
Abstract
In this study, the mitochondrial genome of the stonefly, Oyamia nigribasis Banks, 1920 (Plecoptera: Perlidae), was sequenced and compared with the mtDNA genomes of 38 other stoneflies and two Ephemerae. The O. nigribasis mitogenome is a circular 15,923 bp molecule that encodes a large, noncoding control region (CR) and 37 typical mtDNA genes; these include 13 protein-coding genes (PCGs), 22 transfer RNA genes (tRNAs), and two ribosomal RNA genes (rRNAs), respectively. Most of the PCGs initiated with ATN and terminated with TAN. The dihydrouridine (DHU) arm of tRNASer (AGN) was missing, whereas the other 21 tRNAs all exhibited the typical cloverleaf secondary structure. Stem-loop (SL) structures and tandem repeats were identified in the CR. Phylogenetic analyses using Bayesian inference and maximum likelihood were undertaken to determine relationships between stoneflies. Results indicated that the Antarctoperlaria, which contains Gripopterygidae, was absolutely separated from Arctoperlaria; this finding agrees with morphology. Finally, the overall relationships could be summarized as follows ((((Notonemouridae + Nemouridae) + Leuctridae) + (Scopuridae + (Capniidae + Taeniopterygidae))) + (((Perlodidae + Chloroperlidae) + Perlidae) + (Pteronarcyidae + (Peltoperlidae + Styloperlidae))) + ((Diamphipnoidae + Eustheniidae) + Gripopterygidae)).
Similar content being viewed by others


Introduction
The Plecoptera order of stoneflies is a basal infraorder of the Neoptera; it is dispersed worldwide (except Antarctica) and contains ancient hemimetabolous insects1. Stonefly larvae frequently inhabit clean rivers and streams and are quite sensitive to dirty, polluted environments; thus stoneflies are an important bioindicator of water quality2,3,4.
The typical metazoan mitochondrial genome includes a noncoding sequence called the control region (CR) and 37 genes including 13 protein coding genes (PCGs), 22 tRNAs, two rRNAs5,6. The analysis of mitochondrial genomes has profoundly influenced the genetics and taxonomy of insects7,8,9,10,11,12. The development of whole genome sequencing technologies for insects has been slow in contrast to mtDNA; however, mitochondrial barcodes and sequences are commonly used for insect species identification13,14,15,16. The mtDNA sequences of representative species in all Insecta orders have been deposited in GenBank and exceed 1,500 entries17.
Despite previous studies, conclusions on the phylogeny and biogeography of stoneflies are inconsistent, especially concerning the family composition in the highest systematic categories18,19,20,21,22,23. There is some controversy on the phylogeny of Plecoptera; thus, we obtained the mtDNA sequence of Oyamia nigribasis and constructed phylogenetic trees based on PCG sequences to deduce the phylogenetic relationships of 39 stonefly species.
Results and discussion
Genome annotation and base composition
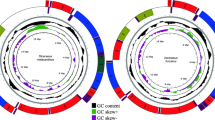
The O. nigribasis mitogenome is a circular 15,923 bp molecule, and contains the typical set of 37 mtDNA genes (13 PCGs, 22 tRNAs, and two rRNAs) along with a noncoding control region (CR) of 1022 bp. Among the 37 genes, nine PCGs and 14 tRNAs were majority strand (J-strand); four PCGs, eight tRNAs, and two rRNAs were minority strand (N-strand) (Fig. 1, Table 1). The gene arrangement in O. nigribasis mtDNA was highly conserved with other sequenced stoneflies and identical to the mitogenome of Drosophila yakuba, which is regarded as the putative ancestral arthropod24. The mitogenome of O. nigribasis contains 11 pairs of adjacent overlapping genes covering 41 nucleotides; 10 pairs were unlinked and encompassed 48 intergenic nucleotides (IGNs). The shortest overlap was 1 bp (multiple sites), whereas the longest was 9 bp and mapped between trnTyr (Y) and cox1 (Table 1). The shortest interval between genes was 1 bp (two sites) while the longest was a 16-bp intergenic region between trnSer2 (UCN) and nad1 (Table 1).

Mitochondrial map of Oyamia nigribasis. Genes outside the map are transcribed clockwise, while genes inside are transcribed counterclockwise. The interior circles show GC content and GC skew, which are plotted as the deviation from the average value of the entire sequence.
