
Abstract
Understanding the physical properties of the organic–inorganic hybrid [NH2(CH3)2]2CuBr4 is essential to expand its applications. The single [NH2(CH3)2]2CuBr4 crystals were grown and their comprehensive properties were investigated. The crystals had a monoclinic structure with the space group P21/n and lattice constants of a = 8.8651 (5) Å, b = 11.9938 (6) Å, c = 13.3559 (7) Å, and β = 91.322°. The transition temperature from phase I to phase II was determined to be 388 K. Variations in the 1H nuclear magnetic resonance chemical shifts of NH2 and 14N NMR chemical shifts according to the temperature changes in the cation were attributed to vibrations of NH2 groups at their localization sites. The 1H and 13C spin–lattice relaxation times (T1ρ) in phase II changed significantly with temperature, indicating that these values are governed by molecular motion. The T1ρ values were much longer in phase I than in phase II, which means energy transfer was difficult. Finally, the activation energies for phases I and II were considered. According to the basic mechanism of [NH2(CH3)2]2CuBr4 crystals, organic–inorganic materials may have potential applications in various fields.
Similar content being viewed by others
Introduction
Significant attention has been paid to organic–inorganic hybrid compounds owing to their diverse applications as catalysts, sensors, functional smart coatings, fuel and solar cells, light-emitting diodes, light-emitting transistors, and perovskite photovoltaic cells1,2,3,4,5,6,7,8,9,10,11,12. Perovskite type organic–inorganic metal halides are lead-based CH3NH3PbX3 (X = Cl, Br, I) compounds with a 3D structure. However, CH3NH3PbX3-based photovoltaics demonstrate high instability under typical environmental conditions, notably moisture, as well as high toxicity owing to the bioaccumulation of Pb13,14,15,16,17,18. Thus, researchers have suggested the substitution of Pb with other low-toxicity or eco-friendly metals such as Cu and Zn to develop Pb-free photovoltaic devices19. The urgent need to develop eco-friendly hybrid perovskite solar cells has recently been highlighted. For example, two-dimensional [NH3(CH2)nNH3]BX4 (n = 2, 3, …, B = Mn, Co, Cu, Zn, Cd; X = Cl, Br, I) and [(CnH2n+1NH3)]2BX4 organic–inorganic hybrids have recently been proposed as an alternative to these materials20,21,22,23,24,25,26,27. In addition, it is needed to study for [NH2(CH3)n]2BX428,29,30,31,32,33, which has a different hydrogen-bond structure from [NH3(CH2)nNH3]BX4, which has three H atoms bonded to one N. The overall structure of these materials consists of a 1D inorganic network. The isolated building blocks in these 1D perovskites provide a large degree of freedom for the dynamic motion of organic ammonium cations, which can trigger a disorder-to-order transition. Such materials are expected to act as proton conductors owing to the availability of hydrogen bonds.
Dimethylammonium tetrabromocuprate (II) [NH2(CH3)2]2CuBr4 is a member of the [NH2(CH3)n]2BX4 family. In this group of compounds, the individual BX4 tetrahedral anions are isolated and surrounded by [NH2(CH3)2]+ cations. However, the properties and characteristics of [NH2(CH3)2]2CuBr4 have not yet been reported.
In this study, the single crystals of [NH2(CH3)2]2CuBr4 were grown using an aqueous solution method, and their structure, the phase-transition temperature (TC), and the thermal property was considered. The coordination geometries around 1H, 13C, and 14N atoms were investigated by obtaining the chemical shifts of the 1H magic-angle spinning (MAS) nuclear magnetic resonance (NMR), 13C MAS NMR, and static 14N NMR as functions of temperature. In addition, the 1H and 13C NMR spin–lattice relaxation times (T1ρ), which represent the energy transfer surrounding the 1H and 13C atoms of the cation, respectively, were discussed, and their activation energies (Ea) were determined. The single-crystal structure and physical properties observed in this study are expected to important information on the basic mechanism for various applications.
Methods
Crystal growth
Single crystals of [NH2(CH3)2]2CuBr4 were prepared by dissolving dimethylammonium bromide (Aldrich, 98%) and CuBr2 (Aldrich, 98%) at a ratio of 2:1 in tertiary distilled water. To make a saturated solution, the mixed material was heated, stirred, and filtered through filter paper. The prepared saturated solution was placed in a beaker, covered with filter paper, to let natural evaporation in an apparatus with a constant temperature of 300 K. Several dark brown colored single crystals with sizes of 6 × 3 × 1 mm were obtained after a few days.
