
Abstract
An in-depth understanding of the physicochemical properties of the organic–inorganic hybrid [NH3(CH2)2NH3]2CdBr6 whose structure corresponds to the formulation [NH3(CH2)2NH3]2CdBr4· 2Br is essential for its application in batteries, supercapacitors, and fuel cells. Therefore, this study aimed to determine the crystal structure, phase transition, structural geometry, and molecular dynamics of these complexes. Considering its importance, a single crystal of [NH3(CH2)2NH3]2CdBr6 was grown; the crystal structure was found to be monoclinic. The phase transition temperatures were determined to be 443, 487, 517, and 529 K, and the crystal was thermally stable up to 580 K. Furthermore, the 1H, 13C, 14N, and 113Cd NMR chemical shifts caused by the local field surrounding the resonating nucleus of the cation and anion varied with increasing temperature, along with the surrounding environments of their atoms. In addition, 1H spin–lattice relaxation time T1ρ and 13C T1ρ, which represent the energy transfer around the 1H and 13C atoms of the cation, respectively, varied significantly with temperature. Consequently, changes in the coordination geometry of Br around Cd in the CdBr6 anion and the coordination environment around N (in the cation) were associated with changes in the N–H···Br hydrogen bond. The structural geometry revealed critical information regarding their basic mechanism of organic–inorganic hybrid compounds.
Similar content being viewed by others
Introduction
Organic–inorganic perovskite-type compounds have been extensively studied in the field of photoelectronics and have been widely applied in systems such as solar cells and light-emitting devices1,2,3,4,5,6. The physicochemical properties and structural phase transitions of organic–inorganic compounds are related to their structure and the interactions between cations and anions7. Recent advances in the development of organic–inorganic hybrid compound-based solar cells have increased the demand for characterization of the dynamics and structures of their various constituents in relation to their potential impact8. In this study, the aforementoined properties were elucidated.
Organic–inorganic compounds based on zero- and two-dimensional perovskites [NH3(CH2)nNH3]BX4 (n = 2, 3, ∙∙∙; B = 55Mn, 59Co, 63Cu, 65Zn, 113Cd; X = Cl, Br)9,10,11,12,13,14,15,16,17 and [CnH2n+1NH3]2BX412, 18,19,20 are of interest owing to their high thermal stability and broad application scope. Studies on [NH3(CH2)nNH3]BX2X2', containing different halogen ions, were reported by Abdel-Aal et al.21,22,23. An interesting group of hybrid compounds include perovskite-type layered compounds containing cations and layered metal-halogen anions. The physical properties of these compounds are attributed to the N‒H···X hydrogen bonds, between their cations and anions13, 14, 24,25,26. The structural flexibility and nonlinear optical properties of these perovskites are attributed to the organic material, whereas their thermal and mechanical properties are related to the inorganic material27, 28. These compounds are attractive because of their diverse crystal structures and phase transitions, which correlate with their cationic and anionic structural dynamics.
[NH3(CH2)2NH3]2CdBr6 crystals, namely [NH3(CH2)2NH3]2CdBr4·2Br, similar to the compounds mentioned above, were grown, and the structure of this single crystal and nuclear quadrupole resonance (NQR) studies of 79Br and 81Br were reported by Krishnan et al.29. The structure at 300 K was reported as monoclinic, space group P21/m with the lattice constants a = 6.69 Å, b = 20.50 Å, c = 6.37 Å, β = 93.4°, and Z = 4. The two N(1) and N(2) atoms of [NH3(CH2)2NH3] cation were crystallographically inequivalent. Although the 79,81Br NQR spectrum experiment according to the temperature change and the crystal structure at 300 K was reported, the nuclear magnetic resonance (NMR) spectrum and spin–lattice relaxation time experiment for other nuclei were not performed.