Similar A + T contents were observed for the entire O. nigribasis mtDNA molecule, PCGs, tRNAs, rRNAs, and CR, which were 70.2%, 69.1%, 70.2%, 71.5%, and 72.7%, respectively (Table 2). The lowest and highest A + T content was 62.4% for cox3 and 88.1% for trnGlu (E), respectively (Table 1). The AT- and GC-skew expressed positively and negatively, respectively, which is consistent with other stonefly mitogenomes (Table 2).
Protein-coding genes
O. nigribasis PCG were similar in size (65–71 bp), whereas the A + T content varied from 62.7–88.1% (Tables 1, 2). The majority of PCGs possessed the standard start codon ATN (ATT, ATC or ATG); however, nad2 started with GTG, which has also previously been observed for other stonefly such as Taeniopteryx ugola10. Furthermore, most PCGs terminated with complete codons (e.g. TAA, TAG); however, the stop colon in cox1, cox2 and nad5 terminated in a single T, which have also been previously reported for many stoneflies like Leuctra sp., Nemoura nankinensis, Taeniopteryx ugola and Doddsia occidentalis9,10,12. Such translation termination could be completed by post-transcriptional polyadenylation9,10. The relative synonymous codon usage (RSCU) values of TTA (Leu), TCT (Ser), and CCT (Pro) were relatively high, whereas TCG (Ser) and ACG (Thr) were used less frequently than other codons (Fig. 2, Table 3). Most species of stoneflies show a high RSCU value of leucine while the usage of other amino acids are diverse from each other8,9,10,11,12.

Relative synonymous codon usage (RSCU) in O. nigribasis.
Transfer RNA genes
The typical set of 22 tRNA genes was observed in the O. nigribasis mitogenome, and the combined length and mean A + T content was 1426 bp and 70.2%, respectively (Table 2). Fourteen tRNAs were encoded in a clockwise orientation, whereas the remaining eight were transcribed counterclockwise (Fig. 1, Table 1). Apart from trnSer (AGN), where the dihydrouridine (DHU) arm was absent, and that is a very common feature of mitochondrial tRNA-Ser conserved in mammals and some insects9,10,11,12,25, the other 21 tRNAs exhibited the representative cloverleaf secondary structure (Fig. 3) that is typical of other metazoan mitogenomes. The tRNAs contained some mismatched base pairs, and many of these contained G-U pairs (Fig. 3).

Predicted secondary structures of tRNAs from O. nigribasis. tRNAs are labelled with the abbreviations of their corresponding amino acids.
Ribosomal RNA genes
Two rRNA genes were predicted from the O. nigribasis mitogenome and the combined length and A + T content was 2181 bp and 71.5%, respectively (Table 2). The two rRNA genes (rrnL, rrnS) generally map between trnLeu (CUN) and the CR, and this location was conserved in the mtDNA of O. nigribasis (Fig. 1). The full-length, intact rrnL was 1362 bp with an A + T content of 72.8%, whereas the 819 bp rrnS was truncated and had a 70.2% A + T content (Table 1).
The non-coding control region
Mitogenome control regions are highly variable and exhibit variable lengths and nucleotide composition. The O. nigribasis CR was slightly larger than the CR in Plecopteran insects, while the A + T content was typical of other sequenced species. The O. nigribasis CR mapped between rrnS and trnIle, a location that is relatively conserved among stoneflies (Fig. 1).
Seven stem-loop (SL) structures mapped in the CR at the following positions: 14,941–15,006 bp; 15,007–15,059 bp; 15,215–15,238 bp; 15,257–15,293 bp; 15,412–15,431 bp; 15,692–15,714 bp; and 15,715–15,742 bp (Fig. 4). Four tandem repeats mapped between 15,525–15,661 bp. The remaining sequences in the CR were A + T rich (Fig. 4).

Potential stem-loop structures in the control region of O. nigribasis. The bilateral nucleotide motifs of each stem-loop structure [(TA)n, CAT, T(A)n, C(T)nA, GTA] are bounded by black rectangles.
Phylogenetic analyses
The concatenated sequences of 13 PCGs from 38 additional Plecopteran species were downloaded from GenBank. The mtDNAs from two Ephemeroptera species, Parafronurus youi and Isonychia ignota, served as outgroups (Table 5) because they are relatively close to stonefly in classification. ClustalX was used to align the amino acid sequences of the 13 PCGs, and MrBayes v. 3.1.2 and IQ-Tree v. 1.6.12 were utilized to generate the topology by Bayesian inference (BI) and maximum likelihood (ML) analysis, respectively.
