
Abstract
Organic–inorganic hybrid perovskites can potentially be used in electrochemical devices, such as batteries and fuel cells. In this study, the structure and phase transition temperatures of the organic–inorganic material [NH3(CH2)5NH3]CdCl4 crystal were confirmed by X-ray diffraction and differential scanning calorimetry. From the nuclear magnetic resonance results, the crystallographic configurations of 1H, 13C, and 14N in the cation changed at temperatures close to TC1 (336 K), whereas that of 113Cd in the anion shows significant changes at temperatures close to TC1 and TC2 (417 K). The activation energy, Ea, values for 1H and 13C obtained from the spin–lattice relaxation time, T1ρ, below and above TC1 were evaluated, where the Ea value for 13C was more flexible at low temperatures than at high temperatures. In addition, the effect on molecular motion was effective at high temperatures. The phase transition at 336 K was associated with the change in the N–H···Cl bond due to the change in the coordination geometry of Cl around Cd in the CdCl6 anion. On the other hand, the phase transition at 417 K was related to the ferroelastic phase transition attributed to the twin domains.
Similar content being viewed by others
Introduction
Recently, many studies with the development of functional materials are being conducted on organic–inorganic hybrid perovskite materials. The organic–inorganic hybrid crystal [NH3(CH2)nNH3]BX4 (n = 2, 3, 4, …), where B is a transition metal, such as Mn, Cd, Fe, Cu …, and X is a halogen ion, crystallizes perovskite-type layer structures1,2,3,4,5,6,7,8,9,10,11,12. The organic part of the hybrid complex determines the optical properties and structural flexibility, whereas the inorganic part affects the mechanical and thermal properties13. The properties and structural phase transitions of organic–inorganic hybrid compounds are affected by their structures and the interactions between cation and anion12. For chains in which n \(\gg \) 4, structural rearrangement by conformational changes in the chains becomes important. An interesting group of hybrid compound is the perovskite-type layer [NH3(CH2)5NH3]CdCl4 (pentylenediammonium cadmium tetrachloride) containing a [NH3(CH2)5NH3] cation and a two-dimensional (2D) layered CdCl6 anion. [NH3(CH2)5NH3]CdCl4 has two structural phase transitions at temperatures near 337 K (TC1) and 417 K (TC2)14,15. It exhibits an unusual phase sequence, in which the phase that is stable at high temperatures exhibits the lowest symmetry.
The phase sequence in the following way16

Phases III (below 337 K) and II (above 337 K) are orthorhombic with the space groups Pnam and Imam, respectively. The lattice constants in phase III (at 293 K) are a = 7.330 Å, b = 7.504 Å, c = 23.862 Å, and Z = 4, while the unit cell parameters in phase II (at 353 K) are a = 7.376 Å, b = 7.561 Å, c = 23.555 Å, and Z = 4. The high-temperature phase I is monoclinic, and the unit cell parameters at 433 K are a = 7.516 Å, b = 7.563 Å, c = 11.22 Å, and β = 98.15° with the space group C12/m1. The [NH3(CH2)5NH3] organic chains are arranged along the longest c-axis. The Cd octahedra is located the edge to form a 2D network, and the diammonium cations are connected to CdCl6 octahedra by hydrogen bonds. In the inorganic layers, the structural geometries around the Cd atoms are described as distorted octahedra. These hybrid perovskite materials have potential applications in various electrochemical devices, such as batteries and fuel cells17,18,19,20,21,22,23,24,25.
The synthesis and characterization of [NH3(CH2)5NH3]CdCl4 were first discussed by Kind et al.26, where the structural phase transitions were studied using 35Cl and 2D nuclear magnetic resonance (NMR), birefringence, dilatation measurements, and optical domain investigations. Negrier et al.15 evaluated the crystal structures via X-ray diffraction (XRD) and Raman scattering experiments at 293 K and 353 K. Our group has also recently reported the effects of 13C length in the cation of [NH3(CH2)2NH3]CdCl4, [NH3(CH2)3NH3]CdCl4, and [NH3(CH2)4NH3]CdCl4 crystals on the thermal and structural dynamic properties13. Meanwhile, a lot of research has been done on the electrical and conductive properties of this type of compound16,27,28,29,30.