Characterization
The structure and lattice parameters of the [NH2(CH3)2]2CuBr4 crystals were determined at 300 K using the single-crystal X-ray diffraction (SCXRD) system of the Seoul Western Center at the Korea Basic Science Institute (KBSI). SCXRD measurements were performed using a diffractometer with a graphite-monochromated Mo-Kα target with a wavelength of 0.71073 Å under a cold nitrogen flow (− 50 °C) (Bruker D8 Venture PHOTON III M14). The data was collected using SMART APEX3 (Bruker 2016) and SAINT (Bruker, 2016). The crystal structure was solved using direct methods and refined using the full-matrix least-squares method34. All hydrogen atoms are presented in their geometric positions. Additionally, powder X-ray diffraction (PXRD) patterns were measured using an XRD system at 300 K with the same target used for SCXRD.
Differential scanning calorimetry (DSC) results were performed in the temperature range of 200–423 K using a DSC instrument (TA Instruments, DSC 25) under a nitrogen atmosphere. The thermogram was measured using the sample of 6.1 mg at a heating rate of 10 °C/min.
Thermogravimetric analysis (TGA) results were performed at a heating rate of 10 °C/min under dry nitrogen gas. The thermogram was collected using a 15.92 mg sample while heating from room temperature to 900 K.
The MAS NMR chemical shifts and spin–lattice relaxation time T1ρ of the [NH2(CH3)2]2CuBr4 crystals were measured using a solid-state NMR spectrometer (AVANCE II + , Bruker) at the Seoul Western Center of the KBSI. The Larmor frequency for the 1H NMR experiment was 400.13 MHz, while that for the 13C NMR experiment was 100.61 MHz. The samples were placed in a cylindrical zirconia rotor and then subjected to MAS NMR measurements at a spinning rate of 10 kHz to reduce the spinning sidebands. Chemical shifts were measured using adamantane and tetramethylsilane as the standards for 1H and 13C, respectively. The 1D NMR spectra of 1H and 13C were obtained with a delay time of 0.2‒2 s. To obtain T1ρ values, the one-pulse method was used, and the delay times were within 0.4‒4 s, and the 90° pulse for 1H and 13C was used to 3.45–7.5 μs and 5–5.5 μs, respectively. The 13C T1ρ values were obtained by varying the duration of the 13C spin-locking pulse applied after cross-polarization (CP) preparation. Static 14N NMR chemical shifts were recorded using the one-pulse method at a Larmor frequency of 28.90 MHz with NH4NO3 as the standard sample. NMR experiments above 430 K were not possible because of the limitations of the NMR instrument. The temperature was maintained nearly constant within the error range of ± 0.5 °C, even when the N2 gas flow rate and heater current were adjusted.
Results and discussions
Crystal structure
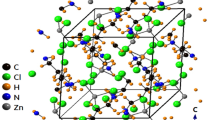
The SCXRD results of the [NH2(CH3)2]2CuBr4 crystals were obtained at 300 K. The crystals had a monoclinic structure with the P21/n space group and lattice constants of a = 8.8651 (5) Å, b = 11.9938 (6) Å, c = 13.3559 (7) Å, β = 91.322 (2)°, and Z = 4. A perspective view of the atomic arrangement in the unit cell of a [NH2(CH3)2]2CuBr4 crystal is shown in Fig. 1a,b. Here, the [CuBr4]2− tetrahedra and [NH2(CH3)2] cations are linked by hydrogen bonds. Specifically, the crystal structure consists of discrete, slightly deformed CuBr4 tetrahedra linked to the organic cations through N‒H∙∙∙Br hydrogen bonds28‒30,33. The SCXRD data for the [NH2(CH3)2]2CuBr4 crystals are shown in Table 1, and the corresponding bond lengths and angles are presented in Table 2. The CIF file result of SCXRD for crystal structure at 300 K is shown in the Supplementary information S1.

(a) Structure of the [NH2(CH3)2]2CuBr4 crystal at 300 K (CCDC No. 2290529). (b) Thermal ellipsoid plot for [NH2(CH3)2]2CuBr4 crystal structure at 300 K.