The two trees showed a high degree of similarity (Fig. 5, 6), excluding individual species such as the ones in Scopuridae and Pteronarcyidae. However, it is important to note that the nodal support values of the BI tree (Fig. 5) were more credible based on a previous analysis by ML11. Sequence data of selected southern hemisphere families were analyzed; the suborder Antarctoperlaria, including Gripopterygidae, Diamphipnoidae and Eustheniidae, was separated from other stonefly families that affiliated with Arctoperlaria, which is distributed in the northern hemisphere. This finding differs from the conclusion that Gripopterygidae could not be separated from other Arctoperlarian families in Shen & Du, 201912, while consistent with Ding, 201923. The two clades of Arctoperlaria, Euholognatha and Systellognatha, were strongly supported at the family level as monophyletic clades. In the infraorder Euholognatha, it was explicit that Leuctridae was clustered with the group of Nemouridae + Notonemouridae and Taeniopterygidae was recovered as the sister group of Capniidae. However, the phylogenetic relationship of Scopuridae, which need more datasets and independent evidence, was difficult to determine. Scopuridae was close to Taeniopterygidae and Capniidae based on BI analysis but clustered with other Euholognatha in the ML tree. From the perspective of Systellognatha, the monophyletic relationships in the superfamily Perloideae could be highly advocated as (Perlidae + (Perlodidae + Chloroperlidae)), even though marginal divergence has been reported11,16. However, the phylogenetic relationship within the superfamily Pteronarcyoidea is more controversial. As shown in the BI tree (Fig. 5), Styloperlidae was more closely related to Peltoperlidae and clustered with Pteronarcyidae, which was consistent with morphology but inconsistent with phylogeny of ((Pteronarcyidae + Styloperlidae) + Peltoperlidae)11. Pteronarcyidae was included in the clade containing Perloideae and clustered with Styloperlidae and Peltoperlidae in the ML tree. Similar discrepancies have been reported in related studies23 and are potentially caused by the use of different algorithms and models.

Phylogenetic relationships among 39 stoneflies based on Bayesian inference (BI). Numbers at the nodes represent posterior probabilities. Family and infraorder names are marked to the right of each species. Parafronurus youi and Isonychia ignota served as outgroup species.

Phylogenetic relationships among 39 stoneflies based on maximum likelihood (ML) analysis. Numbers at the nodes represent bootstrap values. Family and infraorder names are marked to the right of each species. Parafronurus youi and Isonychia ignota served as outgroup species.
Increasing numbers of stonefly mtDNAs are undergoing sequence analysis. Thus, it is likely that controversial phylogenetic relationships will eventually be resolved and the phylogeny of Plecoptera can be more accurately presented based on increased numbers of mitogenomes. It is worth looking forward to that more genes just like nuclear genes can also help to improve the phylogeny of stoneflies.
Methods
Sample preparation and mitogenome amplification
This study was conducted without harming protected or endangered species, and all research activities were authorized. Specimens of O. nigribasis were collected from Benxi (Liaoning Province, China; July, 2018) and preserved in 100% ethanol. DNA extraction was performed using instructions supplied with the Column mtDNAout kit (Tianda Beijing, China). Universal or specifically- designed primers were used to amplify mitochondrial genes in long overlapping fragments (Table 4). LA-PCR and consecutive specific PCR amplifications were conducted using conditions described previously10. PCR products were purified with the Axygen DNA Gel Extraction Kit (Axygen Biotechnology, Hangzhou, China), separated in 1.0% agarose gels, and sequenced by Map Biotech Co. (Shanghai, China).
Mitogenome assembly and annotation
Mitogenome assembly was conducted with CodonCode Aligner (http://www.codoncode.com/aligner/). Genes encoding PCGs and rRNAs were identified using mtDNA sequences of other Plecoptera and boundaries were defined with ORF finder (https://www.ncbi.nlm.nih.gov/orffinder/). CGView Server26 and MITOS27 were used to draft mtDNA maps and predict tRNA secondary structure; nucleotide composition was obtained with MEGA v. 6.028. Formulas for AT-skew [A – T]/[A + T] and GC-skew [G – C]/[G + C]29 were used to derive AT and GC composition, respectively. Tandem Repeats Finder (http://tandem.bu.edu/trf/trf.advanced.submit.html) and DNAMAN v. 6.0.3 was utilized to detect tandem repeats in the putative CR and to predict stem-loop (SL) structures, respectively. The mtDNA sequence of O. nigribasis was deposited in GenBank as accession no. MN548290.
