
Abstract
The mechanisms regulating immune cells recruitment into the heart during healing after an acute myocardial infarction (AMI) have major clinical implications. We investigated whether cholinergic stimulation with pyridostigmine, a cholinesterase inhibitor, modulates heart and spleen immune responses and cardiac remodeling after AMI in spontaneous hypertensive rats (SHRs). Male adult SHRs underwent sham surgery or ligation of the left coronary artery and were randomly allocated to remain untreated or to pyridostigmine treatment (40 mg/kg once a day by gavage). Blood pressure and heart rate variability were determined, and echocardiography was performed at day six after MI. The heart and spleen were processed for immunohistochemistry cellular analyses (CD3+ and CD4+ lymphocytes, and CD68+ and CD206+ macrophages), and TNF levels were determined at day seven after MI. Pyridostigmine treatment increased the parasympathetic tone and T CD4+ lymphocytes in the myocardium, but lowered M1/M2 macrophage ratio towards an anti-inflammatory profile that was associated with decreased TNF levels in the heart and spleen. Treatment with this cholinergic agent improved heart remodeling manifested by lower ventricular diameters and better functional parameters. In summary, cholinergic stimulation by pyridostigmine enhances the parasympathetic tone and induces anti-inflammatory responses in the heart and spleen fostering cardiac recovery after AMI in SHRs.
Similar content being viewed by others

Introduction
Acute myocardial infarction (AMI) triggers a sterile inflammatory response characterized by the recruitment and activation of innate and adaptive immune cells to repair tissue damage1,2,3. AMI also elicits systemic inflammatory responses that result in organism-wide complications reminiscent of that found in sepsis4,5,6. In rodents, AMI triggers hematopoiesis inducing the production of innate immune cells and recruitment of neutrophils and inflammatory monocytes in the damaged tissue7. These mechanisms regulating monocyte recruitment from the bone marrow and spleen into the heart during healing have major clinical implications to design novel therapeutic strategies8, 9. In addition to innate cells, adaptive immune responses also play a fundamental role in cardiac remodeling after AMI9. More specifically, CD4+ T lymphocytes are required for proper healing and might prevent chronic remodeling after AMI. Several studies have shown that CD4+ T regulatory cells (Treg) can modulate inflammation in the myocardium after an ischemic injury10, 11. Patients with acute coronary syndrome show infiltration of CD4+ and CD8+ lymphocytes in MI and non-MI areas, and reduced circulating Treg cells with compromised modulatory function12. These results suggest systemic mechanisms coordinating the autonomic nervous system, bone marrow, and spleen to modulate the immune response from the atherosclerotic plaque to infarcted myocardium13.
Recently, autonomic neural regulation of the immune system has attracted investigators' attention as a novel therapeutic strategy to control inflammation in multiple clinical settings, including myocardial ischemia–reperfusion injury14 and MI15, 16. The main nerve of the parasympathetic part of the autonomic nervous system—the vagus nerve modulates the immune function by regulating innate and acquired immune cell-mediated responses17, 18. Electrical vagus nerve stimulation suppresses aberrant systemic inflammation and decreases serum TNF levels by inhibiting its production in the spleen19. The vagus nerve interacts with the splenic nerve that releases norepinephrine in the spleen20, 21. Norepinephrine activates β2-adrenoceptors on splenic T lymphocytes that contain the enzyme choline acetyltransferase (ChAT), which synthesizes acetylcholine22,23,24. Vagus nerve stimulation causes an increase in splenic acetylcholine levels. In turn, lymphocyte-derived acetylcholine binds to the α7-nicotinic acetylcholine receptors (α7nAChR) on splenic macrophages to inhibit TNF production19, 24, 25. On the other hand, sympathetic nerve hyperactivity in the spleen has been linked to chronic immune-mediated inflammatory diseases26. Moreover, central nervous system angiotensin II infusion and hyperthermia are associated with enhanced level of efferent splenic sympathetic nerve discharge and splenic pro-inflammatory cytokine gene expression in rats27. These findings show the potential of the autonomic nervous system to regulate immune responses and inflammation; in this regulation the vagus nerve plays a specific role in orchestrating cellular and cytokine immune responses directed towards promoting tissue healing.
Pyridostigmine is a cholinesterase inhibitor and a cholinergic drug used to treat myasthenia gravis, a chronic autoimmune, neuromuscular disease that causes muscle weakness28. Pyridostigmine prevents acetylcholine hydrolysis and enhances parasympathetic modulation in normotensive rats29. We have previously reported that pyridostigmine treatment prevents deleterious inflammation and oxidative stress in the ischemic myocardium of these rats30. Pyridostigmine treatment also increased the proportion of T regulatory cells in peripheral circulation, and decreased activated CD8+ lymphocytes in the spleen. These results suggest that pyridostigmine modulates the immune cell response by regulating splenic lymphocytes31. However, the effects of pyridostigmine in infarcted spontaneous hypertensive rats (SHRs) that exhibit autonomic dysfunction and chronic inflammation and may represent an animal model that better mimics the clinical profile of patients with AMI remained unknown. In the present study, we investigated whether pyridostigmine affects splenic lymphocytes and pro-inflammatory cytokines, and its potential to modulate inflammatory, structural, and functional responses after myocardial infarction in SHRs.
Results
Pyridostigmine treatment enhances hemodynamic and autonomic parameters
First, we analyzed the hemodynamic variables as presented in Table 1. Sham animals had very high blood pressure (204/144 mmHg) characteristic of spontaneous hypertensive SHRs. MI significantly lowered (p < 0.05) systolic (SBP), diastolic (DBP), and mean (MBP) blood pressure as compared to sham animals (Fig. 1). These results indicate decreased cardiac output potentially related to reduced left ventricular contraction caused by ischemic injury. Pyridostigmine treatment affected neither of these hemodynamic parameters. Moreover, all groups had similar heart rate (p > 0.05). We also analyzed the autonomic tone via heart rate variability (HRV) and baroreflex sensitivity to determine whether pyridostigmine enhances the parasympathetic modulation (Table 2, Fig. 2). Root mean square of successive differences (RMSSD) reflects the beat-to-beat variance in heart rate and is the primary time domain measurement used to estimate vagal mediated changes reflected in HRV. The RMSSD correlates with HF and reflects self-regulatory capacity. Pyridostigmine treatment significantly increased RMSSD showing its potential to induce cholinergic stimulation (Fig. 2A).

