
Abstract
Successful spermatogenesis and oogenesis are the two genetically independent processes preceding embryo development. To date, several fertility-related proteins have been described in mammalian species. Nevertheless, further studies are required to discover more proteins associated with the development of germ cells and embryogenesis in order to shed more light on the processes. This work builds on our previous software (OOgenesis_Pred), mainly focusing on algorithms beyond what was previously done, in particular new fertility-related proteins and their classes (embryogenesis, spermatogenesis and oogenesis) based on the support vector machine according to the concept of Chou’s pseudo-amino acid composition features. The results of five-fold cross validation, as well as the independent test demonstrated that this method is capable of predicting the fertility-related proteins and their classes with accuracy of more than 80%. Moreover, by using feature selection methods, important properties of fertility-related proteins were identified that allowed for their accurate classification. Based on the proposed method, a two-layer classifier software, named as “PrESOgenesis” (https://github.com/mrb20045/PrESOgenesis) was developed. The tool identified a query sequence (protein or transcript) as fertility or non-fertility-related protein at the first layer and then classified the predicted fertility-related protein into different classes of embryogenesis, spermatogenesis or oogenesis at the second layer.
Similar content being viewed by others


Introduction
Proteins are involved in different aspects of life activities and play critical roles in various biological processes such as the early stages of life development1. Germline developmental events including spermatogenesis and oogenesis, and also other variety of differentiation processes such as embryogenesis and organogenesis are regulated by a number of protein signaling cascades which are critical for normal development2,3,4,5. Gametogenesis is the first stage in sexual reproduction, by which haploid sperm and egg cells are formed from the diploid gamete cells in the ovaries and testes. This process is called oogenesis in the female and spermatogenesis in the male2,3,4,5. Embryogenesis (or embryo development) is the development of a fertilized egg that fuses with a sperm, forming a zygote. After zygote stage, many changes occur and the embryo undergoes several mitotic divisions to generate tissues layers that eventually develop into specific organs6,7. During oogenesis, spermatogenesis and embryogenesis cells initially proliferate and then differentiate into specific tissues. Moreover, oogenesis and spermatogenesis are tightly regulated complex processes critical for fertility8,9. Therefore, because of the importance of proteins related to the fertility, their large-scale identification will provide a knowledge base for detailed understanding of biological processes and the mechanisms underlying each step of spermatogenesis, oogenesis and embryogenesis.
A survey of the UniProtKB/TrEMBL databases showed that a large number of un-reviewed proteins exists, which are not annotated and yet to be reviewed. Furthermore, owing to the availability of large number of proteins generated in postgenomic age, wide varieties of unannotated data sets are accumulated in various species and databases10. On the other hand, an investigation of protein folding, structure, and function has remained experimentally costly, time consuming and requires sophisticated technical equipment. Hence, there seems to be some benefit in developing efficient computational approaches that can predict protein functions timely and precisely8,11,12,13,14,15,16. By applying such computational models, it is possible to provide an advantageous and powerful substitutional strategy for automating whole proteome annotation without costly and time-consuming experiments.
Over the years, different methods have been proposed for predicting the putative function of unannotated proteins17,18. The sequence similarity-based search tools, such as BLAST and PSI-BLAST are among the most robust approaches that have been extensively applied for predicting the unknown protein annotation13,19,20,21. These approaches become more challenging, once the similarity between the input and target sequences is not too much19,21,22,23. To overcome this obstacle, a great deal of attention has been given recently to predict the protein function by applying machine learning based methods. The reliability and efficiency of such methods are well demonstrated in different areas12,13,24,25,26,27. The higher performance of these methods can be attributed to their ability to learn the underlying rules in training datasets by optimizing the related parameters during the model development. Among the variety of machine learning algorithms which have been proposed in the literature, support vector machine (SVM) is one of the state-of-the-art algorithms and well suited. It is widely believed that SVM is a most promising classifier in different disciplines because of its high accuracy, as well as its power of high dimensional data handling12,13,28,29,30,31,32.
In a previous study12, for the first time, a model for identifying proteins related to oogenesis was constructed using SMV. Based on the constructed model, OOgenesis_Pred software was developed, which provides a convenient way to annotate the candidate proteins. For the development of this software, a new algorithm was offered to predict not only the proteins involved in oogenesis, but also those implementing spermatogenesis and embryogenesis processes. It is believed that discrimination of biological functions will become more accurate if a collective approach which considers the different kinds of fertility related proteins and their functions are used. Actually, this kind of multi-prediction systems may lead to deeper informative data. Thus, herein, this study aimed to employ the multi label theory in order to develop a new algorithm based on previous SVM classifier along with informative protein physicochemical features. It is expected that this software will be useful in simultaneously predicting the proteins involved in oogenesis, spermatogenesis and embryogenesis processes. Evaluation through a five-fold cross validation and independent test dataset were applied to prove the validity of this method and to check its efficiency, reliability and robustness for prediction of fertility-related proteins.
Methods
Datasets
To develop a powerful statistical predictor tool and to train and test it, a high quality and objective benchmark dataset is need. This step is the most important concern in any machine learning method33,34. To this end, the following steps were performed:
-
1.
The proteins sequences were collected through searching the UniProtKB database (release 2017_4) with gene ontology terms “oogenesis”, “spermatogenesis” and “embryogenesis”, individually, and then, the initial positive datasets for each fertility-related protein classes were created.
-
2.
Then, only the reviewed proteins which have been experimentally annotated, with the length <6000 or >60 amino acids were selected.
-
3.
The homologous sequences from the datasets using CD-HIT software35 were eliminated to ensure that any two sequences shared a pairwise sequence identity of less than 50%.
-
4.
Thereafter, the protein sequences with non-canonical amino acids such as B, X, and Z were excluded.
In this study and by adopting the aforementioned steps, a total of 345, 641, and 831 proteins for “oogenesis”, “spermatogenesis” and “embryogenesis” classes, respectively were obtained, which constituted the positive datasets in this direction. The protein sequences of the negative dataset were also constructed using the UniProtKB database (release 2017_4)12,13. Briefly, the database was depleted by comprehensive searching of all keywords suspicious of implying fertility functionality. Only reviewed proteins with length of <6000 and >60 and canonical amino acids were retained. The CD-HIT software with a 50% cutoff was used to remove the highly similar sequences.
In the next step, the domains of all proteins (negative and positive datasets) were compared with each other and proteins with a shared domain were removed from negative datasets. To accomplish this, all related domains were extracted through Pfam database. This enabled the construction of a more reliable negative dataset, which is solely constructed by non-fertility related proteins. Eventually, a number of 342,891 non-fertility related protein sequences were obtained as negative dataset.