In this study, single crystals of [NH3(CH2)2NH3]2CdBr6 whose structure corresponds to the formulation [NH3(CH2)2NH3]2CdBr4∙2Br were grown using an aqueous solution method, and their structures and phase transition temperatures (TC) were measured by the single crystal x-ray diffraction (SCXRD), powder x-ray diffraction (PXRD), and differential scanning calorimetry (DSC) experiments. To investigate the role of the [NH3(CH2)2NH3] cation in this single crystal, 1H magic-angle spinning (MAS) NMR, 13C MAS NMR, and 14N static NMR spectra were obtained as a function of temperature. In addition, the role of the CdBr6 anion was considered by the temperature-dependent chemical shift of 113Cd through 113Cd MAS NMR, and from this result, the N‒H∙∙∙Br hydrogen bond between the cation and anion was discussed. 1H T1ρ and 13C T1ρ, which represent the energy transfer around the 1H and 13C atoms, was also considered. The results of the single-crystal structure and physicochemical properties predicted important information on the basic mechanism of organic–inorganic hybrid compounds.
Methods
Materials
Single crystals of [NH3(CH2)2NH3]2CdBr6, namely [NH3(CH2)2NH3]2CdBr4·2Br, were prepared from NH2(CH2)2NH2·2HBr (Aldrich, 98%) and CdBr2∙4H2O (Aldrich, 98%) with a ratio of 2:1 in ultra pure distilled water. The mixed substances were stirred and heated to obtain a homogeneous solution. Subsequently, the resulting solution was filtered, and transparent colorless single crystals were grown by slow evaporation in a thermostat at 300 K over three weeks. The crystals grew into rectangular shapes with dimensions of 5 × 5 × 1 mm.
Characterization
The structures of the [NH3(CH2)2NH3]2CdBr4·2Br crystal at 250, 300, and 350 K were determined by SCXRD at the Korea Basic Science Institute (KBSI) Seoul Western Center. A crystal was mounted on a Bruker D8 Venture PHOTON III M14 diffractometer with a Mo–Ka radiation source. Data was collected and integrated using SMART APEX3 and SAINT (Bruker). The absorption was corrected using the multi-scan method implemented in SADABS. The structure was solved using direct methods and refined by full-matrix least-squares on F2 using SHELXTL30. All non-hydrogen atoms were refined anisotropically and hydrogen atoms were added to their geometrically ideal positions. The PXRD patterns were measured at several temperatures using an XRD system with a Mo–Ka radiation source.
DSC measurements were performed using a DSC instrument (TA Instruments, DSC 25) at a heating rate 10 °C/min, in the 200–553 K temperature range with the amount of the sample of 5.4 mg. The optical observations were measured by an optical polarizing microscope in the 300–573 K at heating stage of a Linkam THM-600. Thermal gravimetric analysis (TGA) was also performed with a heating speed 10 °C/min in the temperature between 300 and 873 K, under nitrogen gas.
The NMR spectra of the [NH3(CH2)2NH3]2CdBr6 crystals were measured using a solid-state 400 MHz NMR spectrometer (AVANCE III, Bruker) at the same facility, KBSI Western Seoul Center. To minimize the spinning sideband, samples in cylindrical zirconia rotors were spun at a spinning rate of 10 kHz for the MAS NMR measurements. Chemical shifts were referenced to adamantane for 1H and tetramethylsilane (TMS) for 13C, as standard materials for accurate NMR chemical shift measurements. MAS spin–lattice relaxation time T1ρ values for 1H and 13C were performed using a π/2 − τ pulse by a spin-lock pulse of duration τ, and the π/2 pulse widths were measured using a previously reported method28. Further, static 14N NMR and 113Cd MAS NMR spectra of a [NH3(CH2)2NH3]2CdBr6 single crystal were measured at Larmor frequencies of 28.90 and 133.13 MHz, respectively, and the chemical shift was referenced with respect to NH3NO3 and CdCl2O8·6H2O as standard samples, respectively. 14N NMR spectra were obtained using a solid-state echo sequence.