Hemodynamic parameters. (A) SBP; Systolic Blood Pressure. (B) DBP; Diastolic Blood Pressure, and (C) MBP; Mean Blood Pressure; HR: Heart rate. *p < 0.05 versus Sham; §p < 0.05 versus AMI. Values expressed as mean ± SEM. Statistical significance was determined by one-way ANOVA followed by a multiple comparisons test Tukey’s was performed using GraphPad Prism version 9.0.1. www.graphpad.com.

Heart rate variability (HRV) and Baroreflex Sensitivity (alpha-index). (A) RMSSD: square root of the mean of the sum of squares of the differences between successive pulse interval values. (B) LF: Low Frequency nu: Normalized units. (C) HF nu: high frequency in normalized units and. (D) LF/HF (E) ALPHA-INDEX: baroreflex sensitivity. *P < 0.05 versus Sham; #P < 0.05 versus AMI and §p < 0.05 versus AMI. Values expressed as mean ± SEM. Statistical significance was determined by one-way ANOVA followed by a multiple comparisons test Tukey’s was performed using GraphPad Prism version 9.0.1. www.graphpad.com.
We also performed the spectral analysis of HRV with the three main spectral components distinguished in short-term recordings: very low frequency (VLF), low frequency (LF), and high frequency (HF). The distribution of the power and the central frequency of LF and HF are not fixed but may vary in relation to changes in autonomic modulations of the heart period. We analyzed LF (Fig. 2B) and HF in both absolute (ms2) and normalized units (n.u), representing sympathetic and parasympathetic modulation, respectively. Normalization tends to minimize the effects of LF and HF in total power and emphasizes the balanced behavior (LF/HF) of the two branches of the autonomic nervous system15, 24, 25). These results showed that AMI decreased HF (nu) (Fig. 2C), and thereby significantly increased the LF/HF ratio (Fig. 2D) consistent with enhanced sympathetic modulation. Conversely, pyridostigmine treatment significantly increased HF (nu) (Fig. 2C), and decreased LF/HF ratio (Fig. 2D). These results concur in showing pyridostigmine potential to enhance the parasympathetic vagal tone. Alterations of the baroreceptor-heart rate reflex contribute to reduced parasympathetic activity and increased sympathetic activity during cardiovascular diseases, including AMI. This baroreflex sensitivity measured by spectral methods assesses the relationship (in terms of gain) between specific oscillatory components of the two signals. Spontaneous oscillations in blood pressure elicit an oscillation at the same frequency in RR interval by the arterial baroreflex activity. BRS is computed as the average value of the transfer function modulus (i.e., the gain) between systolic pressure and RR interval in the frequency range 0.07–0.14 Hz. The analysis of BRS can provide prognostic information in experimental and clinical studies32. We observed that pyridostigmine treatment significantly increased BRS (α-index) indicating an improvement of baroreflex sensitivity (Fig. 2E).
Pyridostigmine treatment improves cardiac morphofunctional analyzes
Next, we performed structural and functional analyses to determine the effects of pyridostigmine on AMI (Table 3). AMI caused an infarcted and akinetic area consistent with moderate infarction, and significantly increased systolic and left diastolic ventricle (LV) diameters as compared to sham animals. AMI also lowered LV systolic function as inferred by LV ejection fraction (LVEF) (Fig. 3C) and fractional area change (LV FAC) (Fig. 3D), but increased E/A ratio (Fig. 3E) (early filling wave/late atrial contraction wave) that was associated with diastolic dysfunction. Together, these results indicate substantial cardiac remodeling after AMI. Pyridostigmine treatment significantly improved structural and functional parameters, but not the infarcted and akinetic area (Table 3). Pyridostigmine treatment significantly decreased systolic (LDV) and left diastolic ventricle (LSV) diameters (Fig. 3A,B, respectively). Although pyridostigmine treatment did not alter LVEF (Fig. 3C), it significantly increased fractional area change (LV FAC), which is an index of global ventricular function (Fig. 3D), and decreased the E/A ratio (Fig. 3E). These results show that pyridostigmine improved systolic and diastolic functions and cardiac remodeling after AMI in SHRs.

EchoDopplercardiographic parameters. (A) LSV Diam. And (B) LDV Diam.: left systolic and diastolic ventricular diameters, respectively. (C) LVEF: left ventricular ejection fraction (D) LV FAC: global left ventricular systolic function estimated by the FAC: fractional area change and (E) E/A: transmitral flow/E/A ratio. *P < 0.05 versus Sham; #P < 0.05 versus AMI; §P < 0.05 Sham versus AMI. Values expressed as mean ± SEM. Statistical significance was determined by one-way ANOVA followed by a multiple comparisons test Tukey’s was performed using GraphPad Prism version 9.0.1. www.graphpad.com.
Pyridostigmine treatment increases T helper and M2 macrophages in the infarcted and peri-infarcted zones, and inhibition of excessive release of TNF in the spleen and heart tissue
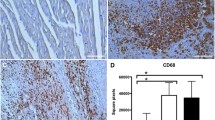
Sham SRH animal have few immune cells in the myocardium. However, AMI induces a significant infiltration of immune cells into the infarcted and peri-infarcted zones at day seven. Among these cells, AMI significantly increased CD68+ (M1) macrophage counts, and pyridostigmine significantly decreased these counts (Fig. 4). Likewise, AMI also increased CD206+ (M2) macrophage counts, but pyridostigmine treatment further increased these cells in the infarcted and peri-infarcted zones (Fig. 5). Therefore, pyridostigmine treatment altered the M1/M2 ratio towards a M2 anti-inflammatory profile. We also detected that sham and AMI groups have similar counts of T (CD3 +) total lymphocytes. However, pyridostigmine treatment increased the number of T cells as compared to those in sham group, and these cells were concentrated in the infarcted and peri-infarcted areas (Fig. 6). Likewise, Sham and AMI animals had similar counts of T helper (CD4 +) lymphocytes, and pyridostigmine treatment increased these cells in the infarcted and peri-infarcted areas (Fig. 7). In agreement with previous studies, we also observed that AMI increased TNFα levels both in heart and spleen, and PY treatment significantly decreased these levels in both organs (Fig. 8A,B). Thus, AMI failed to affect the lymphocyte counts, and pyridostigmine significantly increased T helper counts, decreased the M1/M2 ratio, and decreased TNF levels in the infarcted area but also in the spleen (Fig. 8).