The resulting positive (minority class) and negative (majority class) datasets were extremely unbalanced, an issue which is known as the class imbalance problem. Such an imbalance always has undeniable impact on classification results and would lead to a higher prediction rate in favor of majority class36. To overcome this issue, a commonly used approach, called random sampling solution (reducing the majority class)37, was used. For this purpose, proteins were randomly selected from the negative dataset at the same size of the positive datasets (without replacement). This was performed to generate a balanced benchmark dataset and to minimize influences of the larger negative dataset. However, the efficiency and capability of the predictor cannot be accessed using only one random sample from the negative dataset38. Hence, to increase the confidence which is preserved by the present diversity in the negative dataset in random sampling processes, each positive dataset was mixed with five non-overlapping negative samples (drawn without replacement from negative dataset). In other words, the negative dataset was first divided into five sub-datasets with non-overlapping sequences, where the number of sequences in each sub-dataset was equal to that of the positive dataset. Then, the negative sub-datasets and the positive dataset were combined to form a new dataset. Eventually, five new datasets were constructed for each class. Moreover, to create a general fertility-related protein class, all the positive datasets were incorporated and mixed with five different negative sub-datasets. By so doing, 20 benchmark datasets were constructed, with each being applied to train a different SVM model (three classes including embryogenesis, spermatogenesis and oogenesis along with incorporation of all these classes, and each class was combined with five different negative samples).
It is worth noting that cd-hit-2d tool from CD-HIT package was applied across the positive and negative sequences in each benchmark dataset to remove the sequences with 100% identity. This task was possible due to the incomplete annotation of protein sequences in UniProtKB database. Therefore, it is not too far from expected that some negative sequences are identical with positive sequences in each benchmark dataset. The complete information and details of these benchmark datasets is provided in Supplementary File 1. Also, the exact number of proteins in each of the train and test datasets are displayed in Fig. 1.

Summary of our pipeline for developing PrESOgenesis.
Extraction of protein features
Owing to all the existing machine-learning methods which can only handle vector but not sequence samples, a crucial step in computational biology and in the construction of machine learning-based classifier is the formulation of protein sequences with an effective mathematical vector or a discrete model. In this regard, the pseudo amino acid composition (PseAAC), proposed by Chou (Chou’s PseAAC)39, is an efficient and widely used method for the conversion of a protein sequence to a vector for developing different predictors12,13,33,40,41,42,43,44,45,46,47,48. The PseAAC makes use of a set of more than 20 discrete factors to represent a protein sequence and captures its key features without completely losing its sequence-pattern information19. In the current study, using the concept of Chou’s PseAAC, the protein sequences were encoded with a multi-feature integration strategy to fuse the six different modes of Chou’s PseAAC, using protr package. This package offers a comprehensive and unique tool for generating diverse sequence descriptors of protein sequences49. In total, a number of 1920 features were extracted including a 420 dimensional vector indicating amino acid composition and dipeptide composition, a 720 dimensional vector representing Moreau-Broto autocorrelation, Moran autocorrelation and Geary autocorrelation descriptors, a 147 dimensional vector indicating composition, distribution and transition, a 160 dimensional vector indicating sequence-order-coupling number and quasi-sequence-order descriptors, a 130 dimensional vector indicating Pseudo amino acid composition (Type I PseAAC) and amphiphilic pseudo amino acid composition (Type II PseAAC), and a 343 dimensional vector indicating Conjoint Triad descriptors. Previous studies have shown the efficiency and robustness of these features in predicting the protein function12,13. The detailed description of these features is provided in Supplementary File 2 (S1).
Support vector machine (SVM)
Different methods have been used to predict the protein function, such as decision tree50, random forest51, neural network13 and ensemble learning52. In this study, SVM was applied to develop all possible models for prediction of fertility-related protein, due to its excellent learning ability and good capability for non-linear classification46,53. SVM is a supervised learning hypothesis which can transform the non-linearly separable input vector into a high-dimensional Hilbert space and construct an optimal hyperplane to classify two types of samples54. As SVM can transform the input vector from a low dimensional space to a higher dimensional space, its generalization power is better than other machine learning methods for majority of classification tasks. Accordingly, this method has been extensively used in bioinformatics for pattern recognition as well as protein structure and function classification12,13,31,46,55,56,57,58,59,60,61. LIBSVM package (version 3.22)62 was used to implement SVM using radial basis function (RBF) as kernel function. The kernel function determines the learning ability of SVM and prediction performance can be improved by an appropriate choice of kernel function63. In this study, RBF was used due to its suitability for non-linear classification as well as its good general performance. For achieving the best model, the optimal values of tunable parameter C and the kernel width parameter γ were determined by the grid search method; this method selects the values of parameters with the consideration of the highest accuracy based on five-fold cross validation. To train and test the model, the benchmark datasets were divided into two independent subsets, training (80% of the benchmark dataset) and testing datasets (20% of the benchmark dataset). To carry out five-fold cross-validation, the training dataset was randomly split into five sub-datasets with approximately equal size. For each cross validation, SVM model was trained based on four sub-datasets (called sub-training set) and the other sub-dataset was used as testing set (called sub-testing set). The process was repeated five times to ensure that each sub-dataset was used once as the sub-testing set. The five validation results were then combined to generate a single estimation. Since possibly in the binary classification mode, the constructed model can be overfitted to the training dataset, the testing dataset which is definitely blind to the process of model training, was used to further evaluate the effectiveness of the predictor.
In this study, a novel two-layer classification framework was developed, as the SVM model in the first-layer classifier was trained with all the training datasets (oogenesis, spermatogenesis and embryogenesis), serving to predict a query protein sequence as fertility or non-fertility related protein. The SVM models in the second layer were trained with oogenesis, spermatogenesis and embryogenesis training datasets, separately as binary predictor to further identify the class of the predicted protein in the previous layer (oogenesis, spermatogenesis or embryogenesis).
Performance evaluation
To quantitatively analyze the efficiency of the proposed predictor, all the SVM models were evaluated with four wildly used performance measures, including specificity (Sp, ability to correctly identify non-fertility), sensitivity (Sn, ability to correctly identify fertility), accuracy (Acc, overall accuracy of the discrimination between positive and negative) and Matthew’s correlation coefficient (MCC, a correlation coefficient between the observed and predicted binary classifications, which takes into account both over- and underpredictions). To make these performance measures easily understood by readers, a set of equations were applied as follows64:
In the above equations, N+ and N− represents the total number of the positive and negative protein sequences investigated, respectively, whereas \({{\rm{N}}}_{-}^{+}\) represents the total number of the positive proteins incorrectly predicted to be negative and \({{\rm{N}}}_{+}^{-}\) represents the total number of false prediction which is incorrectly predicted true. For example, when \({{\rm{N}}}_{-}^{+}=0\) implies that none of the fertility-related proteins were mis-predicted to be a non-fertility related protein, so the sensitivity is 1. Also, when \({{\rm{N}}}_{+}^{-}=0\) implies that none of the non-fertility related proteins correctly predicted as fertility-related proteins, so the specificity is 1.