Experimental results
Single-crystal XRD
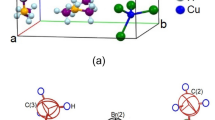
Single-crystal XRD results for the [NH3(CH2)2NH3]2CdBr6 crystals were obtained at 250, 300, and 350 K. At 300 K, the crystal grew a monoclinic system with P21/m space group and lattice constants a = 6.393 (3) Å, b = 20.552 (8) Å, c = 6.710 (4) Å, β = 93.34° (3), and Z = 2. The thermal ellipsoids and atomic numbering for each atom are shown in Fig. 1, and the SCXRD data at 250, 300, and 350 K of the [NH3(CH2)2NH3]2CdBr6 crystal are shown in Table 1. Here, the lattice constants for the single crystals at 250 and 350 K were almost identical to those at 300 K. The two nitrogen atoms of the [NH3(CH2)2NH3] cation were crystallographically inequivalent, and the anion consists of a isolated tetrahedra [CdBr4]2- and separate bromine ions [Br]-. The hydrogen atoms of each formula unit formed hydrogen bonds N − H∙∙∙Br between the anions and cations. The bond lengths and angles at 250, 300, and 350 K are listed in Table 2.

Thermal ellipsoid plot (50% probability) for [NH3(CH2)2NH3]2CdBr4·2Br structure at 300 K.
Phase transition and powder XRD
Figure 2 shows the results of the DSC experiment within the temperature range of 200–553 K with a heating rate of 10 °C/min. Three strong endothermic peaks at 443, 517, and 529 K, and one weak peak at 487 K were observed. The enthalpies of their peaks were 5.78, 0.28, 3.79, and 3.87 kJ/mol, respectively.

Differential scanning calorimetry curve of [NH3(CH2)2NH3]2CdBr4·2Br.
To confirm whether the peaks in the DSC results shown in Fig. 2 are the phase transition temperature or melting point, a PXRD experiment was performed according to the temperature change. The PXRD patterns in the measuring range of 5–65° (2θ) are shown in Fig. 3 at several temperatures. The PXRD patterns below 443 K (olive) differ slightly from those recorded at 473 K (red); this difference is related to TC1 (= 443 K). Furthermore, the PXRD pattern recorded at 473 K varied from that recorded at 493 K (blue), and the PXRD pattern at 493 K differed from those obtained at 523 K (orange) and 543 K (black), related to TC2, TC3, and TC4 exhibiting a clear change in structure. The PXRD result was consistent with that of the DSC experiment, and it was determined that the four peaks corresponded to the phase transition temperatures. Finally, the pattern at 573 K was completely different from that at temperatures below 573 K, and crystallinity was not observed, indicating that it was the melting point.

Powder x-ray diffraction patterns of [NH3(CH2)2NH3]2CdBr4·2Br at different temperatures.
Finally, the appearance of a single crystal was observed using an optical polarizing microscope. The crystal was colorless without significant change until the temperature was raised from 300 to 550 K. The surface of the single crystal began to melt at approximately 570 K. The phase transition and melting temperatures shown in the PXRD and optical polarizing microscope results are consistent with the temperatures shown in the endothermic peaks of the DSC curve. From the DSC, PXRD, and polarizing microscopy experiments, the phase transition and melting temperatures were determined as TC1 = 443 K, TC2 = 487 K, TC3 = 517 K, TC4 = 529 K, and Tm = 570 K.
Thermal property
TGA curve was obtained, and the result is shown in Fig. 4. The TGA results revealed that this crystal is thermally stable up to 580 K. As shown in Fig. 4, the initial weight loss of [NH3(CH2)2NH3]2CdBr6 began at 580 K, representing a partial decomposition temperature with a weight loss of 2%. In the TGA curve, [NH3(CH2)2NH3]2CdBr6 exhibited a two-stage decomposition at high temperatures. The initial weight loss (50%) occurred in the range of 600–650 K, and second-stage decomposition (80%) occurred in the range of 650–800 K due to the inorganic moieties. The amount remaining was calculated from the TGA data and chemical reactions. The weight loss of 45% at approximately 650 K is likely due to the decomposition of the 4HBr moiety. The weight loss of 62% was mainly attributed to organic decomposition. One endothermic peak at 486 K on the DTA curve was assigned to the phase transition temperature obtained from the DSC results.

Thermogravimetry and differential thermal analysis curves of [NH3(CH2)2NH3]2CdBr4·2Br.