CD68+ cell count (M1 macrophages) (A) Photomicrographs showing the M1 macrophages within the infarcted zone from the left ventricle (magnification 400×) and (B) bar graphs of CD68+ positive cells in each group was compared. Fifteen microscopic fields of the infarcted and peri-infarcted zones were analyzed (5–6 animals per group). *P < 0.05; §P < 0.05 statistically significant. §AMI versus Sham; *AMI + PY vs Sham. (Immunohistochemistry, diaminobenzidine: scale bar equals 50 µm). Values expressed as mean ± SEM. Statistical significance was determined by one-way ANOVA followed by a multiple comparisons test Tukey’s was performed using GraphPad Prism version 9.0.1. www.graphpad.com.

CD206+ cell count (M2 macrophages) (A) Photomicrographs showing the M2 macrophages within the infarcted zone from the left ventricle (magnification 400×) and (B) bar graphs of CD206+ positive cells in each group was compared. Fifteen microscopic fields of the infarcted and peri-infarcted zones were analyzed (5–6 animals per group). *P < 0.05; #P < 0.05 statistically significant. *AMI + PY vs Sham; #AMI + PY vs AMI (Immunohistochemistry, diaminobenzidine: scale bar equals 50 µm). Values expressed as mean ± SEM. Statistical significance was determined by one-way ANOVA followed by a multiple comparisons test Tukey’s was performed using GraphPad Prism version 9.0.1. www.graphpad.com.

CD3+ cell count (Total lymphocytes) (A) Photomicrographs showing the Total lymphocytes within the infarcted zone from the left ventricle (magnification 400×) and (B) bar graphs of CD3+ lymphocytes positive cells in each group was compared. Fifteen microscopic fields of the infarcted and peri-infarcted zones were analyzed (5–6 animals per group). *P < 0.05 statistically significant. *AMI + PY vs Sham. (Immunohistochemistry, diaminobenzidine: scale bar equals 50 µm). Values expressed as mean ± SEM. Statistical significance was determined by one-way ANOVA followed by a multiple comparisons test Tukey’s was performed using GraphPad Prism version 9.0.1. www.graphpad.com.

CD4+ cell count (Lymphocytes T helper) (A). Photomicrographs showing the Lymphocytes T helper within the infarcted zone from the left ventricle (magnification 400×) and (B) bar graphs of CD4+ positive cells in each group was compared. Fifteen microscopic fields of the infarcted and peri-infarcted zones were analyzed (5–6 animals per group). *P < 0,05; #P < 0.05 statistically significant. *AMI + PY versus Sham; #AMI + PY versus AMI. (Immunohistochemistry, diaminobenzidine: scale bar equals 50 µm). Values expressed as mean ± SEM. Statistical significance was determined by one-way ANOVA followed by a multiple comparisons test Tukey’s was performed using GraphPad Prism version 9.0.1. www.graphpad.com.

TNF levels in the heart (A) and the spleen (B) in all groups. #p < 0.05; §P < 0.05 statistically significant. Values expressed as mean ± SEM. One-way ANOVA. §AMI versus Sham; #AMI + PY versus AMI. Values expressed as mean ± SEM. Statistical significance was determined by one-way ANOVA followed by a multiple comparisons test Tukey’s was performed using GraphPad Prism version 9.0.1. www.graphpad.com.
Discussion
The main new findings of this study are that pyridostigmine treatment increases parasympathetic modulation and baroreflex sensitivity, promotes anti-inflammatory immune cell modulation in the myocardium, and decreases splenic and cardiac TNF levels after AMI in SHRs. These results also correlate with the potential of pyridostigmine to improve cardiac morphofunctional remodeling after AMI in SHRs. Thus, cholinergic stimulation using pyridostigmine induces cardiac protection after ischemic injury in SHRs.
The bidirectional interplay between the nervous and immune systems has gained great clinical attention to design novel therapeutic strategies for AMI17, 33. The inflammatory reflex and its efferent arm—the cholinergic anti-inflammatory pathway have been successfully explored in therapeutic approaches for sepsis, arthritis and other inflammatory conditions17, 18. Activation of cholinergic signaling in the inflammatory reflex using cholinesterase inhibitors, including the cholinergic drug galantamine suppresses inflammation in preclinical and clinical settings of numerous disorders characterized by immune and metabolic dysregulation33, 34. This is the first study investigating the effects of cholinergic stimulation by the cholinesterase inhibitor pyridostigmine on immune alterations after AMI in SHRs. This is a clinically relevant study, because SHRs have hemodynamic, neural, and target organ damage reminiscent of that found in essential neurogenic human hypertension. Okamoto and Aoki (1963) developed this strain of hypertensive rats35 that are widely used in the literature to study natural history, genetic determinants, and pathophysiological changes in arterial hypertension36, 37. SHRs exhibit immune deficiencies in T and B lymphocytes, decreased T cell proliferative responses36, and lower ChAT mRNA expression in circulating and splenic mononuclear cells as compared to normotensive Wistar rats38. The possibility that hypertension in SHRs is related to immune anomalies is still under investigation36, 37. Enhanced innate immune responses and cytokine release from splenocytes have been demonstrated in pre-hypertensive SHR rats and evoke profound activation of the adaptive immune system contributing to vascular damage and hypertension37. In addition, SHRs exhibit autonomic dysfunction (increased sympathetic and lower parasympathetic vagal modulation) and chronic inflammation, suggesting that they may represent an animal model that better mimics the clinical profile of patients with AMI36, 37.
SH animals have a higher sympathovagal ratio; AMI further increased the sympathetic drive and worsened baroreflex function. Still, pyridostigmine treatment significantly improved the cardiovascular autonomic balance and baroreflex sensitivity. The baroreflex is a fundamental self-regulatory mechanism of the cardiovascular system. La Rovere32 demonstrated a close correlation between post-MI mortality and baroreflex sensitivity (BRS). Although the pathological importance of impaired baroreflex function in high post-MI mortality has already been recognized, no effective long-term pharmacological intervention is available32, which is the direct consequence of the lack of targeted drugs. These findings suggest a mechanistic basis for restoring baroreflex sensitivity with pyridostigmine in AMI.