In addition, PXRD experiments were performed at 300 K. The PXRD patterns obtained in the 2θ measurement range of 8°–50° are shown in red color in Fig. 2. The PXRD pattern simulated by SCXRD structural parameters was consistent with that determined from the PXRD experiment at 300 K. The peaks observed in this diffractogram were indexed using the Mercury program as shown in Fig. 2.

Powder XRD and simulated XRD patterns of [NH2(CH3)2]2CuBr4 at 300 K.
Phase transition temperature
The DSC thermogram of the crystals was measured in the temperature range of 200–423 K at a heating rate of 10 °C/min. Figure 3 shows a strong endothermic peak at 388 K with an enthalpy of 22.26 kJ/mol. The two phases were denoted as phase I, which refers to the region above 388 K, and phase II, which refers to the region below 388 K.

Differential scanning calorimetry (DSC) curve of [NH2(CH3)2]2CuBr4 measured at a heating rate of 10 °C/min.
Thermal property
The TGA curves shown in Fig. 4 were measured as the temperature increased. In the TGA curve, the partial decomposition temperature (Td) represents a weight loss of 2% at 432 K, which means the material is thermally stable up to 432 K. The molecular weight of the [NH2(CH3)2]2CuBr4 crystal decreased abruptly as the temperature increased owing to partial decomposition. The amounts of the sample remaining from the partial decomposition of HBr and 2HBr were obtained from a total molecular weight of 475.37 mg using the TGA data and following chemical reactions35,36.

Thermogravimetric analysis (TGA) and differential thermal analysis (DTA) results of [NH2(CH3)2]2CuBr4 (Inset: Morphology in crystal by polarizing microscopy at (a) 300 K, (b) 373 K, (c) 430 K, and (d) 490 K for [NH2(CH3)2]2CuBr4).
Molecular weight losses of 27% and 34%, which were attributed to the decomposition of HBr and 2HBr, respectively, were observed. The initial weight loss of 27% occurred at 545 K, while the second weight loss of 34% occurred in the range of 577 K.
The endothermic peak at 390 K observed in the DTA curve, which is presented as the differential form of the TGA curve, was in good agreement with the TC shown in the DSC results. Complete weight loss occurred at temperatures above 850 K. Morphology in the crystals with increasing temperature were confirmed by optical polarizing microscopy to understand their thermal properties based on the TGA results. As shown in the inset in Fig. 4, as the temperature increases from 300 to 373 K, the crystal morphology does not change. However, when the temperature reaches 430 K, the single crystal begins to melt, and a considerable amount of it melts at 490 K. From the DSC, TGA, and polarizing microscopy experiments, the TC, Td, and melting temperature of the crystals were determined to be TC = 388 K, Td = 432 K, and Tm = 490 K, respectively.
1H and 13C MAS NMR chemical shifts
The 1H NMR chemical shifts of the [NH2(CH3)2]2CuBr4 crystals were recorded at phases I and II as shown in Fig. 5. As expected, the two 1H signals of NH2 and CH3 in the cation were detected. At low temperatures, the 1H NMR spectra of NH2 and CH3 completely overlapped and only one signal was obtained. The two 1H signals corresponding to NH2 and CH3 began to separate at temperatures above 260 K. The 1H chemical shift for NH2 at 300 K was recorded at 6.81 ppm, while that for CH3 was obtained at 4.67 ppm. In phase II below TC, changes in the chemical shifts with increasing temperature are indicated by dotted lines. The 1H chemical shifts for CH3 were independent of temperature, whereas those for NH2 shifted positively with increasing temperature. The two 1H chemical shifts for NH2 and CH3 changed discontinuously near TC.

1H MAS NMR chemical shifts of NH2 and CH3 in [NH2(CH3)2]2CuBr4 at phases II and I. The open circles and asterisks are denoted sidebands for 1H in NH2 and CH3, respectively.
Furthermore, the linewidths of the 1H spectrum in phase II were very broad because the two signals overlapped, whereas those in phase I were relatively thin owing to their complete separation. The sidebands for 1H in NH2 and CH3 are represented by open circles and asterisks, respectively. A disadvantage of spinning is that it may lead to the presence of spinning sidebands. These are spurious signals (i.e. peaks) that result from the modulation of the magnetic field at the spinning frequency. The peaks always appear on either side of any large genuine peak at a separation equal to the spinning rate. Near TC, the linewidths rapidly changed from a Gaussian shape to a Lorentzian one.