Here, the crystal structures, thermodynamic properties, and ferroelastic domain walls of [NH3(CH2)5NH3]CdCl4 were investigated. The roles of cations and anions in the [NH3(CH2)5NH3]CdCl4 single crystal were discussed, and the chemical shifts and spin-lattice relaxation time, T1ρ, with increasing temperature were measured using 1H magic angle spinning (MAS) NMR, 13C MAS NMR, and static 14N NMR to identify the roles of the [NH3(CH2)5NH3] cation. Furthermore, the 113Cd MAS NMR chemical shifts were recorded to evaluate the coordination geometry of the CdCl6 anion. The results would provide insights into the physicochemical properties of [NH3(CH2)5NH3]CdCl4 crystals, facilitating their various applications in the future.
Methods
A saturated aqueous solution containing NH2(CH2)5NH2·2HCl and CdCl2 was gradually evaporated at 300 K to grow single crystals of [NH3(CH2)5NH3]CdCl4. Colorless single crystals measuring approximately 7 mm × 3 mm × 2 mm were grown for approximately 2–3 weeks in the thermostat.
The structures of the [NH3(CH2)5NH3]CdCl4 crystals at 298 K were analyzed using an XRD system. The lattice parameter and space group was considered by single-crystal XRD at the Seoul Western Center of the Korea Basic Science Institute. Experiments were performed in the same manner as before31.
Differential scanning calorimetry (DSC) (TA, DSC 25) experiments were carried out at a heating rate of 10 K/min from 190 to 550 K in N2 gas. Thermogravimetric analysis (TGA) and differential thermal analysis (DTA) curves were obtained using a thermogravimetric analyzer (TA Instrument) with the same heating rate as in DSC from 300 to 973 K in N2 gas. In addition, the domain patterns were observed using an optical polarizing microscope within the temperature range of 300 to 450 K, where the prepared single crystals were placed on the plate with the temperature sensor of a Linkam THM-600.
NMR spectra of the [NH3(CH2)5NH3]CdCl4 crystals were performed using a Bruker 400 MHz Avance II+ solid-state NMR spectrometer in the same facility. The Larmor frequencies for 1H and 13C MAS NMR experiments were 400.13 and 100.61 MHz, respectively. In MAS NMR experiment, the spinning speed was set to 10 kHz to minimize sideband. And tetramethylsilane (TMS) was used as a standard material to obtain accurate NMR chemical shift. The experimental method to obtain the T1ρ values for 1H and 13C was used in the same way as the previously reported method13. And, static 14N NMR and 113Cd MAS NMR spectra were recorded at Larmor frequencies of 28.90 and 88.75 MHz, respectively. 14N and 113Cd chemical shift measurements were performed using NH4NO3 and CdCl2O8·6H2O as standard materials.
Experimental results
Crystal structure
The powder XRD pattern of the [NH3(CH2)5NH3]CdCl4 crystal at 298 K is shown in Fig. 1. And, the lattice constants analysized from the X-ray crystal diffraction were determined to be a = 7.3292 ± 0.002 Å, b = 7.5058 ± 0.002 Å, and c = 23.9376 ± 0.006 Å with the space group Pnam; this is consistent with the previously reported results14,15.

XRD powder pattern of the [NH3(CH2)5NH3]CdCl4 crystal at 298 K.
Phase transition temperature, thermal property, and ferroelastic twin domain
The DSC curves of the [NH3(CH2)5NH3]CdCl4 crystal at a heating and cooling rate of 10 K/min in N2 gas are presented in Fig. 2. Two endothermic peaks were observed at 336 K (TC1) and 418 K (TC2) during heating, whereas two exothermic peaks were recorded at 327 K (TC1′) and 407 K (TC2′) during cooling. The phase transition enthalpy on heating is 3.17 kJ/mol at 337 K and 0.55 kJ/mol at 417 K, respectively. On the other hand, previous studies reported endothermic peaks at 337 K and 417 K during heating and at 336 K and 407 K during cooling14,15.

DSC curves of [NH3(CH2)5NH3]CdCl4 during heating and cooling.