Pyridostigmine is a potent acetylcholinesterase inhibitor and a clinically-approved cholinergic drug for the treatment of Myasthenia Gravis. Pyridostigmine significantly increases vagal modulation as reported in experimental and human studies. Short-term administration of pyridostigmine increases HRV in healthy humans39 and rats29. In patients with heart failure, pyridostigmine also ameliorates the autonomic and hemodynamic performance during dynamic exercise40 and reduces ventricular arrhythmia density41. Also, pyridostigmine enhances cholinergic modulation of the immune cells by preventing vagal-derived acetylcholine degradation38. We and others have demonstrated that pyridostigmine treatment started just after coronary artery ligation can improve autonomic and cardiocirculatory function in normotensive rats analyzed weeks after the AMI15, 16, 42. A limited number of studies have evaluated the effects of peripheral and centrally-acting cholinesterase inhibitors on autonomic, hemodynamic, and inflammatory indices in SH animals43, 44. Importantly, our findings indicate that pyridostigmine induces autonomic improvement in infarcted SHRs that is associated with anti-inflammatory responses in the heart-spleen axis, and cardiac remodeling markers.
Myocardial infarction triggers a sterile inflammatory response characterized by sequential recruitment and activation of innate and adaptive immune cells to repair the tissue damage1,2,3. Macrophages are among the first responders to local damage. Depending on their phenotype, macrophages may be harmful or protective. M1 macrophages express high levels of inflammatory mediators and dominate the first days after AMI, while M2 macrophages express high levels of anti-inflammatory cytokines involved in tissue repair, gradually appear and remain predominant over five days after the infarction1, 3. Pyridostigmine treatment decreased M1 and increased M2 macrophages, favoring an anti-inflammatory profile with increased T helper (CD4 +) lymphocyte counts and decreased TNF levels in myocardium and spleen. These results provide new insights into the efficacy of pyridostigmine in SHRs subjected to MI and substantially extend the information generated by previous studies using normotensive infarcted animals30, 31. As in these previous studies, a better anti-inflammatory, immune cell profile in the myocardium was associated with decreased levels of pro-inflammatory cytokines.
The sympathetic nervous system enhances macrophage recruitment in the heart after an ischemic event. Sympathetic fibers stimulate the bone marrow to release hematopoietic stem and progenitor cells, as observed in mice and humans after AMI6, 7, 45. The spleen is also a major source of pro-inflammatory cytokines and immune cells. Indeed, within the first day after coronary ligation, the spleen releases monocytes from its subcapsular red pulp into the bloodstream as shown in mice and rats6, 7, 9. As a consequence, bone marrow and splenic leucocyte production lead to monocytosis, and these cells are recruited into the ischemic heart8, 9, 13. This increased metabolic activity of spleen and bone marrow has been shown in patients with acute AMI using 18F-fluorodeoxyglucose PET image46. Our results show that pyridostigmine decreased sympathovagal balance in infarcted rats, improved M1/M2 macrophage ratio, and decreased TNF levels in spleen and heart. Thus, cholinergic stimulation may interfere with local mechanisms preventing macrophage polarization in the ischemic myocardium. The idea of targeting macrophage and lymphocyte polarization to control inflammation following AMI has been recently proposed47. In addition to innate cells, lymphocytes also play a fundamental role after AMI10. More specifically, CD4+ T helper cells are required for proper healing and can attenuate chronic remodeling after AMI. CD4+ T-cells regulate the infiltration of inflammatory monocytes and are required for proper extracellular matrix formation and angiogenesis during post-MI healing48. Furthermore, CD4+ T cell-derived cytokines modulate monocyte differentiation and macrophage activity within the myocardium49.
Our results indicate a link between increased vagal modulation with pyridostigmine and an anti-inflammatory M1/M2 profile, increased T helper counts, and decreased myocardium TNF levels. Given the detrimental potential of TNF on the myocardium, TNF local production may play an important role in ventricular dysfunction and adverse remodeling after infarction50. TNF is produced soon after AMI, regulates myocardial apoptosis and triggers additional cellular inflammatory responses50. Anti-TNF treatment was associated with smaller infarct size and decreased ventricular dysfunction in ischemic-reperfusion and permanent ischemia in animal models51 including AMI. Thus, it is plausible that TNF inhibition by pyridostigmine may trigger a positive remodeling and decrease heart failure in infarcted SHRs.
Targeting cholinergic signaling in the inflammatory reflex provides novel therapeutic opportunities for diseases characterized with inflammatory and cardiovascular derangements, including myocardial ischemia–reperfusion injury and AMI damage17. The vagus nerve can act reflexively on the spleen, reducing systemic inflammation18. Likewise, efferent vagal signals may facilitate the release of lymphocytes from the thymus through nicotinic receptors52. Vagal stimulation can increase acetylcholine release, which activates α7nAChR on macrophages to decrease pro-inflammatory cytokines in the spleen19,22,24. A main source of acetylcholine in the spleen is a subgroup of T lymphocytes expressing β2-adrenoreceptors and ChAT (ChAT-T cells) that synthesize acetylcholine22. Upon vagal activation, the splenic nerve releases norepinephrine (NE) in the spleen to stimulate ChAT-T cells to release acetylcholine, which in turn inhibits TNF production in macrophages via α7nAChR-mediated signaling15, 20, 21, 23.
As both acetylcholine and NE modulate splenic immune cell functions, our study highlights the need to consider the balance between these two mediators, which probably determines the final immunoregulatory outcome53. We show the potential of pyridostigmine to reduce cardiac inflammation and improve heart remodeling after AMI. As expected, AMI caused significant morphofunctional alterations, detected by echocardiography at day 7, which affects circulation and reduces blood pressure. Pyridostigmine treatment significantly improves systolic and diastolic function, but they were insufficient to restore normal blood pressure as seen in sham animals. Noninvasive diastolic function measurements with echocardiography correlated with LV cardiac measurements as determined by catheterization in rat models of myocardial hypertrophy and AMI22; these noninvasive parameters may predict further ventricular dysfunction5. Ventricular dysfunction and heart failure are frequent complications of AMI and are associated with poor prognosis. Some studies have shown the beneficial effects of more prolonged pyridostigmine treatment on cardiac function and remodeling in normotensive infarcted rats15, 54, 55. It can be speculated that this immediate improvement may lead to positive remodeling and decrease the development of heart failure in infarcted SHRs.
