was 35 bp in all evaluated Aeluropus species cp genomes. IRa/LSC was located in intergenic regions between the rps19 and psbA genes. The length of rps19-psbA was 36 bp in all cp genomes of the genus Aeluropus. The IRb/SSC junctions were enclosed in the ndhF gene, and this gene was prolonged by 20 bp in the IRb region. The ndhH gene crossed the SSC/IRa region in all the cp genomes of the genus Aeluropus. Although the IRa, IRb, and SSC regions were conserved in all cp genomes of the genus Aeluropus, slight differences in LSC regions in term of length were revealed (Fig. 4).

Comparison of the boundaries between LSC, SSC, and IR regions among the three Aeluropus species cp genomes.
The divergence hotspots between the three Aeluropus species cp genomes were computed through nucleotide diversity analysis using DnaSP software. As shown in Fig. 5, the nucleotide diversity index (Pi) ranged from 0 to 0.0088 with an average value of 0.0031. A greater number of genetic diversity hotspots were revealed in the LSC region with seven hotspots; however, three hotspots were located in the SSC region. The greatest genetic diversity was located in Rps16-tRNA-Q gene junctions and tRNA-C-rpoB gene junctions with Pi = 0.00889 and Pi = 0.00884, respectively. The IR region had the lowest Pi values, which suggested that it was more conserved than the LSC and SSC regions across the Aeluropus species cp genomes.

Nucleotide divergence analysis across Aeluropus species cp genomes.
Phylogenetic analysis
To uncover more about evolution and phylogenetic positions of A. littoralis a maximum likelihood and Bayesian inference phylogenetic tree with 1000 bootstrap replicates was built using complete cp genomes (Fig. 6) as well as shared amino-acid protein sequences (Fig. S2). These trees regroup A. littoralis and its related members among the Poaceae family, including A. lagopoides, A. sinensis, O. sativa, S. italica, S. bicolor, P. nuttalliana, Z. mays, T. aestivum, and H. vulgare (Fig. 6, Fig. S2). The two generated trees showed similar topologies. In addition, the selected species were subdivided into 16 groups, namely, Triodiinae, Orininae, Cleistogenes, Gouiniinae, Dactylocteniinae, Aeluropodinae, Eleusininae, Tripogoninae, Boutelouodinae, Arundineae, Andropogoneae, Paniceae, Oryzeae, Brachypodieae, Poeae, and Triticeae. The results highlighted that A. littoralis, A. lagopoides, and A. sinensis form a single subtribe, Aeluropodinae, within the Cynodonteae tribe from the Chloridoideae subfamily. The Aeluropodinae and Euleusininae subtribe are sister groups with bootstrap values of 100 and posterior probability values of 1 (Fig. 6). These two subtribes are the third diverged lineage in Cynodonteae. Thus, the species of Aeluropodinae subtribe were clustered with PACMAD species, which are distinguished by their C4 photosynthesis. Additionally, a total of twenty selected species were clustered into four sister groups composed of the tribes Oryzeae, Brachypodieae, Poeae, and Triticeae, which formed the BEP clade harboring species distinguished by their C3 photosynthesis, including O. sativa, P. nuttalliana, B. distachyon, L. chinensis, T. aestivum, and H. vulgare. The Odyssea paucinervis species was shown to be sister to Dactylocterium aegyptium and D. radulans species in the Dactylocteniinae subtribe and not in Aeluropodinae. Moreover, the Eleusine coracana and Eleusine indica species from the Euleusininae subtribe are the sisters nearest to A. littoralis (Fig. 6). Finally, outside the subfamily Chloridoideae, the species nearest to A. littoralis belong to the following tribes: Arundineae (Arundinoideae), Andropogoneae (Panicoideae), and Paniceae (Panicoideae). The O. sativa, P. nuttalliana, B. distachyon, L. chinensis, T. aestivum, and H. vulgare cp genomes, which belong to the BEP clade, exhibited remarkable diversity from the A. littoralis cp genome and formed a C3 photosynthesis-enriched cluster separate from the rest.

Maximum likelihood and Bayesian inference phylogenetic tree based on complete cp genomes of A. littoralis and related-species within the Poaceae family. Bootstrap and posterior probability support values are indicated above each node.
Divergence time estimations
The time of divergence estimation was illustrated in Fig.S3. The nine tribes (Chloridoideae), Arundineae (Arundinoideae), Andropogoneae (Panicoideae), and Paniceae (Panicoideae) diverged from BEP clade the approximately 45 million years ago (Mya). Interestingly, the results showed that in Cynodonteae, Eleusininae and Aeluropodinae are the third diverged lineages from other seven tribes 23.2 Mya. In the Aeluropodinae subtribe, A. littoralis and A. lagopoides diverged from A. sinesis approximately 4.3 Mya (Fig.S3).