To determine the preliminary thermal characteristics, including the structural phase transitions, TGA and DTA results were conducted at the same heating rate as the DSC experiment. Based on the TGA and DTA curves shown in Fig. 3, the crystal exhibited excellent stability up to approximately 600 K. The small inflection points observed at temperatures near 336 K and 417 K in the DTA curve were coincides with the two phase transition temperatures obtained from the DSC results, suggesting that the molecular weight of [NH3(CH2)5NH3]CdCl4 decreased at increasing temperatures. The amount of crystal remaining in the solid state was evaluated from the molecular weights. The 10% and 20% weight losses of the crystal at temperatures of about 617 K and 626 K were attributed to the decomposition of HCl and 2HCl, respectively. On the other hand, the weight loss at approximately 800 K and 900 K shown in Fig. 3 was observed 46% and 87%, respectively.

TGA and DTA curves of [NH3(CH2)5NH3]CdCl4.
A single crystal with ferroelastic properties exhibits two or more orientation states even if mechanical stress does not exist since mechanical stress can change the existing orientation state of the single crystal. Polarized microscopy observations revealed the ferroelastic domain structures of the crystal and their changes at the phase transition temperatures, as shown in Fig. 4. The domain pattern represented by parallel lines was not observed in phases III (300 K, Fig. 4a) and II (403 K, Fig. 4b). No change in the behavior of the crystal was observed at TC1. However, in phase I, twinning occurred in the crystal at temperatures above TC2, resulting in a highly dense domain pattern indicated by the red circle (Fig. 4c). At 433 K, new domain walls indicated by the blue circles were formed next to the parallel domain walls (Fig. 4d). The phase transition at TC2 occurred due to the ferroelastic twin domain. The [NH3(CH2)5NH3]CdCl4 crystal existed in two crystallographic phases: monoclinic (2/m) at temperatures above 417 K, orthorhombic (mmm) at temperatures between 417 and 337 K, and orthorhombic (mmm) at temperatures below 337 K. According to Aizu32 and Sapriel33, for the transition from the mmm space group of the orthorhombic phase II to the 2/m space group of the monoclinic phase I, the domain wall directions were x = 0 and z = 0. The equations of the twin domain walls was expressed as 2/mFmmm, corresponding to the “inverted” mmmF2/m instead of mmmF2/m as reported by Sapriel33.

Optical polarizing microscopy images of [NH3(CH2)5NH3]CdCl4 at (a) phase III (300 K), (b) phase II (403 K), (c) phase I (420 K), and (d) phase I (433 K). The parallel lines represent the ferroelastic twin domain walls.
1H MAS NMR spectrum
The 1H MAS NMR spectra of the [NH3(CH2)5NH3]CdCl4 crystal were obtained, and the 1H chemical shifts are shown in Fig. 5 as a function of temperature. At low temperatures, only one resonance signal was observed. These resonance signals were asymmetric due to the overlapping 1H lines of NH3 and CH2 in [NH3(CH2)5NH3] cations. At 180 K, a single resonance line was present at a chemical shift of 9.04 ppm. The line width and full-width at half-maximum (FWHM) at this temperature were also different from those represented as symbol “1” at 2.97 ppm and as symbol “2” at 6.07 ppm, respectively. At 330 K, which was close to TC1, the NMR spectrum was divided into two resonance lines, showing chemical shifts of 7.56 and 2.58 ppm for NH3 and CH2, respectively. The spinning sidebands were marked with crosses and open circles. Here, phases I, II, and III were plotted in olive, red, and black, respectively. The 1H chemical shifts of NH3 and CH2, presented by dotted lines in Fig. 5, were almost independent of temperature. These results suggested that the surrounding environments of 1H of NH3 and CH2 did not change with temperature.

MAS 1H NMR spectra of [NH3(CH2)5NH3]CdCl4 at phases I, II, and III (olive areas: phase I, red areas: phase II, and black areas: phase III). The spinning sidebands are marked by crosses and open circles.