In the statistical prediction, independent dataset test, subsampling (K-fold cross validation) test, and jackknife cross-validation are often employed to examine the predictive capability of a predictor. As demonstrated in a series of studies65,66,67,68, among the three test methods, the jackknife cross-validation is deemed as the most objective one that always yield a unique result and hence has been widely used to test the quality of various predictors. However, to save computational time, the five-fold cross-validation (during training the model) and independent test (during testing the model) methods were applied to evaluate the models and generate the performance measurements.
Feature importance
In machine-learning methods, not all features are equally important for the performance of the trained model, especially for high dimension data. Hence, some features make key contributions and are more important than the others. Thus, it is imperative to employ feature selection technique in order to discover the top ranked feature set according to their predictive contribution. In this study, to discover the most relevant and informative features for discriminating fertility-related proteins, a feature selection method was employed based on a previous study12. Running towards this, Rapidminer package (Version 5.2) was applied to fuse 10 different feature weighting algorithms including chi squared statistic, information gain rule, information gain ratio, gini index, deviation, uncertainty, relief, principle component analysis (PCA) and SVM (Supplementary Files 2, S2). Important features were initially defined (based on five general benchmark datasets, a combination of all three fertility-related classes) according to their score from each algorithm (a feature with score >0.5 marked as important). Then, only the features that were marked as important by at least five algorithms were considered. Eventually, common important features in five benchmark datasets were reported as optimal feature set. Furthermore, the optimal feature set was applied to train SVM model, based on five general benchmark datasets, and to investigate the performance of this set for discriminating fertility-related proteins.
Results and Discussions
The cascade of molecular and intracellular processes which occur in germ cells during spermatogenesis, oogenesis and later embryogenesis are still far from being fully exploited, and this could be a serious bottleneck in the success of fertilization process. Identifying specific fertility related proteins and elucidating their function in regulation of spermatogenesis, oogenesis and embryogenesis are essential for gaining fundamental biological insight intoclinical practice69. Machine learning methods, such as SVM, have been used in many fields and their applications in bioinformatics are increasing12,13,24,25,26,27. However, these methods are yet to be applied in the study of fertility-related proteins and their relevant classes. Therefore, this study aimed to apply SVM-based approach in combination with a comprehensive physical chemical property set to construct a method, which could be used to predict the probability of a sequence, referred to as fertility related protein, as well as its class. The framework diagram of this proposed method is presented in Fig. 1.
Models performance
To obtain an efficient and robust predictor which can achieve the highest accuracy for identifying fertility-related proteins, it is crucial to apply appropriate training datasets as well as useful physicochemical properties vectors of proteins. Note that no distinct non-fertility related protein dataset exists that can be used here as negative database. Therefore, we took the advantages of our previous method to construct the dataset, which employed a stringency approach to create positive and negative datasets12,13. Moreover, comprehensive and informative protein physicochemical properties were applied for fertility-related protein classification, which has been wildly used to predict the different proteins classes70,71. A total of 20 training datasets were used for SVM models training, which were associated with oogenesis, spermatogenesis, embryogenesis and general classes (which included all three above mentioned classes). The four performance measurements obtained by training and testing SVM models in different classes along with optimized parameter γ for each dataset are listed in Tables 1–4. The experimental results of an independent assessment of SVM as a binary classifier over different training and testing datasets are stored in the rows in the Table. Using five-fold cross validation approach, SVM model achieved an average prediction Acc of 82.97% with average MCC of 66.95 at the first layer (general class).
At the second layer, the average prediction Acc achieved was 84.00% with average MCC value of 79.40 for oogenesis, an average of 83.55% prediction Acc with average MCC value of 67.16 for spermatogenesis and 81.15% average prediction Acc with an average MCC value of 60.61for embryogenesis (Tables 1–4). Also, the maximum prediction Acc of 85.33%, 83.61%, 82.33% and 82.56% was obtained in oogenesis, spermatogenesis, embryogenesis and general classes, respectively. These results showed that the performance of the classifiers was similar in different classes.
The average Sn and Sp achieved for fertility-related protein prediction was nearly equal and greater than 80% in all classes. This finding indicates that there was no bias in classification, implying an equal chance of identifying the fertility and non-fertility-related proteins correctly (Tables 1–4). To examine the efficiency of the proposed model, five different negative samples were adopted, by random sampling, for each positive dataset. The obtained results on different datasets were slightly different, but consistent in general and the average Acc, Sn, Sp and MCC were all higher than 80% by five-fold cross validation. Therefore, down-sampling of the negative dataset was quite useful.
To further emphasize the effectiveness of the proposed models, roughly 20% of total datasets was retained in each class (testing datasets) for independent evaluation of the final model. Similar results were obtained based on the independent blind test, as the positive datasets could be discriminated from negative datasets with acceptable Acc, MCC, Sn and Sp in all classes. This suggests an encouraging capability and robustness of the proposed method for identifying the fertility-related proteins and their classes (Tables 1–4).
Feature importance
It is meaningful to determine the most relevant features critical for fertility-related proteins prediction, so that it could be possible to figure out the value of each feature and better understand these proteins. In this study, a stringency feature selection approach was applied on general benchmark datasets to identify optimal feature set. The results led to identification of 22 important features, listed in Table 5, including two amino acid frequency features (isoleucine and serine frequency). Additionally, another important feature group included one dipeptide frequency feature (IA, isoleucine-alanine), 13 features from the CTD descriptor group and five features from the quasi-sequence-order descriptors group and one feature from the conjoint triad descriptor. This finding was consistent with a previous study, which confirmed serine as an important feature for oogenesis-related proteins prediction12. Interestingly, the importance of serine in fertility-related proteins, especially oogenesis72,73,74, was also highlighted in previous studies75,76,77,78,79,80. Also, how members of the TGF-β superfamily induce the constitution of hetero-oligomeric complexes of two distantly related types of serine/threonine kinases was also highlighted12.