Our study has some limitations. As mentioned above, the specific role of ChAT-positive T cells and possibly B cells in the effects observed in the spleen is unknown and remains to be evaluated in future studies. In addition, there is a lack of specific insight into the role of ChAT-containing immune cells in the heart and cardiac myocytes in mediating local immunomodulatory effects in our model. On a related note, we cannot assess the relative contribution of neural acetylcholine versus non-neuronal acetylcholine in the beneficial immune and cardiometabolic pyridostigmine effects on the heart. Previously, vagus nerve stimulation has been shown to suppress cardiac (myocardial) and systemic TNF levels in rodents during endotoxemia18, 19. Vagus nerve stimulation in animals and patients with heart failure also results in improved cardiac function, indicating the cardioprotective efficacy of vagus nerve cholinergic signaling56,57,58 Previous studies have indicated that cardiac myocytes also contain ChAT and synthesize and secrete acetylcholine, which plays a role in myocardium regulatory functions59, 60. Acetylcholine released from cardiac myocytes reportedly has various protective effects in pathological scenarios, including cardiac hypertrophy and failure as indicated in transgenic mouse models61, 62. Further studies are necessary to evaluate the specific role of non-neuronal cell-derived acetylcholine in addition to the neural vagus nerve-derived cholinergic output in mediating pyridostigmine effects in the heart. In addition, further work using cell-type-specific transcriptomic and epigenomic analysis is necessary to reveal the underlying molecular mechanisms of how pyridostigmine induces the M1-M2 cell type conversion.
In conclusion, cholinergic stimulation with pyridostigmine modulates a heart-spleen axis in the immune response after myocardial infarction and improves heart remodeling. Our results warrant future studies to determine mechanisms of these effects and the consequences in heart failure.
Materials and methods
Experimental design
All animal procedures were approved by the Committee on the Ethics of Animal Experimentation at the University of Nove de Julho (CEUA protocol No. 7612011118). Animal care was performed following the Guide for the Care and Use of Laboratory Animals published by the U.S. National Institutes of Health. Adult male SHR (2–3 months old, 200–250 g) were housed in collective plastic cages (four animals per cage) with controlled temperature (23 °C), a 12:12-h light–dark cycle with rat chow provided ad libitum and unlimited access to water. Animals were randomly assigned to one of three groups, with 10–12 animals in each group: sham rats (Sham), untreated infarcted rats (AMI), and pyridostigmine-treated infarcted rats (AMI + PY). All animals were monitored for 7 days. The AMI + PY Group received pyridostigmine bromide (Sigma-Aldrich, St Louis, MO), as described previously (40 mg/kg once a day, by gavage) started one h after surgery and continued for seven days after this procedure8, 63. According to a prior study, the dose and period of pyridostigmine administration chose were appropriate to inhibit approximately 40% of plasma acetylcholinesterase activity29. All methods were reported in accordance with ARRIVE guidelines.
Myocardial infarction
Rats in the AMI and AMI + PY groups were anesthetized (80 mg/kg ketamine and 12 mg/kg xylazine intraperitoneal injected, I.P.) and underwent induction of AMI by surgical occlusion of the left coronary artery, as previously described30, 31. A left thoracotomy performed by dissecting the third intercostal space and exposing the heart. Then, the left coronary artery was occluded with a single nylon (6.0 mm) suture 1 mm distal to the left atrial appendage. The chest was then sutured. The rats were maintained under ventilation until recovery. The sham group underwent the same procedure, but AMI was not induced. Infarcted rats were randomly allocated to receive or not P.Y. The analytical investigators were blind to the treatment.
Arterial catheterization, hemodynamic measurements, and cardiovascular variability analysis
At day six, rats were anesthetized (80 mg/kg ketamine and 12 mg/kg xylazine, I.P.), and a catheter filled with 0.06 mL of saline solution was implanted into the femoral artery29 for hemodynamic measurements. The arterial cannula was connected to a strain gauge transducer (Blood Pressure XDCR; Kent Scientific, Torrington, CT), and arterial pressure (A.P.) signals and pulse interval heart rate (H.R.) were digitally recorded over a 30-min period in conscious, awake animals using a data acquisition system (WinDaq, 2 kHz; DATAQ, Springfield, OH), as described29. This basal acquisition was used to evaluate heart rate variability (HRV) and systolic arterial pressure variability (as described below). HRV. For time and frequency domains analysis of cardiovascular autonomic modulation, the time series (three time series of 5 Min for each animal) of pulse interval (PI) and systolic arterial pressure (SAP) were cubic spline-interpolated (250 Hz) and cubic spline-decimated to be equally spaced in time after linear trend removal; power spectral density was obtained through the Fast Fourier transformation. Spectral power for low frequency (LF, 0.20–0.75 Hz) and high frequency (HF, 0.75–4.0 Hz) bands was calculated by power spectrum density integration within each frequency bandwidth, using a customized routine (Cardioseries). The time domain variables were: root mean square of the successive differences (RMSSD) and total variance of pulse interval (VAR-PI) for pulse interval (PI); and total variance of systolic arterial pressure (VAR-SAP) for systolic arterial pressure (SAP). The α-index in the low-frequency band was calculated only when the magnitude of the squared coherence between the PI and SAP signals exceeded 0.5 (range, 0–1). After coherence calculation, the α-index was obtained from the square root of the ratio between PI and SAP variability in the two major low frequency (LF) band29.