The 13C NMR chemical shifts of [NH2(CH3)2]2CuBr4 were also measured in phases I and II as a function of temperature, as shown in Fig. 6. Because the signals above 340 K could not be well detected, chemical shifts above this temperature were measured using the one-pulse method. In phases I and II, only one 13C NMR signal was observed, and one signal of 13C at 48.10 ppm was observed at 180 K. At TC values above 380 K, this signal suddenly shifted to 82.32 ppm. This abrupt shift is related to the phase transition caused by structural changes between phases I and II. The changes in the 13C NMR chemical shifts reflect changes in the coordination geometry around 13C.

13C MAS NMR chemical shifts in [NH2(CH3)2]2CuBr4 at phases II and I.
Static 14N NMR chemical shifts
The static NMR spectra of 14N in NH2 at the center of the cation in the [NH2(CH3)2]2CuBr4 single crystals were recorded as a function of temperature in the range of 200–430 K, and the longest direction of the single crystal and the applied magnetic field of 9.4 T were measured in the directions perpendicular to each other. 14N has a spin number of 1, and its signals are expected to show two resonances owing to its quadrupole interactions37. The 14N NMR spectrum was challenging to obtain because of the low Larmor frequency (28.90 MHz). The chemical shifts in the 14N NMR spectra at various temperatures are shown in Fig. 7. The structural geometries of N(1) and N(2) in the two [NH2(CH3)2]+ groups were determined based on the 14N NMR chemical shifts. The resonance pairs for 14N are indicated by the same symbol. The N(1) chemical shifts represented by red squares decrease slightly with increasing temperature, whereas the N(2) chemical shifts represented by blue circles decrease abruptly. The [NH2(CH3)2]2CuBr4 structure consists of complex CuBr4 anions and two [NH2(CH3)2] cations, as shown in Fig. 1. The 14N NMR spectrum of N(2) was difficult to detect at low temperatures because of the wide area outside the observed chemical shift range. In phase I, the four 14N spectra of the two sets of signals were reduced to two 14N spectra of only one set. The abrupt changes in the N(1) and N(2) chemical shifts between phases I and II indicate changes in the coordination geometry of the surrounding environments around 14N; N(1) and N(2) with different surrounding environments exist in phase II, whereas only one N site with the same surroundings exists in phase I; temperature changes in 14N NMR static chemical shifts may be associated with vibrations of NH2 groups at their localization sites.

Static 14N NMR chemical shifts in [NH2(CH3)2]2CuBr4 at phases II and I.
1H and 13C NMR spin–lattice relaxation times
The intensities of the 1H and 13C NMR spectral peaks were measured according to the increase in delay time to obtain T1ρ. The decay curves of the changes in intensities and delay times are represented by the following equation37,38,39:
where I(t) is the intensities of the peaks at time t and I(0) is the intensities of the peaks at time t = 0. The T1ρ values for 1H and 13C in [NH2(CH3)2]2CuBr4 were obtained using Eq. (3), and the results are shown in Figs. 8 and 9, respectively, as functions of the inverse temperature.

1H NMR spin–lattice relaxation times in [NH2(CH3)2]2CuBr4 at phases II and I. The slope of solid line at phases II and I is represented the activation energy by the correlation times and the relaxation times as a function of inverse temperature, respectively.

13C NMR spin–lattice relaxation times in [NH2(CH3)2]2CuBr4 at phases II and I. The slope of solid line at phases II and I is represented the activation energy by the correlation times and the relaxation times as a function of inverse temperature, respectively.