Discussion
The genus Aeluropus consists of 6 species that are distributed mainly in saline habitats from the Mediterranean to the Indian subcontinent through the Mongolian Sahara24. A. littoralis is a perennial plant belonging to the Aeluropus genus from the Poaceae family of flowering plants19. Due to its small genome size, rapid growth rate, high tolerance to salt stress and multiple environmental stressors, high biomass production, and frequent forage use, A. littoralis is considered a model halophyte with increasing attention. The assembled A. littoralis chloroplast genome presents a common quadripartite structure and is similar in size to that of the majority of Poaceae species among angiosperms1,33,34,35,36,37,38,39. The A. littoralis cp genome comprises two IR regions (21,012 bp each) that are distanced by the LSC region (80,823 bp) and the SSC region (12,685 bp), indicating that the assembled cp sequence displayed full coverage with no abnormalities. Generally, the typical terrestrial plant cp genome size is 120 to 180 kb, with IR regions ranging from 10 to 30 kb1,9. The 38.24% GC content and this AT-rich feature of the A. littoralis cp genome are concordant with those reported for other plants, including 38.2% for A. sinensis, A. lagopoides40, S. bicolor (38.5%)35, S. italica (38.9%)38, H. vulgare (38.3%)35, Z. mays (38.5%)33, G. hirsutum (37.2%)41, and A. thaliana (36.3%)42. Interestingly, the A. littoralis cp genome was shown to be present under two chloroplast structural haplotypes based on long-read sequencing data assembly. These results are in agreement with those reported by Wang and Lanfear43, who confirmed the presence of two chloroplast structural haplotypes that occur with equal frequency in most land plant individuals.
Comprehensive analysis of the A. littoralis cp genome revealed that it contains coding regions (54.46% of the genome) harboring 133 genes, 87 of which are protein coding genes (44.59%), 8 are rRNA genes (6.77%), and 38 are tRNA genes (2.1%). Almost 85% of the cp-identified genes were intronless, 14% contained one intron, and rps12 and pafI were the two genes with two introns each. These findings are in line with several cp-structures of angiosperm plants, which include 120–140 genes, 80–90 of which encode proteins, 30–40 of which encode transfer RNA genes, and 4–10 of which encode ribosomal RNA1,44. Likewise, similar Setaria viridis cp genome features were reported by Wang and Gao37. Thus, the cp genome features of land plants seem to be quite universal45. According to multitude studies, cp-SSR and tandem repeats are extremely variable DNA markers and are beneficial for diversity and population genetics analysis studies46,47,48,49. A total of 46 SSRs and 10 long repeats were noticed in the A. littoralis cp genome. Our findings were consistent with previous researches reporting that the common cp SSR markers identified were composed of A or T nucleotides and rarely included C and G nucleotides41. The identified cp SSRs and long tandem repeats could provide useful sequence resources for further molecular genetic studies of A. littoralis, including assessments of species genetic diversity and evolutionary studies.
RNA editing constitutes a common mechanism for cp gene expression modulation in plants through nucleotide insertions, deletions, and substitutions50. Our results indicated that the A. littoralis cp genome contains 78 predicted RNA editing sites dispersed among 31 protein-coding genes. All the predicted RNA editing sites resulted in the conversion of cytosine to uracil predominantly at the 2nd position of the codon. The predominant RNA editing type revealed in the A. littoralis cp genome was comparable to that observed in rice51, proso millet52, wheat53, and maize33. Intriguingly, cytosine—uracil conversion is the most common RNA editing type in plants54. Recently, Ramadan55 reported that differential RNA editing of the ndhB gene of the desert plant Calotropis procera led to the control of photosynthesis across different daylight periods. Moreover, owing to the involvement of chloroplast genes in photosynthesis and metabolite biosynthesis, cp gene expression appears to be crucial for plant responses to environmental stress56. The high number of predicted RNA editing sites in A. littoralis cp genome, particularly in important genes such as the ndh and psb genes, could be one of the keys to tolerance and the dynamic response to environmental stressors. Thus, it was recently reported that Robinia pseudoacacia chloroplastic development and PSI/PSII-related genes, including ndhH, ndhE, psaA, psaB, psbA, psbD, psaC, psbC, ropA, and rps7, are involved in the response to salinity.