1H and 13C chemical shifts and spin–lattice relaxation times
The NMR chemical shifts for 1H in the [NH3(CH2)2NH3]2CdBr6 crystal were recorded by NMR spectroscopy using variable temperature analysis. At all temperatures, the resonance signals for NH3 and CH2 overlap, as shown in Fig. 5. The sidebands for the 1H signal are indicated by the open circles. Owing to the overlap of the 1H signals, the right side of the full width at half maximum (FWHM) appears to be slightly wider than the left side; the 1H chemical shift in NH3 on the left side and the 1H chemical shift in CH2 on the right side appear to overlap. The chemical shift and line width for 1H NMR spectrum at 300 K has 7.79 ppm and 7.85 ppm, respectively. The 1H chemical shifts showed a slight increase with temperature, hence, the coordination geometry around 1H changed with temperature.

In-situ 1H MAS NMR chemical shifts for [NH3(CH2)2NH3]2CdBr4·2Br as a function of temperature.
The 13C MAS NMR chemical shifts in the cation of [NH3(CH2)2NH3]2CdBr6 were also measured with increasing temperature, as shown in Fig. 6. The environments of the two CH2 groups, as shown in the cation structure of the crystal, were identical, thus only one resonance signal for CH2 was obtained. At 300 K, a 13C NMR chemical shift was observed at 47.71 ppm, and the line width was 1.75 ppm, which was very small compared to the 1H line width. The signal intensities at 360 K and 380 K are relatively small, and therefore, they are enlarged and shown again on the left side of Fig. 6. As the temperature increased, the chemical shifts increased without any anomalous change i.e., the coordination geometry of 13C continuously changed with temperature.

In-situ 13C MAS NMR chemical shifts for [NH3(CH2)2NH3]2CdBr4·2Br as a function of temperature (inset: 13C MAS NMR chemical shifts for [NH3(CH2)2NH3]2CdBr4·2Br as a function of temperature).
To obtain the spin–lattice relaxation time T1ρ, the intensities of NMR spectrum for 1H and 13C NMR spectra were measured while increasing the delay times at a given temperature. The decay curves, according to the change in the signal intensities and delay times, are expressed as follows31,32,33,34:
where M(t) is the resonance at time t and M(0) is the intensity of the resonance line at time t = 0. The magnetization decay curves were fitted using Eq. (1), thus, the T1ρ values of 1H and 13C in [NH3(CH2)2NH3]2CdBr6 are shown in Fig. 7 as a function of the inverse temperature. The 1H T1ρ values were strongly dependent on temperature. The 1H T1ρ value increased rapidly from 5 to 70 ms between 193 and 280 K, and it decreases slightly with increasing temperature. T1ρ values of 1H have minimum values at 200 K, indicating molecular motion according to the Bloembergen–Purcell–Pound (BPP) theory. It is clear that the T1ρ minimum is attributable to the reorientational motion of NH3 and CH2. The experimental value of T1ρ is expressed by the correlation time τC for the molecular motion by the BPP theory, where the T1ρ value is given by32, 34:

1H and 13C T1ρ of [NH3(CH2)2NH3]2CdBr4·2Br as a function of 1000/temperature (inset: Correlation times for inverse temperature for 1H at low temperature; the lines represent activation energies).
Here, C is a constant; γH and γC are the gyromagnetic ratios for protons and carbon, respectively; r is the distance of the C-H internucleus; ω1 is the spin-lock field; and ωC and ωH are the Larmor frequencies for carbon and protons, respectively. The data was analyzed assuming that T1ρ was the minimum when ω1τC = 1, and the relationship between T1ρ and the radio frequency power of the spin-lock pulse ω1 was applicable. Because the T1ρ curves were found to exhibit minima, the coefficient C in Eq. (2) could be determined. Using this C, we calculated τC as a function of temperature. The local field fluctuation is governed by the thermal motion of protons and carbons, which are activated by thermal energy. The correlation time τC of motion is generally assumed to have Arrhenius dependence on the activation energy for motion and temperature33, 34
where Ea and kB are the activation energy of the motions and Boltzmann constant, respectively. The magnitude of Ea depends on molecular dynamics. To determine the molecular dynamics, we considered τC on a logarithmic scale vs. 1000/T as shown inset of Fig. 7; it was found to be 18.24 ± 1.25 kJ/mol at low temperature.