In the case of 1H, the chemical shifts for 1H in NH2 and CH3 in phase II were not completely separated, thus, their T1ρ was obtained as a single value. The T1ρ values were strongly dependent on temperature, and T1ρ rapidly decreased as the temperature increased, reaching a minimum value of 0.47 ms at 230 K, as shown in Fig. 8. T1ρ is lengthened again with further increases in temperature, indicating molecular motion according to Bloembergen–Purcell–Pound theory38. The minimum value of T1ρ is clearly due to the reorientational motion of 1H in NH2 and CH3. In the case of 13C, the T1ρ values shown in Fig. 9 decrease as temperature increases and increase abruptly at temperatures above 230 K. Compared with the T1ρ of 1H, the T1ρ of 13C showed a minimum value of 1.70 ms at 230 K, and the 13C T1ρ pattern indicated active molecular motion. In addition, the T1ρ values of 1H and 13C rapidly increased in phase I. However, the T1ρ of 13C was approximately 10 times longer than that of 1H. In phase II, the experimental values of T1ρ can be expressed by the correlation time τC for reorientational motion and are given by39,40:
where R is a constant; ω1 is the spin-lock field; and ωC and ωH are the Larmor frequencies for carbon and protons, respectively. The τC value can be obtained from the condition that T1ρ is at a minimum when ω1 is 1. As the T1ρ curves exhibited minima, the coefficient R in Eq. (4) can be obtained. Using this R, based on the T1ρ values observed over the temperature range investigated and the frequency power of ω1 given in the experiment, the τC value at various temperatures could be obtained. The ω1 values for 1H and 13C were 69.44 and 50 kHz, respectively. Local field fluctuations are caused by thermal motion, which is activated by thermal energy. τC is generally expressed as an Arrhenius-type equation based on the Ea for molecular motion and temperature as follows36,37:
where Ea is the activation energy and kB is the Boltzmann constant. The magnitude of Ea depends on molecular dynamics. The logarithmic scale of τC represented by blue circles versus 1000/T is shown in Figs. 8 and 9 to determine the molecular dynamics of the crystal. In phase II, the activation barriers are 19.38 ± 1.70 kJ/mol (1H) and 23.88 ± 4.48 kJ/mol (13C), and their values are the same within the experimental error. The phenomenon showing the minimum relaxation time for both T1ρ (1H) and T1ρ (13C) is related to the same relaxation process, namely the reorientation of CH3 around its own C3 axis. On the other hand, the changes in the relaxation times for Arrhenius-type random motions as functions of T1ρ for 1H and 13C in phase I are described in terms of slow motions; for τC < < ωC (or ωH), T1ρ ~ τC = τ0exp(‒Ea/kBT), where ωC (or ωH) denotes the Larmor frequency. The Ea values for 1H and 13C obtained from the logarithmic scale of T1ρ in phase I were 5.11 ± 1.24 and 33.36 ± 9.90 kJ/mol, respectively. The difference in Ea values between phases I and II was greater for 1H than for 13C.
Conclusion
The physicochemical properties of organic–inorganic hybrid [NH2(CH3)2]2CuBr4 crystals were investigated in this study. The monoclinic structure of this crystal was determined using SCXRD, and its phase transition temperature TC was determined as 388 K. Its thermal stability at approximately 432 K was not good. 1H, 13C, and 14N NMR spectroscopy provided valuable information on the hydrogen, carbon, and nitrogen environments, as well as their connectivity within the crystal molecule; the 13C and 14N NMR chemical shifts abruptly changed near the TC, thus suggesting that the surrounding environment changes with temperature. By contrast, the 1H chemical shifts for NH2 rather than CH3 changed rapidly over the temperature range investigated. The variations in the 1H NMR chemical shifts for NH2 and 14N NMR chemical shifts according to the changes in temperature in the cation were associated to vibrations of NH2 groups at their localization sites. Finally, the 1H and 13C T1ρ values, which represent the extent of energy transfer surrounding the 1H and 13C atoms of the cation, changed significantly with the temperature in phase II, indicating that these values are governed by the large degree of freedom for the molecular motions of organic cations. The T1ρ values for 1H and 13C indicated similar molecular motions, but the T1ρ values in phase I were much longer than those in phase II, indicating difficulties in energy transfer. The 13C Ea determined from the T1ρ results for molecular motion was larger than the 1H Ea, and the Ea for 1H and 13C near the TC changes greater. Based on their basic mechanism of [NH2(CH3)2]2CuBr4 crystals, it is expected that the potential applications in various fields will be possible.
Data availability
The datasets used and/or analysed during the current study are available from the corresponding author on reasonable request.
References
Dgachi, S., Ben Salah, A. M., Turnbull, M. M., Bataille, T. & Naili, H. Investigations on (C6H9N2)2[MIIBr4] halogenometallate complexes with MII = Co, Cu and Zn: Crystal structure, thermal behavior and magnetic properties. J. Alloy. Compd. 726, 315 (2017).
Elleuch, S., Triki, I. & Abid, Y. Optical and charge transfer properties of a new cadmium based metal-organic-framework material. Mater. Res. Bull. 150, 111754 (2022).
Deng, C. et al. Broadband photoluminescence in 2D organic-inorganic hybrid perovskites: (C7H18N2)PbBr4 and (C9H2N2)PbBr4. J. Phys. Chem. Lett. 11, 2934 (2020).