The pattern of codon usage bias varies among species and between the genes within an organism57. Our results revealed that the AUU nucleotide triplet coding for the isoleucine amino acid was the most abundant while the UGC triplet that encodes cysteine was the least abundant. Thirty codons with RSCU > 1 were frequently used and thirty-two codons showed little usage. Except for methionine and tryptophan, which lack synonymous codons, all amino acids are represented by 2–6 synonymous codons. Twenty-four of the codons with RSCU values greater than one were rich in A/U, indicating that A/T nucleotide bases are preferred at the 3rd codon position in the A. littoralis cp genome. This high preference for A/T nucleotide at the 3rd codon position was similarly noted in numerous terrestrial plant cp genomes1,58,59. Additionally, Somaratne et al.60 pointed to similar codon usage patterns in several analyzed Poaceae cp genomes associated with AT-rich bias particularly in the third codon position.
A phylogenetic tree was built using the entire cp-genome as well as the shared protein sequences of A. littoralis and sixty-nine selected Poaceae species. The inferred phylogenetic tree clearly showed two large distinct clades: the BEP clade and the PACMAD clade. The A. littoralis, A. logopoides, and A. sinensis species form an independent subtribe, Aeluropodinae, in the Cynodonteae tribe of the Chloridoideae subfamily. On the other hand, O. paucinervis, D. aegyptium, and D. radulans were shown to be sister species in the Dactylocteniinae subtribe and not in Aeluropodinae. These results are in agreement with those reported by Peterson et al.26 and Wang et al.17, who used plastid sequences in their phylogenetic analyses. However, when nuclear sequences were used, A. pungens and O. paucinervis were classified into Aeluropodinae subtribe in the Cynodonteae tribe16. Our future work aims to sequence and assemble at chromosome-scale A. littoralis genome will help to clarify this issue. The divergence time estimation revealed that Aeluropodinae and Eleusininae are sister subtribes. This means that E. coracana and E. indica are the nearest species to A. littoralis. Moreover, the Aeluropodinae diverged 45 Mya from the subtribes with C4 plants of Andropogoneae (containing S. bicolor and Z. mays) and Paniceae (containing Panicum capillare, Panicum lycopodioides, Panicum miliaceum, Panicum virgatum, Setaria italica and Setaria viridis). However, the four subtribes belonging to the BEP clade and specified by their C3 photosynthesis plants diverged 59.1 Mya earlier in the large Poaceae family. Our results were in accordance with previous phylogenetic relationships within Poaceae17,35,37,38,40.
Conclusions
In this work, the entire cp genome sequence of A. littoralis was assembled using raw reads generated via PacBio HiFi read sequencing technology. The A. littoralis cp genome was 135,532 bp in length and had a common circular quadripartite structure. This cp genome encodes 133 genes, 85% of which are intronless, along with 64 codons that correspond to 20 amino acids, with the AUU and UGC codons being the most and the least abundant, respectively. Codon bias analysis revealed a marked preferential usage of codons containing A/U in the third position, particularly among those with RSCU values greater than 1. We also identified a total of 46 SSRs and 10 long repeats. A comparison of the A. littoralis cp genome with those of two other Aeluropus species confirmed a highly conserved structure and slight polymorphic spot regions. Phylogenetic analysis based on entire cp genomes demonstrated that A. littoralis, A. lagopoides, and A. sinensis form a single subtribe, Aeluropodinae, within the tribe of Cynodonteae from the subfamily Chloridoideae. The subtribes Aeluropodinae and Euleusininae are sister groups with bootstrap values of 100. These two subtribes are the third diverged lineage in Cynodonteae. Thus, A. littoralis is clustered with PACMAD species, which are mainly distinguished by their C4 photosynthesis. The findings from this study offer valuable genetic information and a framework for further phylogeographic, population genetics, and plastid genetic engineering research on A. littoralis and related species.
Methods
Plant materials and growth conditions
Aeluropus littoralis cuttings and seeds were collected from a salty area (25° 04′ 48.6″ N 46° 20′ 27.7″ E) in Salboukh region, located north of Riyadh, Saudi Arabia. The taxonomic identification was verified by Prof. Dr. Abdulaziz Assaeed, who is affiliated with College of Food and Agriculture Sciences, King Saud University; a specimen under voucher number 69,107 was placed in the herbarium of the college of food and agriculture sciences, King Saud University. A. littoralis cuttings derived from a single seed were rooted in sterile water and subsequently transplanted to a hydroponic system that used the nutritive solution detailed previously by Ben Romdhane et al.61. A. littoralis plants were grown in greenhouse conditions under a 16 h/8 h light/dark cycle. After 2 months, fresh tissues were harvested from A. littoralis plants and immediately ground into a fine powder in a mortar pre-cooled with liquid nitrogen. Tissue samples were then stored at − 80 °C prior to DNA extraction.