Echocardiographic evaluation
Echocardiographic evaluations were performed by a blind observer under the guidelines of the American Society of Echocardiography. Rats were anesthetized (80 mg/kg ketamine and 12 mg/kg xylazine intraperitoneal-I.P.), and images were obtained with a 10–14-MHz linear transducer in a G.E. Vivid 7 Ultra-Definition Clarity Control (G.E. Healthcare, USA). This procedure was performed six days after AMI or sham surgeries in order to analyze AMI area (hipo or acinetic ventricular areas) and LV ejection fraction (LVEF%), and to calculate the following parameters: left atrial diameter, ventricular mass (LV mass); left ventricular end-diameter during systole and diastole (LVSD, LVDD); E wave A wave ratio (E/A); isovolumetric relaxation time (IVRT); fractional area change (FAC), as described in detail elsewhere as described previously15, 63. Through midtransversal and apical transversal views, AMI size was measured by bi-dimensional echocardiogram. In diastole, three measurements of the endocardial perimeter (E.P.) and the length of the infarcted segment (ISe) were obtained for each view. The AMI size for each ISi was calculated by the equation ISi (%) = ISe/E.P. × 100. The total infarct size of each animal was calculated as the mean of ISi (%) of the three segments. AMI was defined as increased echogenicity and change in myocardial systolic movement (hypokinesia, akinesia, or dyskinesia), following Santos et al.28. Our group15 and others63 have demonstrated strong correlations between the AMI area assessed by echocardiogram and postmortem histological analysis, showing that this is a valid method to estimate AMI area in rats.
Immunohistochemistry for immune cells
At day seven, 6–7 animals from each group were anesthetized (80 mg/kg ketamine and 12 mg/kg xylazine, I.P.) and perfused with 0.9% NaCl plus 14 mmol/l KCl solution (IV; with a pressure equal to 13 cmH2O) to arrest the heart in diastole, followed by perfusion of 4% buffered formalin for tissue fixation. Harvested hearts were immersed in formalin for 24 h. Transverse slices were processed and embedded in paraffin. Serial sections of paraffin-embedded tissues (3 μm) were placed on glass slides coated with 2% 3-aminopropyl-triethyl silane (Sigma-Aldrich) and deparaffinized in xylene, then immersed in alcohol and incubated with 3% hydrogen peroxide. The sections were immersed in a citrate buffer (pH 6.0; Sigma- Aldrich) at 95 °C for 20 min for antigen retrieval. Nonspecific signals were blocked using specific antibody diluents (Antibody Diluent, cat. no. S0809; Dako, Glostrup, Denmark)30. The slides were then incubated with the following primary antibodies: anti-rat CD4 (1:500, T Lymphocyte marker, rabbit monoclonal—EPR19514, cat. no. 221775; Abcam, Cambridge, UK), anti-rat CD3 (1:100, T helper Lymphocyte marker, rabbit monoclonal—SP7; cat 16669, Abcam), anti-rat CD68 (1:100, M1 macrophage marker, mouse monoclonal—ED-1 cat. no. 31630, Abcam), and anti-rat CD206 (1:800, M2 macrophage marker cat. no. 64693; Abcam). The samples were kept overnight at 4 °C in a humidified chamber. The sections were incubated with LSAB + System-HRP reagents for 30 min (K0690; Dako Co, Denmark). Finally, the sections were incubated in 3,3-diminobenzidine in a chromogen solution (K346811; Dako Co, Denmark) at room temperature for 2–5 min, and then stained with Mayer's hematoxylin (Sigma-Aldrich) and covered. For the negative controls, the primary antibodies were replaced with 1% PBS/BSA and nonimmune mouse serum (X501-1, Dako).
Cell counts in the infarcted and peri-infarcted zones
Fifteen consecutive microscope fields (magnification: 400×) of the infarcted and peri-infarcted zones were photographed (fluorescence Microscope (Olympus AX70) with a digital camera (Olympus Japan Co, Tokyo, Japan). An investigator blinded to the animal group samples analyzed the images and manually counted them with the aid of the Image J version 1.48v 17 (free software, NIH, Bethesda, Maryland, EUA), using the "cell counter" plug-in31.
Cytokine measurements
A set of 5–6 animals in each group was euthanized by decapitation on day seven after thoracotomy to collect fresh heart and spleen for TNF-α analyses. Measurement of the TNFα was performed in samples of the LV and spleen by ELISA using Duo-set available kits for TNFα (BD Pharmingen, San Jose, CA, USA) as previously describe30. The sensitivity of the assays was 15 pg/mL. The results were normalized by LV or spleen total protein30.
Statistical analysis
All data were represented as means ± the standard error of the mean (SEM). For parametric data, the one-way analysis of variance (one-way ANOVA) was performed with Turkey's multiple comparison tests was performed using GraphPad Prism version 9.0.1 for Windows, GraphPad Software, San Diego, California USA, www.graphpad.com. For nonparametric data, the Kruskal–Wallis test was used. P values less than 0.05 were considered significant.
Accordance statement
All methods were reported in accordance with ARRIVE guidelines.
References
Frangogiannis, N. G. The immune system and cardiac repair. Pharmacol. Res. 58, 88–111 (2008).
Kain, V., Prabhu, S. D. & Halade, G. V. Inflammation revisited: inflammation versus resolution of inflammation following myocardial infarction. Basic Res. Cardiol. 109, 444 (2014).
Yan, X. et al. Temporal dynamics of cardiac immune cell accumulation following acute myocardial infarction. J. Mol. Cell Cardiol. 62, 24–35 (2013).
Anzai, T. Post-infarction inflammation and left ventricular remodeling: a double-edged sword. Circ. J. 77, 580–587 (2013).
Alp, E. et al. Incidence, risk factors and mortality of nosocomial pneumonia in intensive care units: a prospective study. Ann. Clin. Microbiol. Antimicrob. 3, 17 (2004).
Ulloa, L. & Deitch, E. A. Neuroimmune perspectives in sepsis. Crit. Care 13, 133 (2009).
Swirski, F. K. et al. Identification of splenic reservoir monocytes and their deployment to inflammatory sites. Science 325, 612–616 (2009).
Dutta, P. et al. Myocardial infarction accelerates atherosclerosis. Nature 487, 325–329 (2012).
Hoyer, F. F. et al. Tissue-specific macrophage responses to remote injury impact the outcome of subsequent local immune challenge. Immunity 51(899–914), e897 (2019).
Hofmann, U. & Frantz, S. Role of T-cells in myocardial infarction. Eur Heart J. 37, 873–879 (2016).
Li, J., Tan, J., Martino, M. M. & Lui, K. O. Regulatory T-cells: potential regulator of tissue repair and regeneration. Front. Immunol. 9, 585 (2018).