Phylogenetic analysis
The phylogeny of 39 Plecoptera mitogenomes were analyzed, including 18 Euholognathas, 16 Systellognathans, 5 Antarctoperlarias. Parafronurus youi and Isonychia ignota from the family Ephemeroptera were used as outgroup species (Table 5). Thirteen PCGs were ordered and assembled using MAFFT30 and SequenceMatrix v. 1.7.8 31, and stop codons were excluded. Nucleotide saturation was detected using DAMBE v. 5.2 prior to constructing phylogenetic trees; optimal displacement models (GTR + G + I) were deduced using PartitionFinder v. 2.1.1 32 with Bayesian Information Criterion (BIC) and a greedy search algorithm with unlinked branch lengths. Bayesian inference analyses were conducted with MrBayes v. 3.1.2 (http://morphbank.ebc.uu.SE/mrbayes/) and 20 million generations; sampling occurred every 100 generations with four chains (three hot and one cold), and a burn-in of 25% trees33 Tracer v. 1.5 (http://tree.bio.ed.ac.uk/) (effective sample size > 200) was used to examine the stationarity of all runs. For maximum likelihood, 10,000 ultrafast bootstrap (UFBoot) approximations were performed with IQ-Tree v. 1.6.12 (http://www.iqtree.org/) 34,35. Ultimately, TreeView v. 5.1.6 or FigTree v. 1.4.2 was used to transform data into phylogenetic trees and for data annotation.
References
Zwick, P. Insecta: Plecoptera phylogenetisches system and katalog. Das Tierreich. 94, 1–465 (1973).
Stewart, K. W. & Stark, B. P. Nymphs of North American Stonefly Genera (Plecoptera) 2nd edn. (The Caddis Press, London, 2002).
Stewart, K. W. & Stark, B. P. An Introduction to the Aquatic Insects of North America 4th edn, 14 (Kendall-Hunt Publishing Company, Dubuque, IA, 2008).
Qian, Y. H. et al. Mitochondrial genome of thestonefly Kamimuria wangi (Plecoptera: Perlidae) and phylogenetic position of Plecoptera based on mitogenomes. PLoS ONE 9(1), e86328 (2014).
Simon, C. et al. Evolution, weighting, and phylogenetic utility of mitochondrial gene sequences and a compilation of conserved polymerase chain reaction primers. Ann. Entomol. Soc. Am. 87, 651–701 (1994).
Simon, C. et al. Incorporating molecular evolution into phylogenetic analysis, and a new compilation of conserved polymerase chain reaction primers for animal mitochondrial DNA. Annu. Rev. Ecol. Evol. Syst. 37, 545–579 (2006).
Cameron, S. L., Barker, S. C. & Whiting, M. F. Mitochondrial genomics and the new insect order Mantophasmatodea. Mol. Phylogenet. Evol. 38, 274–279 (2006).
Chen, Z. T. & Du, Y. Z. Complete mitochondrial genome of Capnia zijinshana (Plecoptera: Capniidae) and phylogenetic analysis among stoneflies. J. Asia-Pac. Entomol. 20, 305–312 (2017).
Chen, Z. T. & Du, Y. Z. First mitochondrial genome from Nemouridae (Plecoptera) reveals novel features of the elongated control region and phylogenetic implications. Int. J. Mol. Sci. 18(5), 996 (2017).
Chen, Z. T. & Du, Y. Z. The first two mitochondrial genomes from Taeniopterygidae (Insecta: Plecoptera): structural features and phylogenetic implications. Int. J. Biol. Macromol. 111, 70–76 (2018).
Chen, Z. T., Zhao, M. Y., Xu, C. & Du, Y. Z. Molecular phylogeny of Systellognatha (Plecoptera: Arctoperlaria) inferred from mitochondrial genome sequences. Int. J. Biol. Macromol. 111, 542–547 (2018).
Shen, Y. & Du, Y. Z. The mitochondrial genome of Leuctra sp. (Plecoptera: Leuctridae) and itsperformance in phylogenetic analyses. Zootaxa 4671(4), 571–580 (2019).
Ratnasingham, S. & Hebert, P. D. N. BOLD: the barcode of life data system. Mol. Ecol. Not. 7, 355–364 (2007).
Cameron, S. L. Insect mitochondrial genomics: implications for evolution and phylogeny. Annu. Rev. Entomol. 59(1), 95–117 (2014).