DNA extraction, library preparation, and sequencing
The DNA extraction protocol used in this study was based on the conventional CTAB method62. An Epoch microplate spectrophotometer (BioTek, Winooski, VT, USA) was used to measure the gDNA concentration, and two distinct agarose gel concentrations (0.8% for 1 h at 70 mV and 0.6% for 15 h at 35 mV) were employed to examine the sample's quality. The HMW-gDNA was purified using AMPure PB beads (Pacific Biosciences) were employed to purify the HMW-gDNA, which was further eluted via PacBio elution buffer, and inspected for quality through an Agilent 2100 Bioanalyzer (Agilent).
Utilizing the HiFi protocol (PacBio), two libraries for single-molecule real-time (SMRT) sequencing were developed from the extracted gDNA. The whole-genome sequencing (WGS) of A. littoralis was conducted by the DNA Link Sequencing Lab (DNA Link Inc, Seoul, Republic of Korea).
Genome compiling and gene labeling
The chloroplast-related reads were fished from WGS HiFi reads through their alignment to the closest cp-genomes [Oryza sativa (KM088016), Sorghum bicolor (NC-008602), Setaria italica (NC-022850), Zea mays (NC-001666), Aeluropus logopoides (NC_042858), Brachypodium distachyon (NC-011032), and Puccinellia nuttalliana (NC-027485)] via the Minimap2 aligner63. The cp-related reads were subsequently compiled using CLC genomics workbench V22.0 software.
The A. littoralis chloroplast sequence was annotated with the GeSeq pipeline64 using the Chloe V0.1.0 annotation package. The predicted annotation and the start/stop codon were manually inspected using BLAST against the Nr database. Genes encoding transfer RNA (tRNA) were assessed by using tRNAscan-SE 2.0 software with default settings65. The graphical map of the A. littoralis chloroplast genome was drawn by the Organellar Genome DRAW toolkit (https://chlorobox.mpimp-golm.mpg.de/OGDraw.html)66. The A. littoralis chloroplast genome sequence was deposited in the NCBI GenBank database with the accession number ON357749.
Exploration of chloroplast genome repeats
The MISA tool was employed discover simple-sequence-repeats (SSRs) (https://webblast.ipk-gatersleben.de/misa/)67 with the following parameters: ten for mononucleotides, five for dinucleotides, four for trinucleotides and three for tetra, penta, and hexa-nucleotide SSR motifs.
Repeat sequences longer than 20 nucleotides were predicted by the tandem repeats finder program with the following parameters: (2, 7, 7) for alignment parameters (match, mismatch, indels), 80 for minimum alignment score to report repeat, and maximum period size of 500.
Prediction of RNA editing sites
Prediction of putative RNA editing sites in the A. littoralis chloroplast genome was carried out using the plant RNA editing prepact tool (http://www.prepact.de/prepact-main.php). For predicting potential RNA editing sites, the Z. mays (NC_001666.2) full organelle complete record was fixed as a database for BLAST with an E-value cutoff of 0.8.
Examination of codon use
The CodonW program (V1.4.4) was executed to examine the preferred synonymous codons for protein-coding genes and to examine RSCU values.
Phylogenetic analysis
By employing the cp genomes and shared protein sequences of A. littoralis and sixty-nine Poaceae species (Supplementary Table S6), phylogenetic linkage was assessed. The Guaduella macrostachys chloroplast genome (NC_061343) belongs to the Puelioideae subfamily (used as outgroup). The MAFFT program (v7.520)68 was executed to compute the alignment of nucleic acid and protein sequences. The MEGA11 program69 was implemented to determine the best substitution model, and the GTR + G model was selected (Supplementary Table S7). Maximum likelihood analysis was conducted via the RAxML program70 (v8.2.11) with 1000 bootstrap replicates and the GTRGAMMA model. Bayesian inference analysis was carried out in the MrBayes program71 (v3.2.6) with Markov chain Monte Carlo (MCMC) runs for 1,000,000 generations with a random starting tree, and one tree was sampled every 1000 steps. The first 25% of steps were discarded as burn-in.
Divergence time estimations
The divergence time was estimated for each internal node of the generated phylogenetic tree using MEGA1169. The RelTime method was utilized in dating analyses via calibration of the node time to fine-tune the molecular clock. The maximum age of Triodia longiceps and Aegilops tauschii nodes was assigned as 51.9 million years ago (Mya). The minimum age of the Triodia longiceps and Aegilops tauschii nodes was assigned as 41.4 Mya. The minimum and maximum ages of the Oryza sativa and Aegilops tauschii nodes were pointed as 41.5‒62 Mya, respectively.