Roccanova, R. et al. Broadband emission in hybrid organic-inorganic halides of group 12 metals. ACS Omega 3, 18791 (2018).
Klejna, S. et al. Halogen-containing semiconductors: From artificial photosynthesis to unconventional computing. Chem. Rev. 415, 213316 (2020).
Wang, J. et al. Ultrasensitive polarized-light photodetectors based on 2D hybrid perovskite ferroelectric crystals with a low detection limit. Sci. Bull. 66, 158 (2021).
Msalmi, R. et al. Tunable broad-band white-light emission in two-dimensional (110)-oriented lead bromide perovskite (C3H8N6)[PbBr4]: Optical, electronic and luminescence properties. New J. Chem. 45, 20850 (2021).
Hajlaoui, S., Chaabane, I., Oueslati, A., Guidara, K. & Bulou, A. A theoretical study on the molecular structure and vibrational (FT-IR and Raman) spectra of new organic–inorganic compound [N(C3H7)4]2SnCl6. Spectrochim. Acta A Mol. Biomol. Spectrosc. 117, 225 (2014).
Pradeesha, K., Yadava, G. S., Singhb, M. & Prakash, G. V. Synthesis, structure and optical studies of inorganic–organic hybrid semiconductor, NH3(CH2)12NH3PbI4. Mater. Chem. Phys. 124, 44 (2010).
Rhaiem, A. B., Jomni, F., Karoui, K. & Guidara, K. Ferroelectric properties of the [N(CH3)4]2CoCl2Br2 compound. J. Mol. Struct. 1035, 140 (2013).
Gao, Y.-F., Zhang, T., Zhang, W.-Y., Ye, Q. & Da-Wei, F. Great advance in high Tc for hybrid photoelectric-switch bulk/film coupled with dielectric and blue-white light. J. Mater. Chem. C 7, 9840 (2019).
Nie, Y. et al. Flexible thin film and bulk switchable relaxor coexisting most optimal 473 nm blue light without blue-light hazard/visual injury. J. Phys. Chem. C 123, 28385 (2019).
Kojima, A., Teshima, K., Shirai, Y. & Miyasaka, T. Organometal halide perovskites as visible-light sensitizers for photovoltaic cells. JACS Commun. 131, 6050 (2009).
Koh, T. M. et al. Formamidinium-containing metal-halide: An alternative material for near-IR absorption perovskite solar cells. J. Phys. Chem. C 118, 16458 (2014).
Khattak, Y. H., Vega, E., Baig, F. & Soucase, B. M. Performance investigation of experimentally fabricated lead iodide perovskite solar cell via numerical analysis. Mater. Res. Bull. 151, 111802 (2022).
Conings, B. et al. Intrinsic thermal instability of methylammonium lead trihalide perovskite. Adv. Energy Mater. 5, 1 (2015).
Leijtens, T. et al. Stability of metal halide perovskite solar cells. Adv. Energy Mater. 5, 1 (2015).
Babayight, A., Ethirajan, A., Muller, M. & Conings, B. Toxicity of organometal halide perovskite solar cells. Nat. Mater. 15, 247 (2016).
Jellali, H. et al. Zn2+ and Cu2+ doping of one-dimensional lead-free hybrid perovskite ABX3 for white light emission and green solar cell applications. Mater. Res. Bull. 151, 111819 (2022).
Gonzalez-Carrero, S., Galian, R. E. & Perez-Prieto, J. Organometal halide perovskites: Bulk low-dimension materials and nanoparticles. Part. Part. Syst. Char. 32, 709 (2015).
Staskiewicz, B., Czupinski, O. & Czapla, Z. On some spectroscopic properties of a layered 1,3-diammoniumpropylene tetrabromocadmate hybrid crystal. J. Mol. Struct. 1074, 723 (2014).
Staskiewicz, B., Turowska-Tyrk, I., Baran, J., Gorecki, C. & Czapla, Z. Structural characterization, thermal, vibrational properties and molecular motions in perovskite-type diaminopropanetetrachlorocadmate NH3(CH2)3NH3CdCl4 crystal. J. Phys. Chem. Solids 75, 1305 (2014).
Czapla, Z. et al. Structural phase transition in a perovskite-type NH3(CH2)3NH3CuCl4 crystal—X-ray and optical studies. Phase Transit. 90, 637 (2017).