In order to provide more evidence regarding the important role of serine in spermatogenesis, first the role of protamine in maintaining spermatogenesis and spermatozoa quality was highlighted. Protamine is well known to function as an essential protein for sperm nuclear condensation. It is a very simple and specialized protein which comprises 44 amino acid residues that belongs to three amino acid types: arginine, glycine, and serine. In all vertebrate, two structural elements have been identified in protamines. The first, which is facilitating binding of protein to DNA, is a series of small ‘anchoring’ domains containing multiple arginine or lysine amino acids. The second one is a multiple residues of serine and threonine, which could potentially act as phosphorylation sites81. There is also enough evidence that phosphorylation-dephosphorylation events control the deposition of protamines on sperm chromatin and the subsequent chromatin condensation. Protamines are highly phosphorylated, shortly after their synthesis and before binding to DNA, whereas they become largely dephosphorylated during sperm maturation82. Sperm function is regulated by the activation of intracellular signaling systems during fertilization, which control protein phosphorylation. Protein phosphorylation is involved in modification of proteins, post-translationally, that allows the control of various cellular processes by the cell. Phosphorylation mostly occurs on serine or threonine residues, but it is also encountered on tyrosine residues. Protein kinases and phosphatases are enriched in sperm and regulate the phosphorylation state of phosphoproteins. Serine/threonine phosphorylation are known to occur in spermatozoa and has a pivotal role in the regulation of sperm motility83. As a consequence of improved understanding about serine amino acid, perturbation of the cell cycle at the G1–S and/or G2–M transitions is likely to occur just following serine deficiency. It was recently demonstrated that in the absence of L-serine, the fibroblast cells prepared from knockout embryos are unable to proliferate. Therefore, the serine synthesized within cells of embryos plays a crucial role in cell cycle progression of a variety of cell types including radial glia during fetal development. With regard to cell cycle dysregulation in the knockout spinal cord, it is notable that radial glia cells in the ventricular zone expresses cell proliferation markers PCNA and Ki67, and are not assumed to enter neuronal differentiation or a mitotically quiescent G0-like state84.
Isoleucine is believed to be genetically related to male fertility through its synthetic and metabolic activities85. For instance, mutation of encoding gene of ubiquitin-specific protease 26 (responsible for a valine to isoleucine change) has been reported to cause male infertility and adversely affect the testicular function86. Cytochrome P4501A1 participates in isoleucine–valine exchange; mutation of its heme-binding region is also associated with infertile men87. Haqq et al., highlighted the importance of isoleucine in sex-determining region Y (SRY) protein, specifically the orientation of isoleucine side chain in DNA minor groove88. Isoleucine plays essential roles in embryogenesis, particularly during fetal development89. Isoleucine is among the branched chain amino acids (BCAA), which have been considered as one of the vital elements in fetus development. In this regard, it has been shown that BCAA supplemented diets can improve the gene and protein expression of IGF-1 and IGF-2 in fetal liver, consequently leading to amelioration of fetal growth restrictions90.
A SVM-based machine learning method was also built using the general datasets for the prediction of fertility-related proteins based on optimal feature set. Similarly, five-fold cross validation and independent tests were applied to estimate the performance. The performances of the trained model using optimal feature set on general datasets are shown in Table 6. When optimal feature set were used, the model could reach (on average) 81.68%, 79.44%, 79.98% and 59.9 (evaluated by five-fold cross validation) for Sn, Sp, Acc and MCC, respectively. Also, the average Sn, Sp, Acc and MCC were 78.53%, 76.15%, 78.32% and 55 based on independent test, respectively (Table 6). As shown in Table 6, once optimal feature set was used for training the SVM model compared to the original feature set, there was decrease in the Acc by 3% and 4.6% by five-fold cross validation and independent test, respectively. This can be somehow attributed to the stringent feature selection method, which led to finding only 22 very important features. However, the models trained by SVM using the optimal feature set could classify fertility-related proteins and their classes, with a relatively high accuracy as well as with a relatively high and equal sensitivity and specificity (Table 6). Overall, the results in this study imply that a comprehensive feature set is more efficient than selected features in recognizing fertility-related protein. It is in complete agreement with previous studies suggesting that using a comprehensive and proper protein feature set gives the better result12,13,91.
Software development
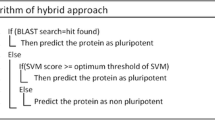
A software support is required to make the development of new classification models publicly available. In order to enhance the value of our evolving software into practical applications, a two-layer classifier called PrESOgenesis (Predict Embryo-, Spermato- and Oogenesis) has been provided freely at https://github.com/mrb20045/PrESOgenesis. The best model in each class was selected based on five-fold cross validation results for the development of the predictor. The first layer predicts the input sequence, whether it is fertility-related or not, using a binary SVM classifier. If not, the classifier is automatically stopped. If yes, the sequence is considered as a fertility-related protein candidate and is subsequently submitted into the second layer. Then, in this layer, three binary SVM classifiers (for oogenesis, spermatogenesis, and embryogenesis classes) are applied to determine to which classes of fertility-related proteins they are assigned. The class is designated as one of the three categories (oogenesis, spermatogenesis or embryogenesis), on the basis of highest SVM score. In this study, a two-layer classifier was proposed for predicting fertility-related protein. The high efficiency of this method has been reported in previous studies such as predicting membrane proteins92, enhancer prediction93, remote protein homology detection94, identifying piwi-interacting RNAs34 and miRNA Drosha processing site detection95.
PrESOgenesis can be used by a wide variety of researchers with limited knowledge of the SVM computing environment, since it just requires simply upload sequence(s) in FASTA format for prediction. The user receives the prediction reports as output and the estimated probability scores. Probability score (ranging from 0 to 1), which reflect the confidence of decisions, is assigned to each predicted protein. PrESOgenesis marks inputted sequences with probability score >0.5 as fertility-related protein (first layer) or one of the fertility-related classes (second layer). However, the threshold can be adjusted by users to adjust the false positive results (higher score can lead to lower false positive).
Protein or mRNA transcript sequences can be used as input sequences to PrESOgenesis. Accordingly, the software was equipped with TransDecoder tool (version 3.0.1, http://transdecoder.sourceforge.net), which obtained the candidate protein region based on the open reading frame (ORF) and nucleotide composition. Then, the predicted protein sequences were automatically inputted to the first layer of classifier for predicting their potential as fertility-related proteins. This capability can be applied to annotate the unknown transcripts that have been generated from deep sequencing projects such as RNA-Seq studies.
In this study, to address the issue of whether the new PrESOgenesis has a better or at least comparable performance to the previously introduced OOgenesis_Pred, a comparison of the two softwares was made. Towards a fair performance comparison, a negative sample including 1000 protein sequences were randomly selected from the negative dataset and were used as query. To avoid bias, none of the sequences in the negative sample appeared in the datasets used to train both of software. The results showed that PrESOgenesis achieved better performance in predicting the protein sequences as non-oogenesis related protein than OOgenesis_Pred. Using these two software, 140 (by PrESOgenesis) and 184 (by OOgenesis_Pred) proteins as non-oogenesis-related proteins were identified (Supplementary File 3). The higher performance of PrESOgenesis can be attributed solely with certainty the two-layer prediction architecture of this software, which is equivalent to making full use of the interclass relationships between fertility and non-fertility related proteins. Since PrESOgenesis is the first classifier ever developed for identifying fertility related proteins, it is not possible to compare its accuracy precisely against its counterparts for exactly the same purpose. However, its power can be compared with some related tools in other areas.