Ethical approval and consent to participate
The authors have respected the relevant institutional, national and international guidelines in collecting biological materials for this work. This research contributes to facilitating future studies in species identification, phylogeny, and transplastomic research.
Data availability
The A. littoralis cp genome was deposited into NCBI database (ON357749). The PacBio sequencing reads utilized during the study are available in the SRA (Sequence Read Archive) of NCBI under the accession number PRJNA1075656.
References
Dobrogojski, J., Adamiec, M. & Luciński, R. The chloroplast genome: A review. Acta Physiol. Plant. https://doi.org/10.1007/s11738-020-03089-x (2020).
Green, B. R. Chloroplast genomes of photosynthetic eukaryotes. Plant J 66, 34–44. https://doi.org/10.1111/j.1365-313X.2011.04541.x (2011).
Finkeldey, R. & Gailing, O. Brenner’s Encyclopedia of Genetics 525–527 (Elsevier, 2013).
Rascio, N. Encyclopedia of Biological Chemistry 506–510 (Elsevier, 2013).
Chen, Y. et al. Formation and change of chloroplast-located plant metabolites in response to light conditions. Int. J. Mol. Sci. 19, 654. https://doi.org/10.3390/ijms19030654 (2018).
Ohyama, K. et al. Chloroplast gene organization deduced from complete sequence of liverwort Marchantia polymorpha chloroplast DNA. Nature 322, 572–574. https://doi.org/10.1038/322572a0 (1986).
Shinozaki, K. et al. The complete nucleotide sequence of the tobacco chloroplast genome: Its gene organization and expression. EMBO J. 5, 2043–2049. https://doi.org/10.1002/j.1460-2075.1986.tb04464.x (1986).
Daniell, H., Lin, C. S., Yu, M. & Chang, W. J. Chloroplast genomes: Diversity, evolution, and applications in genetic engineering. Genome Biol. 17, 134. https://doi.org/10.1186/s13059-016-1004-2 (2016).
Park, I. et al. The complete chloroplast genomes of six Ipomoea species and indel marker development for the discrimination of authentic Pharbitidis semen (seeds of I. nil or I. purpurea). Front. Plant Sci. 9, 965. https://doi.org/10.3389/fpls.2018.00965 (2018).
Li, D. M., Zhao, C. Y. & Liu, X. F. Complete chloroplast genome sequences of Kaempferia Galanga and Kaempferia Elegans: Molecular structures and comparative analysis. Molecules 24, 474. https://doi.org/10.3390/molecules24030474 (2019).
Asaf, S. et al. The complete chloroplast genome of wild rice (Oryza minuta) and its comparison to related species. Front. Plant Sci. 8, 304. https://doi.org/10.3389/fpls.2017.00304 (2017).
Kellogg, E. A. Evolutionary history of the grasses. Plant Physiol. 125, 1198–1205. https://doi.org/10.1104/pp.125.3.1198 (2001).
GII Grass Phylogeny Working. New grass phylogeny resolves deep evolutionary relationships and discovers C4 origins. New Phytol. 193, 304–312. https://doi.org/10.1111/j.1469-8137.2011.03972.x (2012).
Soreng, R. J. et al. A worldwide phylogenetic classification of the Poaceae (Gramineae) II: An update and a comparison of two 2015 classifications. J. Syst. Evol. 55, 259–290. https://doi.org/10.1111/jse.12262 (2017).
Soreng, R. J. et al. A worldwide phylogenetic classification of the Poaceae (Gramineae). J. Syst. Evol. 53, 117–137. https://doi.org/10.1111/jse.12150 (2015).
Huang, W. et al. A well-supported nuclear phylogeny of Poaceae and implications for the evolution of C(4) photosynthesis. Mol. Plant 15, 755–777. https://doi.org/10.1016/j.molp.2022.01.015 (2022).
Wang, R. et al. Plastid phylogenomics and morphological character evolution of Chloridoideae (Poaceae). Front. Plant Sci. 13, 1002724. https://doi.org/10.3389/fpls.2022.1002724 (2022).
Christin, P. A. et al. Anatomical enablers and the evolution of C4 photosynthesis in grasses. PNAS 110, 1381–1386. https://doi.org/10.1073/pnas.1216777110 (2013).
Zouari, N. et al. Identification and sequencing of ESTs from the halophyte grass Aeluropus littoralis. Gene 404, 61–69. https://doi.org/10.1016/j.gene.2007.08.021 (2007).
Modarresi, M., Nematzadeh, G. A. & Moradian, F. Salinity response pattern and isolation of catalase gene from halophyte plant Aeluropus littoralis. Photosynthetica 51, 621–629. https://doi.org/10.1007/s11099-013-0060-z (2013).