Abdel-Aal, S. K., Kocher-Oberlehner, G., Ionov, A. & Mozhchil, R. N. Effect of organic chain length on structure, electronic composition, lattice potential energy, and optical properties of 2D hybrid perovskites [(NH3)(CH2)n(NH3)]CuCl4, n = 2–9. Appl. Phys. A 123, 531 (2017).
Wei, Y., Audebert, P., Galmiche, L., Lauret, J.-S. & Deleporte, E. Photostability of 2D organic-inorganic hybrid perovskites. Materials 7, 4789 (2014).
Ahmad, S., Hanmandlu, C., Kanaujia, P. K. & Prakash, G. V. Direct deposition strategy for highly ordered inorganic organic perovskite thin films and their optoelectronic applications. Opt. Mater. Express 4, 1313 (2014).
Abdel-Aal, S. K. & Abdel-Rahman, A. S. Synthesis, structure, lattice energy and enthalpy of 2D hybrid perovskite [NH3(CH2)4NH3]CoCl4, compared to [NH3(CH2)nNH3]CoCl4, n=3–9. J. Cryst. Growth 457, 282 (2017).
Waskowska, A. Crystal structure of dimethylammonium tetrabromocadmate(II). Z. Krist. 209, 752 (1994).
Mahfoudh, N., Karoui, K., Jomni, F. & Rhaiem, A. B. Structural phase transition, thermal analysis, and spectroscopic studies in an organic–inorganic hybrid crystal: [(CH3)2NH2]2ZnBr4. Appl. Organo. Chem. 34, e5656 (2020).
Mahfoudh, N., Karoui, K., Gargouri, M. & Rhaiem, A. B. Optical and electrical properties and conduction mechanism of [(CH3)2NH2]2CoCl4. Appl. Organo. Chem. 34, e5404 (2020).
Mahfoudh, N., Karoui, K. & Rhaiem, A. B. Optical studies and dielectric response of [DMA]2MCl4 (M = Zn and Co) and [DMA]2ZnBr4. RSC Adv. 11, 24526 (2021).
Kim, N. H., Choi, J. H. & Lim, A. R. Tumbling motions of NH2(CH3)2 ions in [NH2(CH3)2]2ZnCl4 studied using 1H MAS NMR and 13C CP/MAS NMR. Solid State Sci. 38, 103 (2014).
Horiuchi, K., Ishihara, H. & Terao, H. Differential scanning calorimetry studies on structural phase transitions and molecular motions in (CH3NH3)2MBr4 and [(CH3)2NH2]2MBr4 (M = Zn, Cd, and Hg). J. Phys. Condens. Matter 12, 4799 (2000).
SHELXTL v6.10, Bruker AXS, Inc., Madison, Wisconsin, USA (2000).
Lim, A. R. & Kim, S. H. Physicochemical property investigations of perovskite-type layer crystals [NH3(CH2)nNH3]CdCl4 (n = 2, 3, and 4) as a function of length n of CH2. ACS Omega 6, 27568 (2021).
Lim, A. R. & Cho, J. Growth, structure, phase transition, thermal properties, and structural dynamics of organic–inorganic hybrid [NH3(CH2)5NH3]ZnCl4 crystal. Sci. Rep. 12, 16901 (2022).
Abragam, A. The Principles of Nuclear Magnetism (Oxford University press, 1961).
Bloembergen, N., Purcell, E. M. & Pound, R. V. Relaxation effects in nuclear magnetic resonance absorption. Phys. Rev. 73, 679 (1948).
Koenig, J. L. Spectroscopy of Polymers (Elsevier, 1999).
Harris, R. K. Nuclear Magnetic Resonance Spectroscopy (Pitman Pub., UK, 1983).
Acknowledgements
This work was supported by the National Research Foundation of Korea (NRF) grant, funded by the Korean government (MSIT) (2023R1A2C2006333). The work was supported by the Basic Science Research Program of the National Research Foundation of Korea (NRF), funded by the Ministry of Education, Science, and Technology (2016R1A6A1A03012069).
Author information
Authors and Affiliations
Contributions
A.R.L. designed the project, NMR experiment, and wrote the manuscript. C.N. performed X-ray experiment.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Na, C., Lim, A.R. Investigation on the organic–inorganic hybrid crystal [NH2(CH3)2]2CuBr4: structure, phase transition, thermal property, structural geometry, and dynamics. Sci Rep 13, 21008 (2023). https://doi.org/10.1038/s41598-023-48015-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-48015-6
- Springer Nature Limited