13C MAS NMR spectrum
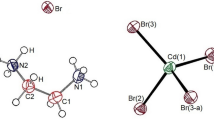
The 13C chemical shifts at increasing temperature for the in situ MAS NMR spectra are shown in Fig. 6. The TMS reference signal at 300 K recorded at 38.3 ppm was used as the standard for the 13C chemical shift. In the [NH3(CH2)5NH3] cation, CH2 located close to NH3 was designated as C-3, CH2 located at the center was designated as C-1, and CH2 located between C-3 and C-1 was designated as C-2. The structure of the cation for this crystal is shown in the inset of Fig. 6. At 300 K, the 13C chemical shifts were recorded at 28.26, 25.90, and 41.67 ppm for C-1, C-2, and C-3, respectively. The FWHM for 13C NMR at 300 K were 6.20, 5.72, and 9.06 ppm for C-1, C-2, and C-3, respectively. The line width of C-3 located close to N was wider than those of C-1 and C-2. The chemical shifts changed at temperatures close to TC1 (336 K), but not at temperatures close to TC2 (417 K). Below TC1, all 13C positions showed positive chemical shifts with increasing temperatures. Above TC1, the chemical shift of C-2 was almost independent of temperature, while the shifts in C-1 and C-3 progressed in a negative and positive direction, respectively. The results proved that below TC1, the surrounding environments of all 13C ions would change with temperature. At temperatures above TC1, the surrounding environments of C-2 did not change. However, the chemical shifts of C-1 and C-3 continuously changed in all temperature ranges, including TC1 and TC2.

MAS 13C NMR spectra of [NH3(CH2)5NH3]CdCl4 at phases I, II, and III (olive areas: phase I, red areas: phase II, and black areas: phase III).
Static 14N NMR
The 14N NMR spectra of the [NH3(CH2)5NH3]CdCl4 single crystal in the temperature range of 180–420 K were recorded by the solid-state echo method with static NMR. Since 14N has quadrupole interactions with spin number I = 1, two 14N NMR signals were expected34. The 14N resonance frequency at increasing temperatures is shown in Fig. 7. Despite the presence of intense background noise due to the very low NMR frequency (28.90 MHz), the 14N spectrum was obtained without difficulty. Here, the crystal demonstrated an arbitrary direction with respect to the magnetic field. The six resonance lines of the three pairs at increasing temperatures were below TC1. At temperatures close to 336 K (TC1), the number of resonance lines and resonance frequencies of the NMR spectra showed abrupt changes. At TC1, a reduction from three pairs to two pairs of NMR lines was observed. At TC2, another pair of NMR lines reappeared. Below TC1, as the temperature increased, the resonance frequencies increased, and above TC1, as the temperature increased, the resonance frequencies decreased. At TC2, only the number of resonance lines changed, and the resonance frequency showed almost continuous values. Symbols with the same color indicated the same pairs of 14N. Changes in the 14N resonance frequencies due to the change in temperature were related to the changes in the crystallographic configuration of the crystal.

Static 14N NMR spectra of [NH3(CH2)5NH3]CdCl4 at phases I, II, and III.
113Cd MAS NMR
The 113Cd MAS NMR experiments were measured to detect the structural environments around Cd when the temperature in the CdCl6 anions of the [NH3(CH2)5NH3]CdCl4 single crystal were varied. This information was crucial to demonstrate the anion coordination environments around Cd2+ in CdCl6 using 113Cd NMR spectroscopy. The changes in the in situ 113Cd MAS NMR spectra are shown in Fig. 8. The 113Cd chemical shift at 300 K was 323.19 ppm. As the temperature increased, the 113Cd chemical shifts slightly moved in the negative direction, but these chemical shifts changed discontinuously near TC1 and TC2. In particular, more changes were observed at temperatures near TC2 than at temperatures near TC1, suggesting that temperature affected the environments around Cd. This proved that the coordination geometry of 6Cl around Cd ions in the CdCl6 octahedra, as shown in the inset of Fig. 8, would change at the phase transition temperatures.

MAS 113Cd NMR spectra of [NH3(CH2)5NH3]CdCl4 at phases I, II, and III (olive areas: phase I, red areas: phase II, and black areas: phase III).