Ben Saad, R. et al. Improved drought and salt stress tolerance in transgenic tobacco overexpressing a novel A20/AN1 zinc-finger “AlSAP” gene isolated from the halophyte grass Aeluropus littoralis. Plant Mol. Biol. 72, 171–190. https://doi.org/10.1007/s11103-009-9560-4 (2010).
Ahmed, M. Z., Gul, B., Khan, M. A. & Watanabe, K. N. Halophytes for Food Security in Dry Lands 1–16 (Elsevier, 2016).
Rad, M. S., Rad, J. S., da Silva, J. T. & Mohsenzadeh, S. Forage quality of two halophytic species, Aeluropus lagopoides and Aeluropus littoralis, in two phenological stages. Int. J. Agron. Plant Prod. 4, 998–1005 (2013).
Hasanuzzaman, M. et al. Potential use of halophytes to remediate saline soils. Biomed. Res. Int. 2014, 589341. https://doi.org/10.1155/2014/589341 (2014).
Fisher, A. E. et al. Evolutionary history of chloridoid grasses estimated from 122 nuclear loci. Mol. Phylogenet. Evol. 105, 1–14. https://doi.org/10.1016/j.ympev.2016.08.011 (2016).
Peterson, P. M., Romaschenko, K. & Herrera Arrieta, Y. A molecular phylogeny and classification of the Cynodonteae (Poaceae: Chloridoideae) with four new genera: Orthacanthus, Triplasiella, Tripogonella, and Zaqiqah; three new subtribes: Dactylocteniinae, Orininae, and Zaqiqahinae; and a subgeneric classification of Distichlis. Taxon 65, 1263–1287. https://doi.org/10.12705/656.4 (2016).
Ben Saad, R. et al. Marker-free transgenic durum wheat cv. Karim expressing the AlSAP gene exhibits a high level of tolerance to salinity and dehydration stresses. Mol. Breed. 30, 521–533. https://doi.org/10.1007/s11032-011-9641-3 (2011).
Orton, L. M. et al. A 313 plastome phylogenomic analysis of Pooideae: Exploring relationships among the largest subfamily of grasses. Mol. Phylogenet. Evol. 159, 107110. https://doi.org/10.1016/j.ympev.2021.107110 (2021).
Lian, C. et al. Comparative analysis of chloroplast genomes reveals phylogenetic relationships and intraspecific variation in the medicinal plant Isodon rubescens. PLoS One 17, e0266546. https://doi.org/10.1371/journal.pone.0266546 (2022).
Wu, L. et al. Comparative and phylogenetic analyses of the chloroplast genomes of species of Paeoniaceae. Sci. Rep. 11, 14643. https://doi.org/10.1038/s41598-021-94137-0 (2021).
Chen, Q., Hu, H. & Zhang, D. DNA barcoding and phylogenomic analysis of the genus Fritillaria in China based on complete chloroplast genomes. Front. Plant Sci. 13, 764255. https://doi.org/10.3389/fpls.2022.764255 (2022).
Zhao, K. et al. Comparative analyses of chloroplast genomes from 14 Zanthoxylum species: Identification of variable DNA markers and phylogenetic relationships within the genus. Front. Plant Sci. 11, 605793. https://doi.org/10.3389/fpls.2020.605793 (2020).
Maier, R. M., Neckermann, K., Igloi, G. L. & Kossel, H. Complete sequence of the maize chloroplast genome: Gene content, hotspots of divergence and fine tuning of genetic information by transcript editing. J. Mol. Biol. 251, 614–628. https://doi.org/10.1006/jmbi.1995.0460 (1995).
Yu, Y., Lee, H. O., Chin, J. H., Park, H. Y. & Yoo, S. C. The complete chloroplast genome sequence of Oryza sativa aus-type variety Nagina-22 (Poaceae). Mitochondrial DNA B Resour. 2, 819–820. https://doi.org/10.1080/23802359.2017.1407710 (2017).
Saski, C. et al. Complete chloroplast genome sequences of Hordeum vulgare, Sorghum bicolor and Agrostis stolonifera, and comparative analyses with other grass genomes. Theor. Appl. Genet. 115, 571–590. https://doi.org/10.1007/s00122-007-0567-4 (2007).
Cao, X. et al. The complete chloroplast genome of Panicum miliaceum. Mitochondrial DNA B Resour. 2, 43–45. https://doi.org/10.1080/23802359.2016.1157773 (2017).