Santos-Zas, I., Lemarie, J., Tedgui, A. & Ait-Oufella, H. Adaptive immune responses contribute to post-ischemic cardiac remodeling. Front. Cardiovasc. Med. 5, 198 (2018).
Libby, P., Nahrendorf, M. & Swirski, F. K. Leukocytes link local and systemic inflammation in ischemic cardiovascular disease: an expanded “cardiovascular continuum”. J. Am. Coll. Cardiol. 67, 1091–1103 (2016).
Calvillo, L. et al. Vagal stimulation, through its nicotinic action, limits infarct size and the inflammatory response to myocardial ischemia and reperfusion. J. Cardiovasc. Pharmacol. 58, 500–507 (2011).
de La Fuente, R. N. et al. Cholinergic stimulation with pyridostigmine improves autonomic function in infarcted rats. Clin. Exp. Pharmacol. Physiol. 40, 610–616 (2013).
Durand, M. T. et al. Pyridostigmine restores cardiac autonomic balance after small myocardial infarction in mice. PLoS ONE 9, e10447 (2014).
Pavlov, V. A., Chavan, S. S. & Tracey, K. J. Molecular and functional neuroscience in immunity. Annu. Rev. Immunol. 36, 783–812 (2018).
Olofsson, P. S., Rosas-Ballina, M., Levine, Y. A. & Tracey, K. J. Rethinking inflammation: neural circuits in the regulation of immunity. Immunol. Rev. 248, 188–204 (2012).
Borovikova, L. V. et al. Vagus nerve stimulation attenuates the systemic inflammatory response to endotoxin. Nature 405, 458–462 (2000).
Besedovsky, H. O. & del Rey, A. Immune-neuro-endocrine interactions: facts and hypotheses. Endocr. Rev. 17, 64–102 (1996).
Bratton, B. O. et al. Neural regulation of inflammation: no neural connection from the vagus to splenic sympathetic neurons. Exp. Physiol. 97, 1180–1185 (2012).
Rosas-Ballina, M. et al. Acetylcholine-synthesizing T cells relay neural signals in a vagus nerve circuit. Science 334, 98–101 (2011).
Vida, G. et al. beta2-Adrenoreceptors of regulatory lymphocytes are essential for vagal neuromodulation of the innate immune system. FASEB J. 25, 4476–4485 (2011).
Olofsson, P. S. et al. alpha7 nicotinic acetylcholine receptor (alpha7nAChR) expression in bone marrow-derived non-T cells is required for the inflammatory reflex. Mol. Med. 18, 539–543 (2012).
Fujii, T. et al. Physiological functions of the cholinergic system in immune cells. J. Pharmacol. Sci. 134, 1–21 (2017).
Bellinger, D. L., Lorton, D., Hamill, R. W., Felten, S. Y. & Felten, D. L. Acetylcholinesterase staining and choline acetyltransferase activity in the young adult rat spleen: lack of evidence for cholinergic innervation. Brain Behav. Immun. 7, 191–204 (1993).
Ganta, C. K. et al. Central angiotensin II-enhanced splenic cytokine gene expression is mediated by the sympathetic nervous system. Am. J. Physiol. Heart Circ. Physiol. 289, H1683-1691 (2005).
Breyer-Pfaff, U., Maier, U., Brinkmann, A. M. & Schumm, F. Pyridostigmine kinetics in healthy subjects and patients with myasthenia gravis. Clin. Pharmacol. Ther. 37, 495–501 (1985).
Soares, P. P., da Nobrega, A. C., Ushizima, M. R. & Irigoyen, M. C. Cholinergic stimulation with pyridostigmine increases heart rate variability and baroreflex sensitivity in rats. Auton. Neurosci. 113, 24–31 (2004).
Bezerra, O. C. et al. Cholinergic stimulation improves oxidative stress and inflammation in experimental myocardial infarction. Sci. Rep. 7, 13687 (2017).
Rocha, J. A. et al. Increase in cholinergic modulation with pyridostigmine induces anti-inflammatory cell recruitment soon after acute myocardial infarction in rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 310, R697-706 (2016).
La Rovere, M. T., Pinna, G. D. & Raczak, G. Baroreflex sensitivity: measurement and clinical implications. Ann. Noninvasive Electrocardiol. 13, 191–207 (2008).
Pavlov, V. A. & Tracey, K. J. Neural circuitry and immunity. Immunol. Res. 63, 38–57 (2015).
Metz, C.N. & Pavlov, V.A. Treating disorders across the lifespan by modulating cholinergic signaling with galantamine. J. Neurochem. (2020). pp 1– 22
Okamoto, K. & Aoki, K. Development of a strain of spontaneously hypertensive rats. Jpn. Circ. J. 27, 282–293 (1963).
Rodriguez-Iturbe, B., Pons, H. & Johnson, R. J. Role of the immune system in hypertension. Physiol. Rev. 97, 1127–1164 (2017).
Harwani, S. C., Chapleau, M. W., Legge, K. L., Ballas, Z. K. & Abboud, F. M. Neurohormonal modulation of the innate immune system is proinflammatory in the prehypertensive spontaneously hypertensive rat, a genetic model of essential hypertension. Circ. Res. 111, 1190–1197 (2012).
Fujimoto, K., Matsui, M., Fujii, T. & Kawashima, K. Decreased acetylcholine content and choline acetyltransferase mRNA expression in circulating mononuclear leukocytes and lymphoid organs of the spontaneously hypertensive rat. Life Sci. 69, 1649–1638 (2001).
Nobrega, A. C. et al. Enhancement of heart rate variability by cholinergic stimulation with pyridostigmine in healthy subjects. Clin. Auton. Res. 11, 11–17 (2001).
Androne, A. S., Hryniewicz, K., Goldsmith, R., Arwady, A. & Katz, S. D. Acetylcholinesterase inhibition with pyridostigmine improves heart rate recovery after maximal exercise in patients with chronic heart failure. Heart 89, 854–858 (2003).
Behling, A. et al. Cholinergic stimulation with pyridostigmine reduces ventricular arrhythmia and enhances heart rate variability in heart failure. Am. Heart J. 146, 494–550 (2013).
Santos-Almeida, F. M., Girao, H., da Silva, C. A., Salgado, H. C. & Fazan, R. Jr. Cholinergic stimulation with pyridostigmine protects myocardial infarcted rats against ischemic-induced arrhythmias and preserves connexin43 protein. Am. J. Physiol. Heart Circ. Physiol. 308, H101-107 (2015).