In contrast, the 13C T1ρ value, unlike 1H T1ρ, shows an almost constant value below 300 K, but the T1ρ value shows a tendency to decrease rapidly at temperatures above 300 K. The behaviors of the T1ρ for Arrhenius-type molecular motions with relaxation time are split into fast- and slow-motion parts. Fast motion is represented as ω1τC « 1, T1ρ–1 ~ exp(Ea/kBT), and the slow motion as ω1τC » 1, T1ρ ~ ω1–2 exp(–Ea/kBT). Different limits were satisfied for ω1τC in each temperature range, separated by T = 300 K. The 1H T1ρ and 13C T1ρ values at high temperatures were in the slow-motion regime, whereas the 13C T1ρ values at low temperatures were attributed to the fast-motion regime. As represented by the blue lines in Fig. 7, Ea = 2.85 ± 0.16 kJ/mol for 1H at high temperature, and Ea = 0.48 ± 0.29 kJ/mol for 13C at low temperature, whereas Ea = 50.98 ± 3.33 kJ/mol for 13C at high temperature (see Table 3).
Static 14N NMR chemical shifts
To investigate the structural geometry of 14N in the [NH3(CH2)2NH3] cation, static 14N NMR experiments were performed over a temperature range of 180–410 K using the solid-state echo method. The advantage of 14N NMR is its relatively small quadrupole coupling constant and the relatively simple structure of the NMR spectra by the spin quantum number (I = 1)35, 36. The applied magnetic field was measured in an arbitrary direction in the single crystal. The 14N NMR spectrum expected two 14N NMR signals because the spin number I = 1. Figure 8 shows two sets of signals, represented as squares and triangles, which are due to the crystallographically different N(1) and N(2) sites. At 300 K, the separation of the pair of lines was approximately 5405 and 4816 ppm for N(1) and N(2), respectively. As the temperature increased, the separation became narrower. This indicates that the quadrupole constant decreased as the temperature increased. N(1) is connected to Br(1), Br(2), and Br(3) by hydrogen bonds, whereas N(2) is connected to the surrounding Br via hydrogen bonds. The changes in the surrounding environment of N(1) and N(2), according to temperature change, were similar. In addition, this phenomenon was attributed to the changes in the structural coordinates and indicated the changes in atomic configurations around the 14N nuclei. The linewidth at 300 K was approximately 41.94 ppm, which was broader than those observed in the 1H and 13C NMR spectra. At temperatures above 330 K, it was difficult to obtain a signal because the intensity of the 14N signal was small and the line width was wide.

Static 14N NMR chemical shifts of [NH3(CH2)2NH3]2CdBr4·2Br single crystal as a function of temperature.
113Cd NMR chemical shifts
The chemical shift among the NMR parameters is very useful for clarifying the coordination environments around Cd2+ with unknown structures using 113Cd NMR spectroscopy37. In-situ 113Cd MAS NMR experiments, as a function of temperature, were employed to examine the structural environment in CdBr6 anions of [NH3(CH2)2NH3]2CdBr6, as shown in Fig. 9. NMR chemical shifts were recorded using CdCl2O8.6H2O as the standard. The sidebands of the 113Cd NMR signal are marked with asterisks as small signals located at equal intervals on both sides of the central peak. Unlike the 1H and 13C chemical shifts, the chemical shifts of 113Cd continuously decreased with increasing temperature from 200 to 420 K, as shown in Fig. 10. These are related to the change in the position of Br around Cd from the SCXRD results at 250, 300, and 350 K. The line width for 113Cd at 300 K is 6.23 ppm, and the 113Cd line width sharply narrows at temperatures above 260 K, which means that molecular motion becomes very active at high temperatures (Fig. 10). This result indicates that the environment of the Cd atom surrounded by Br atoms continuously changes with increasing temperature.