The trained models in this study could achieve an accuracy more than 81%8,11,12,13,14,15,16,96. The accuracy and robustness of the model could also be evaluated using new fertility-related proteins belonging to different classes which were added into Uniprot databases (release 2017_10) since after PrESOgenesis had been developed. Therefore, to test the prediction power of PrESOgenesis, proteins sequences were collected again by searching the UniProtKB database (release 2017_10) with gene ontology terms “oogenesis”, “spermatogenesis” and “embryogenesis” and new added protein sequences were retrieved from the datasets. 18, 39 and 144 new protein sequences were obtained in oogenesis, spermatogenesis and embryogenesis classes, respectively. Interestingly, PrESOgenesis could properly predict 15 of 18 (83.33%), 35 of 39 (89.74%) and 117 of 144 (81.25%) sequences. These results further proved the reliability of PrESOgenesis for identifying fertility-related proteins (Supplementary File 4).
Limitation and future work
Three classes of fertility-related proteins have been focused on in this study; though there are other relevant protein classes to fertility. Incorporating such proteins data and thus complementing the training datasets, may well improve the accuracy of predictors and help to reduce the false positive rates. Therefore, in future work it is necessary to attempt to add other fertility-related protein classes to the training datasets, which can be used in combination to further improve the reliability of the predictor.
Since both user-friendly and publicly accessible web-servers97,98,99 and databases100,101 represent the direction of developing new prediction method, efforts shall be made in future work to provide a web-server for the prediction method presented in this paper.
Conclusions
With the advent of post-genomic era and increasing use of computational techniques, the computational annotation of proteins has become a priority research area nowadays. In this study, the hypothesis was that fertility-related proteins possess some characteristics which distinguish their sequences from their non-fertility counterpart proteins. To this end, six sequence-based feature descriptors were integrated with a vector of 1,920 dimensions to facilitate the analysis and identification of fertility-related proteins and their classes. Here, for the first time, a two-layer classification framework was developed based on the SVM method, called PrESOgenesis. At the first layer, each protein was classified by SVM classifier to determine whether it is a fertility-related protein or not. If so, it was further classified by three SVM models to determine to which functional classes it belongs. Five-fold cross-validations along with independent test indicated that the proposed method is very powerful and promising. Also, an in-depth feature analysis was used to identify the most important features for identifying fertility-related proteins. A total of 22 important features were identified such as serine and isoleucine frequency and showed that they significantly contribute to the prediction. It is anticipated that PrESOgenesis will become a very useful bioinformatics tool for predicting fertility-related proteins.
References
Anifandis, G., Messini, C., Dafopoulos, K., Sotiriou, S. & Messinis, I. Molecular and cellular mechanisms of sperm-oocyte interactions opinions relative to in vitro fertilization (IVF). Int J Mol Sci 15, 12972–12997, https://doi.org/10.3390/ijms150712972 (2014).
Johnson, J. et al. Oocyte generation in adult mammalian ovaries by putative germ cells in bone marrow and peripheral blood. Cell 122, 303–315, https://doi.org/10.1016/j.cell.2005.06.031 (2005).
Johnson, J., Canning, J., Kaneko, T., Pru, J. K. & Tilly, J. L. Germline stem cells and follicular renewal in the postnatal mammalian ovary. Nature 428, 145–150, https://doi.org/10.1038/nature02316 (2004).
Johnson, L., Petty, C. S. & Neaves, W. B. Further Quantification of Human Spermatogenesis - Germ-Cell Loss during Postprophase of Meiosis and Its Relationship to Daily Sperm Production. Biology of Reproduction 29, 207–215, https://doi.org/10.1095/biolreprod29.1.207 (1983).
Larry, J., Hung, B. N., Charles, S. P. & William, B. N. Quantification of human spermatogenesis: germ cell degeneration during spermatocytogenesis and meiosis in testes from younger and older adult men. Biology of reproduction 37, 739–747 (1987).
Carlson, B. M. Chapter 4: Formation of germ layers and initial derivatives. Human Embryology & Developmental Biology, 62–68 (1999).
Moore, K. L. & Persaud, V. Chapter 3: Formation of the bilaminar embryonic disc: second week. The Developing Human, Clinically Oriented Embryology, 47–51 (2003).
Ng, X. Y., Rosdi, B. A. & Shahrudin, S. Prediction of antimicrobial peptides based on sequence alignment and support vector machine-pairwise algorithm utilizing LZ-complexity. Biomed Res Int 2015, 212715, https://doi.org/10.1155/2015/212715 (2015).
Rahman, A., Abdullah, R. & Wan-Khadijah, W. Gametogenesis, fertilization and early embryogenesis in mammals with special reference to goat: A review. J. Biol. Sci 8, 1115–1128, https://doi.org/10.3923/jbs.2008.1115.1128 (2008).
Grabherr, M. G. et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol 29, 644–652, https://doi.org/10.1038/nbt.1883 (2011).
Thakur, N., Qureshi, A. & Kumar, M. AVPpred: collection and prediction of highly effective antiviral peptides. Nucleic Acids Res 40, W199–204, https://doi.org/10.1093/nar/gks450 (2012).
Rahimi, M., Bakhtiarizadeh, M. R. & Mohammadi-Sangcheshmeh, A. OOgenesis_Pred: A sequence-based method for predicting oogenesis proteins by six different modes of Chou’s pseudo amino acid composition. J Theor Biol 414, 128–136, https://doi.org/10.1016/j.jtbi.2016.11.028 (2017).
Bakhtiarizadeh, M. R., Moradi-Shahrbabak, M., Ebrahimi, M. & Ebrahimie, E. Neural network and SVM classifiers accurately predict lipid binding proteins, irrespective of sequence homology. J Theor Biol 356, 213–222, https://doi.org/10.1016/j.jtbi.2014.04.040 (2014).
Lata, S., Sharma, B. K. & Raghava, G. P. Analysis and prediction of antibacterial peptides. BMC Bioinformatics 8, 263, https://doi.org/10.1186/1471-2105-8-263 (2007).
Chen, W., Feng, P. M., Lin, H. & Chou, K. C. iRSpot-PseDNC: identify recombination spots with pseudo dinucleotide composition. Nucleic Acids Res 41, e68, https://doi.org/10.1093/nar/gks1450 (2013).
Dehzangi, A. et al. Gram-positive and Gram-negative protein subcellular localization by incorporating evolutionary-based descriptors into Chous general PseAAC. J Theor Biol 364, 284–294, https://doi.org/10.1016/j.jtbi.2014.09.029 (2015).
Feng, P.-M., Lin, H. & Chen, W. Identification of antioxidants from sequence information using Naive Bayes. Computational and Mathematical Methods in Medicine 2013, https://doi.org/10.1155/2013/567529 (2013).
Feng, P.-M., Ding, H., Chen, W. & Lin, H. Naive Bayes classifier with feature selection to identify phage virion proteins. Computational and mathematical methods in medicine 2013, https://doi.org/10.1155/2013/530696 (2013).