1H and 13C spin-lattice relaxation times
The 1H MAS NMR and 13C MAS NMR spectra were obtained with increasing delay times, and the plot of spectral intensities against increasing delay times was expressed as an exponential function. The decay rates of the spin-locked proton and carbon magnetization are expressed as the spin-lattice relaxation time, T1ρ, as34,35:
where PH(C)(τ) and PH(C)(0) are the signal intensities for the proton (carbon) at time τ and τ = 0, respectively. The 1H T1ρ values of NH3 and CH2 at several temperatures were determined by the slope of the logarithmic plots of intensities against delay times. From the slope of their recovery curves, the 13C T1ρ values for C-1, C-2, and C-3 were determined. The 1H T1ρ and 13C T1ρ values are shown in Fig. 9 as a function of the inversed temperature. The 1H T1ρ values increased rapidly from 100 to 1000 ms. While the slope of the T1ρ values at temperatures near TC1 changed, the slope at temperatures near TC2 exhibited a rather continuous value. Above TC1, the 1H T1ρ value for NH3 showed a decreasing trend. The activation energy, Ea, values for 1H in NH3 were evaluated from the slopes (represented by the solid lines in Fig. 9) of their log T1ρ versus 1000/T plots. The Ea values below TC1 were 6.65 ± 0.40 kJ/mol and 8.60 ± 2.32 kJ/mol for NH3 and CH2, respectively, while the Ea values above TC1 were 2.85 ± 0.96 kJ/mol and 3.49 ± 1.47 kJ/mol for NH3 and CH2, respectively. And, the 13C T1ρ values below TC1 increased gradually with increasing temperature and then increased rapidly above TC1. Near TC2, the T1ρ values were almost continuous, showing no significant changes. The Ea values of C-1, C-2, and C-3 below TC obtained from the plot of log T1ρ versus 1000/T were 1.73 ± 0.58 kJ/mol, 1.33 ± 0.49 kJ/mol, and 1.36 ± 0.76 kJ/mol, respectively. The Ea values of C-1, C-2, and C-3 above TC1 were 3.04 ± 1.38 kJ/mol, 5.57 ± 1.04 kJ/mol, and 0.97 ± 1.43 kJ/mol, respectively. The behavior of T1ρ for random motions with a correlation time, τC, could be described as fast- and slow-motion zones. The 1H and 13C T1ρ values at low and high temperatures correspond to the fast-motion region, where ω1τC \(\ll \) 1 and T1ρ−1 α exp(Ea/kBT). In contrast, the 1H T1ρ values in NH3 at high temperatures were attributed to the slow-motion region, where ω1τC \(\gg \) 1 and T1ρ−1 α ω1−2exp(Ea/kBT).

Temperature dependences of 1H and 13C NMR spin–lattice relaxation times, T1ρ, in [NH3(CH2)5NH3]CdCl4 near phase transition temperatures. Solid lines represent the activation energies.
Conclusion
The structure and phase transition temperatures of the [NH3(CH2)5NH3]CdCl4 crystal were confirmed using XRD and DSC. Based on the NMR analysis of the crystal, we deduced that the crystallographic surroundings of 1H, 13C, and 14N in the cation at temperatures close to TC1 changed, whereas that of 113Cd in the anion at temperatures close to TC1 and TC2 exhibited significant changes. The changes in the NMR chemical shifts near TC1 and TC2 also suggested that the N–H···Cl hydrogen bond was affected.
On the other hand, the T1ρ values of 1H in NH3 changed from fast to slow motion near TC1. The T1ρ values of 13C in CH2 increased rapidly at TC1, and the Ea values for 13C were more flexible at low temperatures than at high temperatures. By evaluating the T1ρ values, we deduced that the effect on the molecular motion was effective at high temperatures.
Consequently, the phase transition at 336 K was associated with the change in the N–H···Cl bond due to the change in the coordination geometry of Cl around Cd in the CdCl6 anion. The phase transition at 417 K was related to the ferroelastic phase transition attributed to the twin domains.
The thermodynamic properties, ferroelastic domain walls, coordination geometries, and molecular motions of [NH3(CH2)5NH3]CdCl4 in this study are thought to be helpful in the study of hybrid perovskite types for their various applications in batteries and fuel cells.
References
Pradeesh, K., Yadav, G. S., Singh, M. & Vijaya Prakash, G. Synthesis, structure and optical studies of inorganic–organic hybrid semiconductor, NH3(CH2)12NH3PbI4. Mater. Chem. Phys. 124, 44 (2010).