In-situ 113Cd MAS NMR chemical shifts of [NH3(CH2)2NH3]2CdBr4·2Br single crystal as a function of temperature.

113Cd MAS NMR chemical shifts and line widths of [NH3(CH2)2NH3]2CdBr4·2Br single crystal as a function of temperature.
Conclusion
We analyzed the crystal structures, phase transition temperatures, thermal behavior, and structural dynamics of the organic–inorganic hybrid [NH3(CH2)2NH3]2CdBr6, namely [NH3(CH2)2NH3]2CdBr4·2Br, crystals to investigate their physicochemical properties. Firstly, the monoclinic structure of this crystal was confirmed by single-crystal XRD, and the TC of 443, 487, 517, and 529 K were determined using DSC and powder XRD results. This crystal had a good thermal stability of approximately 580 K, and mass loss was observed with increasing temperature owing to thermal decomposition, resulting in the loss of the 2HBr and 4HBr moieties. Secondly, the chemical shifts were caused by the local field around the resonating nucleus, and the 1H, 13C, 14N, and 113Cd NMR chemical shifts of the [NH3(CH2)2NH3] cation and CdBr6 anion varied with increasing temperature, suggesting that the surrounding environment changes with temperature; changes in the chemical shifts of 1H, 13C, 14N, and 113Cd were explained as affecting the coordination geometry of the hydrogen bond N‒H···Br connecting the cation and anion. From the SCXRD results, the position of Br around Cd was changed more than other atoms at 250, 300, and 350 K shown in CIF file, and the changes of the coordination geometry of Br around Cd in the CdBr6 and the coordination environments around N were related with changes in the hydrogen bond N‒H···Br. And, the displacement parameters of H bonded to N(1) in NH3 as shown in the supplementary data were the largest in relation to the change of the hydrogen bond N(1)-H-Br. In addition, 1H T1ρ and 13C T1ρ, which represent the energy transfer around the 1H and 13C atoms of the cation, vary significantly with temperature. As show in Table 3, the Ea for molecular motion was found to be very high at 13C at high temperature compared to 1H. And, the fact that the line width of 14N is wider than those of 1H, 13C, and 113Cd means that the molecular motion of 14N is relatively rigid. This work serves to provide an understanding of these fundamental properties to broaden the application of organic– inorganic hybrid compounds.
Data availability
The datasets generated and/or analysed during the current study are available in the CCDC 2246614, 2246616, and 2246615. For ESI and crystallographic data in CIF or other electronic format see https://doi.org/.
References
Mahfoudh, N., Karoui, K., Jomni, F. & Rhaiem, A. B. Structural phase transition, thermal analysis, and spectroscopic studies in an organic–inorganic hybrid crystal: [(CH3)2NH2]2ZnBr4. Appl. Organo. Chem. 34, e5656 (2020).
Gao, Y.-F., Zhang, T., Zhang, W.-Y., Ye, Q. & Da-Wei, F. Great advance in high Tc for hybrid photoelectric-switch bulk/film coupled with dielectric and blue-white light. J. Mater. Chem. C 7, 9840 (2019).
Yu, N. et al. Flexible thin film and bulk switchable relaxor coexisting most optimal 473 nm blue light without blue-light hazard/visual injury. J. Phys. Chem. C 123, 28385 (2019).
Mathlouthi, M., Valkonen, A., Rzaigui, M. & Smirani, W. Structural characteri-zation, spectroscopic, thermal, AC conductivity and dielectric properties and antimicrobial studies of (C8H12N)2[SnCl6]. Phase Transit. 90, 399 (2017).
Hua, X.-N. et al. Switchable dielectric phase transition with drastic symmetry breaking in a Sn(IV)-based perovskite-type halide semiconductor. J. Phys. Chem. C 123, 21161 (2019).
Ishihara, H. et al. Isolated versus condensed anion structure: The influence of the cation size and hydrogen bond on structure and phase transition in MX42- complex salts. 2,2-Dimethyl-1,3-propanediammonium Tetrabromocadmate (II) Monohydrate, Di-methylammonium Tetrabromozincate (II), and Dimethylammonium Tetra bromocadmate (II). Z. Naturforsch A 51, 1027 (1996).