Wang, S. & Gao, L. Z. Complete chloroplast genome sequence of green foxtail (Setaria viridis), a promising model system for C4 photosynthesis. Mitochondrial DNA A DNA Mapp. Seq. Anal. 27, 3707–3708. https://doi.org/10.3109/19401736.2015.1079867 (2016).
Wang, S. & Gao, L. Z. The complete chloroplast genome of an irreplaceable dietary and model crop, foxtail millet (Setaria italica). Mitochondrial DNA A DNA Mapp. Seq. Anal. 27, 4442–4443. https://doi.org/10.3109/19401736.2015.1089562 (2016).
Raveendar, S. et al. The complete chloroplast genome of pearl millet (Pennisetum glaucum (L.) R. Br.) and comparative analysis within the family poaceae. Cereal Res. Commun. 47, 1–10. https://doi.org/10.1556/0806.46.2018.064 (2019).
Wang, R. et al. Comparative plastomes and phylogenetic analysis of Cleistogenes and closely related genera (Poaceae). Front. Plant Sci. 12, 638597. https://doi.org/10.3389/fpls.2021.638597 (2021).
Lee, S. B. et al. The complete chloroplast genome sequence of Gossypium hirsutum: Organization and phylogenetic relationships to other angiosperms. BMC Genom. 7, 61. https://doi.org/10.1186/1471-2164-7-61 (2006).
Sato, S., Nakamura, Y., Kaneko, T., Asamizu, E. & Tabata, S. Complete structure of the chloroplast genome of Arabidopsis thaliana. DNA Res. 6, 283–290. https://doi.org/10.1093/dnares/6.5.283 (1999).
Wang, W. & Lanfear, R. Long-reads reveal that the chloroplast genome exists in two distinct versions in Most Plants. Genome Biol. Evol. 11, 3372–3381. https://doi.org/10.1093/gbe/evz256 (2019).
Bock, R. Cell and Molecular Biology of Plastids 29–63 (Springer, 2007).
Wicke, S., Schneeweiss, G. M., dePamphilis, C. W., Muller, K. F. & Quandt, D. The evolution of the plastid chromosome in land plants: Gene content, gene order, gene function. Plant Mol. Biol. 76, 273–297. https://doi.org/10.1007/s11103-011-9762-4 (2011).
Powell, W. et al. Hypervariable microsatellites provide a general source of polymorphic DNA markers for the chloroplast genome. Curr. Biol. 5, 1023–1029. https://doi.org/10.1016/s0960-9822(95)00206-5 (1995).
Ebert, D. & Peakall, R. Chloroplast simple sequence repeats (cpSSRs): Technical resources and recommendations for expanding cpSSR discovery and applications to a wide array of plant species. Mol. Ecol. Resour. 9, 673–690. https://doi.org/10.1111/j.1755-0998.2008.02319.x (2009).
Wheeler, G. L., Dorman, H. E., Buchanan, A., Challagundla, L. & Wallace, L. E. A review of the prevalence, utility, and caveats of using chloroplast simple sequence repeats for studies of plant biology. Appl. Plant Sci. 2, 1400059. https://doi.org/10.3732/apps.1400059 (2014).
Reinar, W. B., Lalun, V. O., Reitan, T., Jakobsen, K. S. & Butenko, M. A. Length variation in short tandem repeats affects gene expression in natural populations of Arabidopsis thaliana. Plant Cell 33, 2221–2234. https://doi.org/10.1093/plcell/koab107 (2021).
Hao, W. et al. RNA editing and its roles in plant Organelles. Front. Genet. 12, 757109. https://doi.org/10.3389/fgene.2021.757109 (2021).
Corneille, S., Lutz, K. & Maliga, P. Conservation of RNA editing between rice and maize plastids: Are most editing events dispensable?. Mol. Gen. Genet. 264, 419–424. https://doi.org/10.1007/s004380000295 (2000).
Nie, X. et al. Complete chloroplast genome sequence of broomcorn millet (Panicum miliaceum L.) and comparative analysis with other Panicoideae species. Agronomy 8, 159. https://doi.org/10.3390/agronomy8090159 (2018).
Ogihara, Y. et al. Structural features of a wheat plastome as revealed by complete sequencing of chloroplast DNA. Mol. Genet. Genom. 266, 740–746. https://doi.org/10.1007/s00438-001-0606-9 (2002).
Gerke, P. et al. Towards a plant model for enigmatic U-to-C RNA editing: the organelle genomes, transcriptomes, editomes and candidate RNA editing factors in the hornwort Anthoceros agrestis. New Phytol. 225, 1974–1992. https://doi.org/10.1111/nph.16297 (2020).