Saikumar, S., Ahmad, J. J., Baumberg, G. & Vijaya Prakash, G. Fabrication of excitonic luminescent inorganic–organic hybrid nano- and microcrystals. Scr. Mater. 67, 834 (2012).
Staskiewicz, B., Czupinski, O. & Czapla, Z. On some spectroscopic properties of a layered 1,3-diammoniumpropylene tetrabromocadmate hybrid crystal. J. Mol. Struct. 1074, 723 (2014).
Staskieqicz, B., Turowska-Tyrk, I., Baran, J., Gorecki, C. & Czapla, Z. Structural characterization, thermal, vibrational properties and molecular motions in perovskite-type diamono-propanetetrachlorocadmate NH3(CH2)3NH3CdCl4 crystal. J. Phys. Chem. Solids 75, 1305 (2014).
Sourisseau, C., Lucazeau, G. & Dianoux, A. J. Neutron scattering study of the reorientational motions for NH3 groups in (NH3(CH2)3NH3)MnCl4. J. Phys. 44, 967 (1983).
Eremenko, V. V., Fomin, V. I. & Kurnosov, V. S. Spin wave spectrum of quasi-two-dimensional antiferromagnet NH3(CH2)2NH3MnCl4. Physica B 194–196, 187 (1994).
Bissey, J.-C., Filloleau, N., Chanh, N.-B., Berger, R. & Flandrois, S. Exchange interaction as studied by EPR in a two-dimensional molecular composite [NH3–(CH2)4–NH3]MnCl4. Solid State Commun. 106, 385 (1998).
Bogdan, M. M., Kobets, M. I. & Khats’ko, E. N. Chaotic regimes of antiferromagnetic resonance in a quasi-two-dimensional easy-axis antiferromagnet (NH3)2(CH2)4MnCl4. Low. Temp. 25, 192 (1999).
Lv, X.-H. et al. Dielectric and photoluminescence properties of a layered perovskite-type organic–inorganic hybrid phasetransition compound: NH3(CH2)5NH3MnCl4. J. Mater. Chem. C 4, 1881 (2016).
Chhor, K., Abello, L., Pommier, C. & Sourisseau, C. Reorientational motions in a perovskite-type layer compound [NH3(CH2)5NH3]MnCl4. A calorimetric study. J. Phys. Chem. Solids 49, 1079 (1988).
Abello, L., Chhor, K., Pommier, C. & Sourisseau, C. Thermodynamic study on perovskite-type layer compounds: [(NH3(CH2)nNH3]MnCl4 (cr) (M = Mn, Cd; n = 3, 5) at low temperatures. J. Chem. Thermodyn. 20, 1433 (1988).
Przeslawski, J., Czapla, Z., Crofton, M. & Dacko, S. On the “inverted” phase transitions in ferroic crystals containing propylenediammonium cations. Ferroelectrics 534, 220 (2018).
Lim, A. R. & Kim, S. H. Physicochemical property investigations of perovskite-type layer crystals [NH3(CH2)nNH3]CdCl4 (n = 2, 3, and 4) as a function of length n of CH2. ACS Omega 6, 27568 (2021).
Negrier, P., Couzi, M., Chanh, N. B., Hauw, C. & Meresse, A. Structural phase transitions in the perovskite-type layer compound NH3(CH2)5NH3CdCl4. J. Phys. France 50, 405 (1989).
Negrier, P. et al. The Imma ↔ Pnma Phase transition of NH3(CH2)5NH3CdCl4 studied by X-ray diffraction. Phys. Stat. Sol. (a) 100, 473 (1987).
Mostafa, M. F. & Hassen, A. Phase transition and electric properties of long chain Cd(II) layered perovskite. Phase Transit. 79, 305 (2006).
Rao, C. N. R., Cheetham, A. K. & Thirumurugan, A. Hybrid inorganic–organic materials: A new family in condensed matter physics. J. Phys. Condens. Matter 20, 83202 (2008).
Arkenbout, H., Uemura, T., Takeya, J. & Palstra, T. T. M. Charge-transfer induced surface conductivity for a copper based inorganic–organic hybrid. Appl. Phys. Lett. 95, 17314 (2009).