Czupinski, O., Ingram, A., Kostrzewa, M., Przeslawski, J. & Czapla, Z. On the structural phase transition in a perovskite-type diaminopropanetetrachloro-cuprate(II) NH3(CH2)3NH3CuCl4 crystal. Acta Phys. Pol., A 131, 304 (2017).
Franssen, W. M. J. & Kentgens, A. P. M. Solid-state NMR of hybrid halide perovskites. Solid State Nucl. Magn. Reson. 100, 36 (2019).
Rao, C. N. R., Cheetham, A. K. & Thirumurugan, A. Hybrid inorganic-organic materials: a new family in condensed matter physics. J. Phys. Condens. Matter 20, 83202 (2008).
Cheng, Z. & Lin, J. Layered organic–inorganic hybrid perovskites: structure, optical properties, film preparation, patterning and templating engineering. Cryst. Eng. Com. 12, 2646 (2010).
Mostafa, M. F. & El-khiyami, S. S. Crystal structure and electric properties of the organic-inorganic hybrid: [(CH2)6(NH3)2]ZnCl4. J. Solid State Chem. 209, 82 (2014).
Gonzalez-Carrero, S., Galian, R. E. & Perez-Prieto, J. Organometal halide perov-skites: Bulk low-dimension materials and nanoparticles. Part. Syst. Charact. 32, 709 (2015).
Abdel-Adal, S. K., Kocher-Oberlehner, G., Ionov, A. & Mozhchil, R. N. Effect of organic chain length on structure, electronic composition, lattice potential energy, and optical properties of 2D hybrid perovskites [(NH3)(CH2)n(NH3)]CuCl4, n = 2–9. Appl. Phys. A 123, 531 (2017).
Liu, W. et al. Giant two-photon absorption and its saturation in 2D organic-inorganic perovskite. Adv. Opt. Mater. 5, 1601045 (2017).
Mondal, P., Abdel-Aal, S. K., Das, D. & Manirul Islam, S. K. Catalytic activity of crystallographically characterized organic-inorganic hybrid containing 1,5-di-amino-pentane tetrachloro manganate with perovskite type structure. Cat. Let. 147, 2332 (2017).
Elseman, A. M. et al. Copper-substituted lead perovskite materials constructed with different halides for working (CH3NH3)2CuX4-based perovskite solar cells from experimental and theoretical view. ACS Appl. Mater. Interfaces 10, 11699 (2018).
Aramburu, J. A., Garcia-Fernandez, P., Mathiesen, N. R., Garcia-Lastra, J. M. & Moreno, M. Changing the usual interpretation of the structure and ground state of Cu2+ layered perovskites. J. Phys. Chem. C 122, 5071 (2018).
Staskiewicz, B., Czupinski, O. & Czapla, Z. On some spectroscopic properties of a layered 1,3-diammoniumpropylene tetrabromocadmate hybrid crystal. J. Mol. Struct. 1074, 723 (2014).
Staskiewicz, B., Turowska-Tyrk, I., Baran, J., Gorecki, C. & Czapla, Z. Structural characterization, thermal, vibrational properties and molecular motions in perovskite-type diaminopropanetetrachlorocadmate NH3(CH2)3NH3CdCl4 crystal. J. Phys. Chem. Solids 75, 1305 (2014).
Czapla, Z. et al. Structural phase transition in a perovskite-type NH3(CH2)3NH3 CuCl4 crystal X-ray and optical studies. Phase Transit. 90, 637 (2017).
Abdel-Aal, S. K. & Ouasri, A. Crystal structure, Hirshfeld surfaces and vibrational studies of tetrachlorocobaltate hybrid perovskite salts NH3(CH2)nNH3CoCl4 (n = 4, 9). J. Mol. Struct. 1251, 131997 (2022).
Abdel-Aal, S. K., Kandeel, M. F., El-Sherif, A. F. & Abdel-Rahman, A. S. Synthesis, characterization, and optical properties of new organic–inorganic hybrid perovskites [(NH3)2(CH2)3]CuCl4 and [(NH3)2(CH2)4]CuCl2Br2. Phys. Stat. Solidi 218, 2100036 (2021).