Chou, K. C. & Shen, H. B. Recent progress in protein subcellular location prediction. Anal Biochem 370, 1–16, https://doi.org/10.1016/j.ab.2007.07.006 (2007).
Yuan, M., Yang, Z., Huang, G. & Ji, G. Feature selection by maximizing correlation information for integrated high-dimensional protein data. Pattern Recognition Letters 92, 17–24, https://doi.org/10.1016/j.patrec.2017.03.011 (2017).
Ding, C. H. Q. & Dubchak, I. Multi-class protein fold recognition using support vector machines and neural networks. Bioinformatics 17, 349–358, https://doi.org/10.1093/bioinformatics/17.4.349 (2001).
Cheng, J. & Baldi, P. A machine learning information retrieval approach to protein fold recognition. Bioinformatics 22, 1456–1463, https://doi.org/10.1093/bioinformatics/btl102 (2006).
Hoglund, A., Donnes, P., Blum, T., Adolph, H. W. & Kohlbacher, O. MultiLoc: prediction of protein subcellular localization using N-terminal targeting sequences, sequence motifs and amino acid composition. Bioinformatics 22, 1158–1165, https://doi.org/10.1093/bioinformatics/btl002 (2006).
Li, K. et al. Prediction and identification of the effectors of heterotrimeric G proteins in rice (Oryza sativa L.). Briefings in bioinformatics 18, 270–278, https://doi.org/10.1093/bib/bbw021 (2016).
Zuo, Y. C. et al. Discrimination of membrane transporter protein types using K-nearest neighbor method derived from the similarity distance of total diversity measure. Mol Biosyst 11, 950–957, https://doi.org/10.1039/c4mb00681j (2015).
Liu, B., Wang, X., Lin, L., Dong, Q. & Wang, X. A discriminative method for protein remote homology detection and fold recognition combining Top-n-grams and latent semantic analysis. BMC Bioinformatics 9, 510, https://doi.org/10.1186/1471-2105-9-510 (2008).
Lin, C. et al. LibD3C: Ensemble classifiers with a clustering and dynamic selection strategy. Neurocomputing 123, 424–435, https://doi.org/10.1016/j.neucom.2013.08.004 (2014).
Cinelli, M. et al. Feature selection using a one dimensional naïve Bayes’ classifier increases the accuracy of support vector machine classification of CDR3 repertoires. Bioinformatics 33, 951–955, https://doi.org/10.1093/bioinformatics/btw771 (2017).
Yu, B. et al. Prediction subcellular localization of Gram-negative bacterial proteins by support vector machine using wavelet denoising and Chou’s pseudo amino acid composition. Chemometrics and Intelligent Laboratory Systems 167, 102–112, https://doi.org/10.1016/j.chemolab.2017.05.009 (2017).
Manavalan, B. & Lee, J. SVMQA: support-vector-machine-based protein single-model quality assessment. Bioinformatics 33, 2496–2503, https://doi.org/10.1093/bioinformatics/btx222 (2017).
Tang, H., Chen, W. & Lin, H. Identification of immunoglobulins using Chou’s pseudo amino acid composition with feature selection technique. Molecular Biosystems 12, 1269–1275, https://doi.org/10.1039/c5mb00883b (2016).
Guo, H., Liu, B., Cai, D. & Lu, T. Predicting protein–protein interaction sites using modified support vector machine. International Journal of Machine Learning and Cybernetics, 1–6, https://doi.org/10.1007/s13042-015-0450-6 (2016).
Cheng, X., Xiao, X. & Chou, K. C. pLoc-mPlant: predict subcellular localization of multi-location plant proteins by incorporating the optimal GO information into general PseAAC. Molecular Biosystems 13, 1722–1727, https://doi.org/10.1039/c7mb00267j (2017).
Liu, B., Yang, F. & Chou, K. C. 2L-piRNA: A Two-Layer Ensemble Classifier for Identifying Piwi-Interacting RNAs and Their Function. Mol Ther-Nucl Acids 7, 267–277, https://doi.org/10.1016/j.omtn.2017.04.008 (2017).
Fu, L., Niu, B., Zhu, Z., Wu, S. & Li, W. CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics 28, 3150–3152, https://doi.org/10.1093/bioinformatics/bts565 (2012).
He, H. & Ma, Y. Imbalanced learning: foundations, algorithms, and applications. (John Wiley & Sons 2013).
Batista, G. E., Prati, R. C. & Monard, M. C. A study of the behavior of several methods for balancing machine learning training data. ACM Sigkdd Explorations Newsletter 6, 20–29, https://doi.org/10.1145/1007730.1007735 (2004).
Sun, Y., Wong, A. K. C. & Kamel, M. S. Classification of Imbalanced Data: A Review. International Journal of Pattern Recognition and Artificial Intelligence 23, 687–719, https://doi.org/10.1142/s0218001409007326 (2009).
Chou, K. C. Prediction of protein cellular attributes using pseudo‐amino acid composition. Proteins: Structure, Function, and Bioinformatics 43, 246–255, https://doi.org/10.1002/prot.1035 (2001).
Chen, X. X. et al. Identification of Bacterial Cell Wall Lyases via Pseudo Amino Acid Composition. Biomed Res Int 2016, 1654623, https://doi.org/10.1155/2016/1654623 (2016).
Yang, H. et al. Identification of Secretory Proteins in Mycobacterium tuberculosis Using Pseudo Amino Acid Composition. Biomed Res Int 2016, 5413903, https://doi.org/10.1155/2016/5413903 (2016).
Tang, H. et al. Identification of Secretory Proteins of Malaria Parasite by Feature Selection Technique. Letters in Organic Chemistry 14, 621–624 (2017).
Zhao, Y. W. et al. IonchanPred 2.0: A Tool to Predict Ion Channels and Their Types. Int J Mol Sci 18, https://doi.org/10.3390/ijms18091838 (2017).
Chen, W., Feng, P., Ding, H. & Lin, H. PAI: Predicting adenosine to inosine editing sites by using pseudo nucleotide compositions. Sci Rep 6, 35123, https://doi.org/10.1038/srep35123 (2016).
Cheng, X., Xiao, X. & Chou, K.-C. pLoc-mVirus: predict subcellular localization of multi-location virus proteins via incorporating the optimal GO information into general PseAAC. Gene 628, 315–321, https://doi.org/10.1016/j.gene.2017.07.036 (2017).
Meher, P. K., Sahu, T. K., Saini, V. & Rao, A. R. Predicting antimicrobial peptides with improved accuracy by incorporating the compositional, physico-chemical and structural features into Chou’s general PseAAC. Scientific Reports 7, 42362, https://doi.org/10.1038/srep42362 (2017).
Tang, H., Su, Z. D., Wei, H. H., Chen, W. & Lin, H. Prediction of cell-penetrating peptides with feature selection techniques. Biochem Biophys Res Commun 477, 150–154, https://doi.org/10.1016/j.bbrc.2016.06.035 (2016).