Lataro, R. M., Silva, C. A., Tefe-Silva, C., Prado, C. M. & Salgado, H. C. Acetylcholinesterase inhibition attenuates the development of hypertension and inflammation in spontaneously hypertensive rats. Am. J. Hypertens. 28, 1201–2120 (2015).
Lataro, R. M. et al. Chronic treatment with acetylcholinesterase inhibitors attenuates vascular dysfunction in spontaneously hypertensive rats. Am. J. Hypertens. 32, 579–587 (2019).
Katayama, Y. et al. Signals from the sympathetic nervous system regulate hematopoietic stem cell egress from bone marrow. Cell 124, 407–421 (2006).
Kim, E. J., Kim, S., Kang, D. O. & Seo, H. S. Metabolic activity of the spleen and bone marrow in patients with acute myocardial infarction evaluated by 18f-fluorodeoxyglucose positron emission tomograpic imaging. Circ. Cardiovasc. Imaging 7, 454–460 (2014).
Harel-Adar, T. et al. Modulation of cardiac macrophages by phosphatidylserine-presenting liposomes improves infarct repair. Proc. Natl. Acad. Sci. U. S. A. 108, 1827–1832 (2011).
Tang, T. T. et al. Regulatory T cells ameliorate cardiac remodeling after myocardial infarction. Basic Res. Cardiol. 107, 232 (2012).
Weirather, J. et al. Foxp3+ CD4+ T cells improve healing after myocardial infarction by modulating monocyte/macrophage differentiation. Circ. Res. 115, 55–67 (2014).
Nian, M., Lee, P., Khaper, N. & Liu, P. Inflammatory cytokines and postmyocardial infarction remodeling. Circ. Res. 94, 1543–1553 (2004).
Hartman, M. H. T., Groot, H. E., Leach, I. M., Karper, J. C. & van der Harst, P. Translational overview of cytokine inhibition in acute myocardial infarction and chronic heart failure. Trends Cardiovasc. Med. 28, 369–379 (2018).
Antonica, A., Magni, F., Mearini, L. & Paolocci, N. Vagal control of lymphocyte release from rat thymus. J. Auton. Nerv. Syst. 48, 187–197 (1994).
Ratcliffe, E. M., deSa, D. J., Dixon, M. F. & Stead, R. H. Choline acetyltransferase (ChAT) immunoreactivity in paraffin sections of normal and diseased intestines. J. Histochem. Cytochem. 46, 1223–1231 (1998).
Lu, Y. et al. Pyridostigmine ameliorates cardiac remodeling induced by myocardial infarction via inhibition of the transforming growth factor-beta1/TGF-beta1-activated kinase pathway. J. Cardiovasc. Pharmacol. 63, 412–420 (2014).
Barboza, C. A. et al. Cholinergic stimulation by pyridostigmine bromide before myocardial infarction prevent cardiac and autonomic dysfunction. Sci. Rep. 9, 2481 (2019).
Zhang, Y. et al. Chronic vagus nerve stimulation improves autonomic control and attenuates systemic inflammation and heart failure progression in a canine high-rate pacing model. Circ. Heart Fail. 2, 692–699 (2009).
De Ferrari, G. M. et al. Chronic vagus nerve stimulation: a new and promising therapeutic approach for chronic heart failure. Eur. Heart J. 32, 847–855 (2011).
Schwartz, P. J. & De Ferrari, G. M. Vagal stimulation for heart failure: background and first in-man study. Heart Rhythm 6, S76-81 (2009).
Kakinuma, Y., Akiyama, T. & Sato, T. Cholinoceptive and cholinergic properties of cardiomyocytes involving an amplification mechanism for vagal efferent effects in sparsely innervated ventricular myocardium. FEBS J. 276, 5111–5125 (2009).
Rocha-Resende, C. et al. Non-neuronal cholinergic machinery present in cardiomyocytes offsets hypertrophic signals. J. Mol. Cell Cardiol. 53, 206–216 (2012).
Roy, A. et al. Cardiac acetylcholine inhibits ventricular remodeling and dysfunction under pathologic conditions. FASEB J. 30, 688–770 (2016).
Kakinuma, Y. et al. Heart-specific overexpression of choline acetyltransferase gene protects murine heart against ischemia through hypoxia-inducible factor-1alpha-related defense mechanisms. J. Am. Heart Assoc. 2, e004887 (2013).
Azevedo, P. S. et al. Early echocardiographic predictors of increased left ventricular end-diastolic pressure three months after myocardial infarction in rats. Med. Sci. Monit. 18, BR253–BR258 (2012).
Acknowledgements
The all authors and the Coordenação de Aperfeiçoamento de Pessoal de Nivel Superior (CAPES), CNPq (435495/2018-6) and Universidade Nove de Julho.
Funding
This study was supported by the Coordenação de Aperfeiçoamento de Pessoal de Nivel Superior (CAPES) for the support to Post-Graduation Programs, CNPq (435495/2018–6) and Universidade Nove de Julho.
Author information
Authors and Affiliations
Contributions
R.L.B., P.N.B.C., H.D., T.L.M., M.H.M.P., B.D.S., G.A.N., M.C.I., K.A., V.A.P., L.U., and F.M.C.C contributed to the conception and design of the study. P.N.B.C., R.L.B., H.D., T.L.M., and performed the experiments, and analyzed the data and designed the figures; V.A.P., L.U., and F.M.C.C, analyzed the data and wrote the manuscript and final revision. All authors provided critical feedback, edited, and finalized the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Bandoni, R.L., Bricher Choque, P.N., Dellê, H. et al. Cholinergic stimulation with pyridostigmine modulates a heart-spleen axis after acute myocardial infarction in spontaneous hypertensive rats. Sci Rep 11, 9563 (2021). https://doi.org/10.1038/s41598-021-89104-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-021-89104-8
- Springer Nature Limited
This article is cited by
-
Aerobic exercise attenuates dysautonomia, cardiac diastolic dysfunctions, and hemodynamic overload in female mice with atherosclerosis
Scientific Reports (2024)
-
B cells regulate hematopoietic stem cells via cholinergic signaling
Nature Immunology (2022)