Elbrecht, V. et al. The complete mitochondrial genome of the stonefly Dinocras cephalotes (Plecoptera, Perlidae). Mitochondrial DNA. 26, 469–470 (2015).
Wang, Y. et al. The first mitochondrial genome from Scopuridae (Insecta: Plecoptera) reveals structural features and phylogenetic implications. Int. J. Biol. Macromol. 122, 893–902 (2019).
Chen, Z. T. Molecular Phylogeny of Plecoptera and Taxonomy of Euholognatha (Plecoptera: Arctoperlaria) from China (Yangzhou University, Yangzhou, 2018).
Illies, J. Phylogeny and zoogeography of the Plecoptera. Annu. Rev. Entomol. 10, 117–140 (1965).
Thomas, M. A. et al. Molecular phylogenetic analysis of evolutionary trends in stonefly wing structure and locomotor behavior. Proc. Natl Acad. Sci. 97(24), 13178–13183 (2000).
Zwick, P. Phylogenetic system and zoogeography of the Plecoptera. Annu. Rev. Entomol. 45, 709–746 (2000).
Terry, M. D. & Whiting, M. F. Mantophasmatodea and phylogeny of lower neopterous insects. Cladistics 21(3), 240–257 (2005).
McCulloch, G. A., Wallis, G. P. & Waters, J. M. A time-calibrated phylogeny of southern hemisphere stoneflies: testing for Gondwanan origins. Mol. Phylogenet. Evol. 96, 150–160 (2016).
Ding, S. M. et al. The phylogeny and evolutionary timescale of stoneflies (Insecta: Plecoptera) inferred from mitochondrial genomes. Mol. Phylogenet. Evol. 135, 123–135 (2019).
Clary, D. O. & Wolstenholme, D. R. The ribosomal RNA genes of Drosophila mitochon-drial DNA. Nucl. Acids Res. 13, 4029–4045 (1985).
Suzuki, T. et al. Human mitochondrial tRNAs: biogenesis, function, structural aspects, and diseases. Ann. Rev. Genet. 45(1), 299–329 (2011).
Grant, J. R. & Stothard, P. The CGView server: a comparative genomics tool for circular genomes. Nucl. Acids Res. 36, 181–184 (2008).
Bernt, M. et al. MITOS: Improved de novo metazoan mitochondrial genome annotation. Mol. Phylogenet. Evol. 69, 313–319 (2013).
Tamura, K. et al. MEGA6: molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 30, 2725–2729 (2013).
Perna, N. T. & Kocher, T. D. Patterns of nucleotide composition at fourfold degenerate sites of animal mitochondrial genomes. J. Mol. Evol. 41, 353–358 (1995).
Katoh, K. & Standley, D. M. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780 (2013).
Vaidya, G., Lohman, D. J. & Meier, R. SequenceMatrix: concatenation software for the fast assembly of multi-gene datasets with character set and codon information. Cladistics 27, 171–180 (2010).
Lanfear, R. et al. PartitionFinder 2: new methods for selecting partitioned models of evolution for molecular and morphological phylogenetic analyses. Mol. Biol. Evol. 34, 772–773 (2016).
Ronquist, F. & Huelsenbeck, J. P. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 19, 1572–1574 (2003).
Nguyen, L. T. et al. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum likelihood phylogenies. Mol. Biol. Evol. 32, 268–274 (2014).
Gao, F. L., Shen, J. G. & Liao, F. R. The first complete genome sequence of narcissus latent virus from Narcissus. Arch. Virol. 163, 1383–1386 (2018).
Acknowledgments
We sincerely thank Carol for help in the improvement of manuscript and the editors for their valuable suggestions and comments. We are also very grateful to the National Natural Science Foundation of China (31872266; 31071958) for financial support.
Author information
Authors and Affiliations
Contributions
M.Y.Z. and Y.Z.D. conceived and designed the experiment; M.Y.Z. and Q.B.H. performed the experiments, completed genome annotation; M.Y.Z. analyzed the data and wrote the manuscript; Y.Z.D. provided financial support, revised the manuscript, and approved the final version.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhao, MY., Huo, QB. & Du, YZ. Molecular phylogeny inferred from the mitochondrial genomes of Plecoptera with Oyamia nigribasis (Plecoptera: Perlidae). Sci Rep 10, 20955 (2020). https://doi.org/10.1038/s41598-020-78082-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-78082-y
- Springer Nature Limited
This article is cited by
-
Mitochondrial genomes provide insights into the Euholognatha (Insecta: Plecoptera)
BMC Ecology and Evolution (2024)