Lai, H. Y., Chen, X. X., Chen, W., Tang, H. & Lin, H. Sequence-based predictive modeling to identify cancerlectins. Oncotarget 8, 28169–28175, https://doi.org/10.18632/oncotarget.15963 (2017).
Xiao, N., Cao, D. S., Zhu, M. F. & Xu, Q. S. protr/ProtrWeb: R package and web server for generating various numerical representation schemes of protein sequences. Bioinformatics 31, 1857–1859, https://doi.org/10.1093/bioinformatics/btv042 (2015).
Zhou, C., Yu, H., Ding, Y. J., Guo, F. & Gong, X. J. Multi-scale encoding of amino acid sequences for predicting protein interactions using gradient boosting decision tree. Plos One 12, e0181426, https://doi.org/10.1371/journal.pone.0181426 (2017).
Vigil, A. Building explainable random forest models with applications in protein functional analysis, San Francisco State University (2016).
Qiao, S., Yan, B. & Li, J. Ensemble learning for protein multiplex subcellular localization prediction based on weighted KNN with different features. Applied Intelligence, 1–12, https://doi.org/10.1007/s10489-017-1029-6 (2017).
Weng, S. L. et al. Investigation and identification of protein carbonylation sites based on position-specific amino acid composition and physicochemical features. BMC Bioinformatics 18, 66, https://doi.org/10.1186/s12859-017-1472-8 (2017).
Cortes, C. & Vapnik, V. Support-Vector Networks. Machine Learning 20, 273–297, https://doi.org/10.1007/Bf00994018 (1995).
Lin, H., Chen, W. & Ding, H. AcalPred: a sequence-based tool for discriminating between acidic and alkaline enzymes. PLoS One 8, e75726, https://doi.org/10.1371/journal.pone.0075726 (2013).
Jiang, Y. et al. An expanded evaluation of protein function prediction methods shows an improvement in accuracy. Genome biology 17, 184 (2016).
Cao, R. & Cheng, J. Integrated protein function prediction by mining function associations, sequences, and protein–protein and gene–gene interaction networks. Methods 93, 84–91 (2016).
Zhu, P. P. et al. Predicting the subcellular localization of mycobacterial proteins by incorporating the optimal tripeptides into the general form of pseudo amino acid composition. Molecular Biosystems 11, 558–563, https://doi.org/10.1039/c4mb00645c (2015).
Cao, R. et al. ProLanGO: Protein Function Prediction Using Neural Machine Translation Based on a Recurrent Neural Network. Molecules 22, 1732 (2017).
Cao, R. et al. QAcon: single model quality assessment using protein structural and contact information with machine learning techniques. Bioinformatics 33, 586–588 (2017).
Wang, Z., Cao, R. & Cheng, J. In BMC bioinformatics. S3 (BioMed Central).
Chang, C. C. & Lin, C. J. LIBSVM: A Library for Support Vector Machines. Acm Transactions on Intelligent Systems and Technology 2, 27, https://doi.org/10.1145/1961189.1961199 (2011).
Amari, S. & Wu, S. Improving support vector machine classifiers by modifying kernel functions. Neural Netw 12, 783–789, https://doi.org/10.1016/S0893-6080(99)00032-5 (1999).
Chou, K. C. Using subsite coupling to predict signal peptides. Protein Engineering 14, 75–79, https://doi.org/10.1093/protein/14.2.75 (2001).
Chen, W., Xing, P. & Zou, Q. Detecting N 6-methyladenosine sites from RNA transcriptomes using ensemble Support Vector Machines. Scientific reports 7, 40242, https://doi.org/10.1038/srep40242 (2017).
Lin, H., Liang, Z.-Y., Tang, H. & Chen, W. Identifying sigma70 promoters with novel pseudo nucleotide composition. IEEE/ACM transactions on computational biology and bioinformatics, https://doi.org/10.1109/TCBB.2017.2666141 (2017).
Chen, W., Yang, H., Feng, P., Ding, H. & Lin, H. iDNA4mC: identifying DNA N4-methylcytosine sites based on nucleotide chemical properties. Bioinformatics 33, 3518–3523, https://doi.org/10.1093/bioinformatics/btx479 (2017).
Chen, W., Feng, P.-M., Lin, H. & Chou, K.-C. iSS-PseDNC: identifying splicing sites using pseudo dinucleotide composition. BioMed research international 2014, https://doi.org/10.1155/2014/623149 (2014).
Thonig, A. The effect of variation in developmental mode on the population dynamics of a spionid polychaete (Pygospio elegans) in a heterogeneous environment. Jyväskylä studies in biological and environmental science 335 (2018).
Tiwari, A. K. Prediction of G-protein coupled receptors and their subfamilies by incorporating various sequence features into Chou’s general PseAAC. Computer Methods and Programs in Biomedicine 134, 197–213, https://doi.org/10.1016/j.cmpb.2016.07.004 (2016).
Ibrahim, W. & Abadeh, M. S. Extracting features from protein sequences to improve deep extreme learning machine for protein fold recognition. J Theor Biol 421, 1–15, https://doi.org/10.1016/j.jtbi.2017.03.023 (2017).
Mino, M. & Sawada, H. Follicle cell trypsin-like protease HrOvochymase: Its cDNA cloning, localization, and involvement in the late stage of oogenesis in the ascidian Halocynthia roretzi. Mol Reprod Dev 83, 347–358, https://doi.org/10.1002/mrd.22627 (2016).
Tanigawa, M. et al. Participation of D-serine in the development and reproduction of the silkworm Bombyx mori. J Insect Physiol 87, 20–29, https://doi.org/10.1016/j.jinsphys.2016.01.006 (2016).
Brand, A. H. & Perrimon, N. Raf acts downstream of the EGF receptor to determine dorsoventral polarity during Drosophila oogenesis. Genes Dev 8, 629–639, https://doi.org/10.1101/gad.8.5.629 (1994).
ten Dijke, P., Miyazono, K. & Heldin, C. H. Signaling via hetero-oligomeric complexes of type I and type II serine/threonine kinase receptors. Curr Opin Cell Biol 8, 139–145, https://doi.org/10.1016/S0955-0674(96)80058-5 (1996).
Murugasu-Oei, B., Rodrigues, V., Yang, X. & Chia, W. Masquerade: a novel secreted serine protease-like molecule is required for somatic muscle attachment in the Drosophila embryo. Genes & Development 9, 139–154, https://doi.org/10.1101/gad.9.2.139 (1995).
Klemm, U., Muller-Esterl, W. & Engel, W. Acrosin, the peculiar sperm-specific serine protease. Hum Genet 87, 635–641, https://doi.org/10.1007/BF00201716 (1991).
Kohno, N. et al. Two novel testicular serine proteases, TESP1 and TESP2, are present in the mouse sperm acrosome. Biochem Biophys Res Commun 245, 658–665, https://doi.org/10.1006/bbrc.1998.8501 (1998).