Ramadan, A. M. Light/heat effects on RNA editing in chloroplast NADH-plastoquinone oxidoreductase subunit 2 (ndhB) gene of Calotropis (Calotropis procera). J. Genet. Eng. Biotechnol. 18, 49. https://doi.org/10.1186/s43141-020-00064-4 (2020).
Zhang, Y., Zhang, A., Li, X. & Lu, C. The role of chloroplast gene expression in plant responses to environmental stress. Int. J. Mol. Sci. 21, 6082. https://doi.org/10.3390/ijms21176082 (2020).
Parvathy, S. T., Udayasuriyan, V. & Bhadana, V. Codon usage bias. Mol. Biol. Rep. 49, 539–565. https://doi.org/10.1007/s11033-021-06749-4 (2022).
Chakraborty, S., Yengkhom, S. & Uddin, A. Analysis of codon usage bias of chloroplast genes in Oryza species: Codon usage of chloroplast genes in Oryza species. Planta 252, 67. https://doi.org/10.1007/s00425-020-03470-7 (2020).
He, L. et al. Complete chloroplast genome of medicinal plant Lonicera japonica: Genome rearrangement, intron gain and loss, and implications for phylogenetic studies. Molecules 22, 249. https://doi.org/10.3390/molecules22020249 (2017).
Somaratne, Y. et al. Comparison of the complete Eragrostis pilosa chloroplast genome with its relatives in Eragrostideae (Chloridoideae; Poaceae). Plants (Basel) 8, 485. https://doi.org/10.3390/plants8110485 (2019).
Ben Romdhane, W. et al. Expression of an A20/AN1 stress-associated protein from Aeluropus littoralis in rice deregulates stress-related genes. J. Plant Growth Regul. 41, 848–862. https://doi.org/10.1007/s00344-021-10344-z (2021).
Murray, M. G. & Thompson, W. F. Rapid isolation of high molecular weight plant DNA. Nucleic Acids Res. 8, 4321–4325. https://doi.org/10.1093/nar/8.19.4321 (1980).
Li, H. Minimap2: Pairwise alignment for nucleotide sequences. Bioinformatics 34, 3094–3100. https://doi.org/10.1093/bioinformatics/bty191 (2018).
Tillich, M. et al. GeSeq—Versatile and accurate annotation of organelle genomes. Nucleic Acids Res. 45, W6–W11. https://doi.org/10.1093/nar/gkx391 (2017).
Chan, P. P. & Lowe, T. M. tRNAscan-SE: Searching for tRNA genes in genomic sequences. Methods Mol. Biol. 1–14, 2019. https://doi.org/10.1007/978-1-4939-9173-0_1 (1962).
Greiner, S., Lehwark, P. & Bock, R. Organellargenomedraw (OGDRAW) version 1.3.1: Expanded toolkit for the graphical visualization of organellar genomes. Nucleic Acids Res. 47, W59–W64. https://doi.org/10.1093/nar/gkz238 (2019).
Beier, S., Thiel, T., Munch, T., Scholz, U. & Mascher, M. MISA-web: A web server for microsatellite prediction. Bioinformatics 33, 2583–2585. https://doi.org/10.1093/bioinformatics/btx198 (2017).
Katoh, K. & Standley, D. M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. https://doi.org/10.1093/molbev/mst010 (2013).
Tamura, K., Stecher, G. & Kumar, S. MEGA11: Molecular evolutionary genetics analysis version 11. Mol. Biol. Evol. 38, 3022–3027. https://doi.org/10.1093/molbev/msab120 (2021).
Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313. https://doi.org/10.1093/bioinformatics/btu033 (2014).
Ronquist, F. et al. MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 61, 539–542. https://doi.org/10.1093/sysbio/sys029 (2012).
Acknowledgements
This project was funded by the National Plan for Science, Technology and Innovation (MAARIFAH), King Abdul Aziz City for Science and Technology, Kingdom of Saudi Arabia (Award number 2-17-04-001-0046).
Author information
Authors and Affiliations
Contributions
Conceptualization, A.H. and W.B.R; methodology, A.H. and W.B.R; software, W.B.R and A.A.D; validation, A.H., W.B.R and A.A.D; writing—original draft preparation, W.B.R; writing—review and editing, A.H.; supervision, A.A.D; funding acquisition, A.H. All authors have read and agreed to the published version of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ben Romdhane, W., Al-Doss, A. & Hassairi, A. The newly assembled chloroplast genome of Aeluropus littoralis: molecular feature characterization and phylogenetic analysis with related species. Sci Rep 14, 6472 (2024). https://doi.org/10.1038/s41598-024-57141-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-57141-8
- Springer Nature Limited