Abdel-Aal, S. K. et al. Crystal structure, vibrational spectroscopy and optical properties of a one-dimensional organic–inorganic hybrid perovskite of [NH3CH2CH(NH3)CH2]BiCl5. Acta Cryst. B 75, 880 (2019).
Przeslawski, J., Czapla, Z., Crofton, M. & Dacko, S. On the “inverted” phase transitions in ferroic crystals containing propylenediammonium cations. Ferroelectrics 534, 220 (2018).
Bissey, J.-C., Filloleau, N., Chanh, N.-B., Berger, R. & Flandrois, S. Exchange interaction as studied by EPR in a two-dimensional molecular composite [NH3-(CH2)4-NH3]MnCl4. Solid State Commun. 106, 385 (1998).
Lim, A. R., Kim, S. H. & Joo, Y. L. Physicochemical properties and structural dynamics of organic–inorganic hybrid [NH3(CH2)3NH3]ZnX4 (X = Cl and Br) crystals. Sci. Rep. 11, 8408 (2021).
Zhang, W. & Xiong, R.-G. Ferroelectric metal–organic frameworks. Chem. Rev. 112, 1163 (2012).
Lim, A. R. Effect of methylene chain length of perovskite-type layered [NH3(CH2)nNH3]ZnCl4 (n=2, 3, and 4) crystals on thermodynamic properties, structural geometry, and molecular dynamics. RSC Adv. 11, 37824 (2021).
Krishnan, V. G., Dou, S.-Q., Paulus, H. & Weiss, A. Solid phases from the liquid system H2N(CH2)2NH2-CdBr2-HBr-H2O. An X-ray and 79,81Br NQR study of (H3N(CH2)2NH3)CdBr4, (H3N(CH2)2NH3)(CdBr3)2·H2O, and (H3N(CH2)2NH3)2 CdBr6. Ber. Bunsenges. Phys. Chem. 95, 1256 (1991).
SHELXTL ν 6.10, Bruker AXS, Inc., Madison, Wisconsin, USA, (2000).
Lim, A. R. & Kim, S. H. Physicochemical property investigations of perovskite-type layer crystals [NH3(CH2)nNH3]CdCl4 (n=2, 3, and 4) as a function of length n of CH2. ACS Omega 6, 27568 (2021).
Abragam, A. The Principles of Nuclear Magnetism (Oxford University Press, 1961).
Koenig, J. L. Spectroscopy of Polymers (Elsevier, 1999).
Harris, R. K. Nuclear Magnetic Resonance Spectroscopy (Pitman Pub, 1983).
Seliger, J., Blinc, R., Arend, H. & Kind, R. Proton-14N double resonance study of the structural phase transitions in the perovskite type layer compound (CH3NH3)2CdCl4. Z. Phys. B 25, 189 (1976).
Mulla-Osman, S., Michel, D. & Czapla, Z. 14N NMR study of the domain structure of tetramethylammonium cadmium chloride (TMCC). Phys. Stat. Sol. 236, 173 (2003).
Sakida, S. & Kawamoto, Y. J. 113Cd MAS and static NMR study of halogeno-cadmate crystals. Phys. Chem. Solid 63, 151 (2002).
Acknowledgements
This research was supported by the Basic Science Research program through the National Research Foundation of Korea (NRF), funded by the Ministry of Education, Science, and Technology (2018R1D1A1B07041593 and 2016R1A6A1A03012069).
Author information
Authors and Affiliations
Contributions
A.R.L. designed the project, performed NMR and XRD experiments, and wrote the manuscript.
Corresponding author
Ethics declarations
Competing interests
The author declares no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Lim, A.R. Investigating the physicochemical properties, structural attributes, and molecular dynamics of organic–inorganic hybrid [NH3(CH2)2NH3]2CdBr4·2Br crystals. Sci Rep 13, 6133 (2023). https://doi.org/10.1038/s41598-023-33192-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-33192-1
- Springer Nature Limited