Zolfaghari, P., de Wijs, G. A. & de Groot, R. A. The electronic structure of organic-inorganic hybrid compounds: (NH4)2CuCl4, (CH3NH3)2CuCl4 and (C2H5NH3)2CuCl4. J. Phys. Condens. Matter 25, 295502 (2013).
Yadav, R. et al. Dielectric and Raman investigations of structural phase transitions in (C2H5NH3)CdCl4. Phys. Chem. Chem. Phys. 17, 12207 (2015).
Elseman, M. et al. Copper-substituted lead perovskite materials constructed with different halides for working (CH3NH3)2CuX4-based perovskite solar cells from experimental and theoretical view. ACS Appl. Mater. Interfaces 10, 11699 (2018).
Aramburu, J. A., Garcia-Fernandez, P., Mathiesen, N. R., Garcia-Lastra, J. M. & Moreno, M. Changing the usual interpretation of the structure and ground state of Cu2+ layered perovskites. J. Phys. Chem. C 122, 5071 (2018).
Al-Amri, A. M., Leung, S.-F., Vaseem, M., Shamin, A. & He, J.-H. Fully inkejet-printed photodector using a graphene/perovskite/graphene heterostructure. IEEE Trans. Electron. Dev. 66, 2657 (2019).
Al-Amri, A. M., Cheng, B. & He, J.-H. Perovskite methylammonium lead trihalide heterostructures: Progress and challenges. IEEE Trans. Nanotechnol. 18, 1 (2019).
Lin, C.-H. et al. Giant optical anisotropy of perovskite nanowire array films. Adv. Funct. Mater. 30, 1909275 (2020).
Kind, R., Plesko, S., Gunter, P., Ross, J. & Fousek, J. Structural phase transitions in the perovskite-type layer compounds NH3(CH2)3NH3CdCl4, NH3(CH2)4NH3MnCl4, and NH3(CH2)5NH3CdCl4. Phys. Rev. B 23, 5301 (1981).
Khechoubi, M. et al. Thermal conformational changes in a bidimensional molecular composite material: A thermodynamic and crystallographic study of NH3–(CH2)4–NH3 CdCl4. J. Phys. Chem. Solids 55, 1277 (1994).
Mostafa, M. F. & El-hakim, S. A. Structural phase transition and the dielectric permittivity of the model lipid bilayer [(CH2)12(NH3)2]CuCl4. Phase Transit. 76, 587 (2003).
Mostafa, M. F. & Youssef, A. A. A. Magnetic and electric studies of a new Cu(II) perovskite-like material. Z. Naturforsch. 59a, 35 (2004).
Mostafa, M. F. & El-khiyami, S. S. Crystal structure and electric properties of the organic–inorganic hybrid: [(CH2)6(NH3)2]ZnCl4. J. Solid State Chem. 209, 82 (2014).
Lee, S. J., Choi, M. Y. & Lim, A. R. Effect of methylene chain length on the thermodynamic properties, ferroelastic properties, and molecular dynamics of the perovskite-type layer crystal [NH3(CH2)nNH3]MnCl4 (n = 2, 3, and 4). ACS Omega 6, 15392 (2021).
Aizu, K. Determination of the state parameters and formulation of spontaneous strain forferroelastics. J. Phys. Soc. Jpn. 28, 706 (1970).
Sapriel, J. Domain-wall orientations in ferroelastics. Phys. Rev. B 12, 5128 (1975).
Abragam, A. The Principles of Nuclear Magnetism (Oxford University Press, 1961).
Koenig, J. L. Spectroscopy of Polymers (Elsevier, 1999).
Acknowledgements
This research was supported by the Basic Science Research program through the National Research Foundation of Korea (NRF), funded by the Ministry of Education, Science, and Technology (2018R1D1A1B07041593 and 2016R1A6A1A03012069).
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Lim, A.R., Joo, Y.L. Study on structural geometry and dynamic property of [NH3(CH2)5NH3]CdCl4 crystal at phases I, II, and III. Sci Rep 12, 4251 (2022). https://doi.org/10.1038/s41598-022-08246-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-022-08246-5
- Springer Nature Limited