Jha, K. N. et al. Evidence for the involvement of proline-directed serine/threonine phosphorylation in sperm capacitation. Mol Hum Reprod 12, 781–789, https://doi.org/10.1093/molehr/gal085 (2006).
Chasan, R. & Anderson, K. V. The role of easter, an apparent serine protease, in organizing the dorsal-ventral pattern of the Drosophila embryo. Cell 56, 391–400, https://doi.org/10.1016/0092-8674(89)90242-0 (1989).
Balhorn, R. The protamine family of sperm nuclear proteins. Genome Biol 8, 227, https://doi.org/10.1186/gb-2007-8-9-227 (2007).
Han, Y., Haines, C. J. & Feng, H. L. Role(s) of the serine/threonine protein phosphatase 1 on mammalian sperm motility. Arch Androl 53, 169–177, https://doi.org/10.1080/01485010701314032 (2007).
Urner, F. & Sakkas, D. Protein phosphorylation in mammalian spermatozoa. Reproduction 125, 17–26, https://doi.org/10.1530/rep.0.1250017 (2003).
Kawakami, Y. et al. Impaired neurogenesis in embryonic spinal cord of Phgdh knockout mice, a serine deficiency disorder model. Neurosci Res 63, 184–193, https://doi.org/10.1016/j.neures.2008.12.002 (2009).
Kumar, A., Kroetsch, T., Blondin, P. & Anzar, M. Fertility-associated metabolites in bull seminal plasma and blood serum: 1H nuclear magnetic resonance analysis. Mol Reprod Dev 82, 123–131, https://doi.org/10.1002/mrd.22450 (2015).
Zhang, J. et al. Novel mutations in ubiquitin‐specific protease 26 gene might cause spermatogenesis impairment and male infertility. Asian journal of andrology 9, 809–814, https://doi.org/10.1111/j.1745-7262.2007.00305.x (2007).
Fritsche, E. et al. Increased frequencies of cytochrome P4501A1 polymorphisms in infertile men. Andrologia 30, 125–128, https://doi.org/10.1111/j.1439-0272.1998.tb01387.x (1998).
Haqq, C. M. et al. Molecular basis of mammalian sexual determination: activation of Mullerian inhibiting substance gene expression by SRY. Science 266, 1494–1500, https://doi.org/10.1126/science.7985018 (1994).
Zhang, S., Zeng, X., Ren, M., Mao, X. & Qiao, S. Novel metabolic and physiological functions of branched chain amino acids: a review. Journal of animal science and biotechnology 8, 10, https://doi.org/10.1186/s40104-016-0139-z (2017).
Mogami, H. et al. Isocaloric high-protein diet as well as branched-chain amino acids supplemented diet partially alleviates adverse consequences of maternal undernutrition on fetal growth. Growth Hormone & IGF Research 19, 478–485, https://doi.org/10.1016/j.ghir.2009.03.002 (2009).
Ong, S. A., Lin, H. H., Chen, Y. Z., Li, Z. R. & Cao, Z. Efficacy of different protein descriptors in predicting protein functional families. BMC Bioinformatics 8, 300, https://doi.org/10.1186/1471-2105-8-300 (2007).
Wan, S., Mak, M. W. & Kung, S. Y. Mem-ADSVM: A two-layer multi-label predictor for identifying multi-functional types of membrane proteins. J Theor Biol 398, 32–42, https://doi.org/10.1016/j.jtbi.2016.03.013 (2016).
Herman-Izycka, J., Wlasnowolski, M. & Wilczynski, B. Taking promoters out of enhancers in sequence based predictions of tissue-specific mammalian enhancers. BMC Med Genomics 10, 34, https://doi.org/10.1186/s12920-017-0264-3 (2017).
Bedoya, Ó. Remote protein homology detection using physicochemical properties. Revista EIA 14, 111–125 (2017).
Hu, X., Ma, C. & Zhou, Y. A novel two-layer SVM model in miRNA Drosha processing site detection. BMC Syst Biol 7(Suppl 4), S4, https://doi.org/10.1186/1752-0509-7-S4-S4 (2013).
Ahmad, K., Waris, M. & Hayat, M. Prediction of Protein Submitochondrial Locations by Incorporating Dipeptide Composition into Chou’s General Pseudo Amino Acid Composition. Journal of Membrane Biology 249, 293–304, https://doi.org/10.1007/s00232-015-9868-8 (2016).
Feng, P. et al. iDNA6mA-PseKNC: Identifying DNA N6-methyladenosine sites by incorporating nucleotide physicochemical properties into PseKNC. Genomics, https://doi.org/10.1016/j.ygeno.2018.01.005 (2018).
Feng, P.-M., Chen, W., Lin, H. & Chou, K.-C. iHSP-PseRAAAC: Identifying the heat shock protein families using pseudo reduced amino acid alphabet composition. Analytical Biochemistry 442, 118–125, https://doi.org/10.1016/j.ab.2013.05.024 (2013).
Chen, W. et al. iNuc-PhysChem: a sequence-based predictor for identifying nucleosomes via physicochemical properties. PloS one 7, e47843, https://doi.org/10.1371/journal.pone.0047843 (2012).
Feng, P., Ding, H., Lin, H. & Chen, W. AOD: the antioxidant protein database. Scientific reports 7, 7449, https://doi.org/10.1038/s41598-017-08115-6 (2017).
Liang, Z.-Y. et al. Pro54DB: a database for experimentally verified sigma-54 promoters. Bioinformatics 33, 467–469, https://doi.org/10.1093/bioinformatics/btw630 (2017).
Acknowledgements
This work was supported by University of Tehran, Iran.
Author information
Authors and Affiliations
Contributions
M.R.B., M.R. and V.S. conceived the ideas and designed study. M.R.B. and M.R. analyzed the data and developed the PrESOgenesis model. M.R.B. developed the software. M.R.B., A.M.S. and S.A.S. participated in the interpretation the data and discussion of the results and writing of the article. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Bakhtiarizadeh, M.R., Rahimi, M., Mohammadi-Sangcheshmeh, A. et al. PrESOgenesis: A two-layer multi-label predictor for identifying fertility-related proteins using support vector machine and pseudo amino acid composition approach. Sci Rep 8, 9025 (2018). https://doi.org/10.1038/s41598-018-27338-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-27338-9
- Springer Nature Limited
This article is cited by
-
HormoNet: a deep learning approach for hormone-drug interaction prediction
BMC Bioinformatics (2024)
-
A computational model to identify fertility-related proteins using sequence information
Frontiers of Computer Science (2024)
-
AptaNet as a deep learning approach for aptamer–protein interaction prediction
Scientific Reports (2021)
-
NeuroPIpred: a tool to predict, design and scan insect neuropeptides
Scientific Reports (2019)