
Abstract
Limited data is available on phenotypic variations with the same genotype in hypertrophic cardiomyopathy (HCM). The present study aims to explore the relationship between genotype and phenotype characterized by cardiovascular magnetic resonance (CMR) in a large Chinese family. A proband diagnosed with HCM from a multigenerational family underwent next-generation sequencing based on a custom sureSelect panel, including 117 candidate pathogenic genes associated with cardiomyopathies. All genetic results were confirmed by the Sanger sequencing method. All confirmed mutation carriers underwent CMR exam and myocardial tissue characterization using T1 mapping and late gadolinium enhancement (LGE) on a 3T scanner (Siemens Trio, Gemany). After clinical and genetic screening of 36 (including the proband) members of a large Chinese family, nineteen family members are determined to carry the single p.T1377M (c.4130C>T) mutation in the MYH7 gene. Of these 19 mutation carriers, eight are diagnosed with HCM, one was considered as borderline affected and ten are not clinically or phenotypically affected. Different HCM phenotypes are present in the nine affected individuals in this family. In addition, we have found different tissue characteristics assessed by T1 mapping and LGE in these individuals. We describe a family that demonstrates the diverse HCM phenotypes associated with a single MYH7 mutation.
Similar content being viewed by others

Introduction
Hypertrophic cardiomyopathy (HCM) is an autosomal dominant genetic disease where nearly 60% of the disease is caused by mutations of myocardial sarcomere proteins. Of those, myosin heavy chain beta isoform (MYH7) mutations account for 35~50%1, especially the mutations located in the head and neck domains of the MYH7 gene2. More than 200 mutations were described in MHY73 and different mutations could lead to other cardiomyopathies, including restrictive cardiomyopathy (RCM), dilated cardiomyopathy (DCM), and left ventricular non-compaction (LVNC)3. In addition, MYH7 with different point mutations could lead to different clinical manifestations and disease progression. For instance, HCM patients with Arg453Cys mutation in the MYH7 gene might have a higher incidence of end stage heart failure and premature death4. Similarly, Gly716Arg, Arg719Trp, Arg719Gln, Arg723Gly, or Gly741Arg mutations have malignant clinical manifestations5,6,7,8. On the contrary, patients with MYH7 point mutation of Val606Met have a good prognosis9. Due to the autosomal dominant inheritance pattern of HCM, familial forms which result from a single genetic mutation with a Mendelian inheritance pattern are increasingly recognized10. There are a few case reports of families with single MYH7 mutations that have diverse clinical manifestations and disease progression. For instance, Arg787His11 or Gly425Arg12 mutation in MYH7 gene showed significant differences in clinical manifestations.
Recently, targeted gene panels based on next-generation sequencing (NGS) method has been validated in multiple studies13,14 and they are expected to provide increased depth of coverage with higher sensitivity and specificity by using a limited number of genes to detect genetic changes in patients with cardiomyopathies15,16.
Cardiovascular magnetic resonance (CMR) is considered the reference noninvasive standard for the evaluation of left ventricular (LV) volumes, ejection fraction (EF), mass, and tissue characterization using T1 mapping and late gadolinium enhancement (LGE). CMR has been increasingly utilized as the standard imaging modality for HCM patients. However, scarce data exists on the genotype–phenotype association in patients with HCM as assessed by CMR. In this report, we describe a large HCM family with a single MYH7 mutation and diverse HCM phenotypes as characterized by CMR.
Methods
Clinical evaluation
We conducted clinical evaluations which consisted of family history, previous medical history, physical examination, 12-lead electrocardiography (ECG), and echocardiography. Contrast enhanced chest computed tomography (CT) and digital subtraction angiography (DSA) were performed when necessary for clinical diagnosis. Diagnosis of HCM was made according to the AHA/ACC guideline17, and all methods were performed in accordance with the approved guidelines. This study was approved by the ethics committee of West China Hospital, and written informed consents were obtained from all participants.
Genotyping
The screening genetic panel included 117 candidate genes reported to be causative of cardiomyopathy, according to Online Mendelian Inheritance in Man (http://omim.org) and PubMed literature review. The gene list is shown in Supplemental Table S1. After obtaining informed consent, genomic DNA was extracted and captured with TruSeq DNA sample preparation kit and sequenced on a Hiseq platform. Genetic results were confirmed by the Sanger sequencing method subsequently. Pathogenicity determination of gene mutation was performed following guideline recommendations18.
Image acquisition and analyses
We obtained echocardiographic parameters according to the American College of Cardiology/American Heart Association guidelines19, including measurements of cardiac chamber dimensions, ventricular wall thickness, and measurements of LVEF.
ECG gated CMR imaging was performed on a 3.0-T Magnetom scanner (Trio Tim; Siemens Healthineers, Erlangen, Germany) with a with a 32-channel dedicated cardiac phase-array receiver coil. SSFP cine images of the LV from the base to apex in consecutive short-axis views and long-axis views (two-, three-, and four-chamber) were acquired during breath-holds with the following parameters: temporal resolution: 42 ms; repetition time (TR), 3.4ms; echo time (TE), 1.3ms; flip angle (FA), 50 degrees; field of view, 320–340 mm; matrix size, 256 × 144; Reconstructed in plane spatial resolution was 1.4 mm*1.3 mm and slice thickness of 8 mm with no gap. Native T1 measurements were acquired at the basal, mid, and apical short axis before injection of gadolinium using motion corrected modified Look-Locker inversion recovery sequence (MOLLI) (Siemens Healthcare works-in-progress 448; protocol: 5(3)3). Parameters for MOLLI was as follows: TR 740ms, TE 1.06ms, FA 35°, bandwidth 930 Hz/pixel, initial T1 100ms with 80ms increments, parallel imaging factor 2, matrix 192*144, in plane spatial resolution 2.4*1.8 mm, total acquisition time 11 heart beats. T1 measurements were repeated in the identical short-axis slices at about 15 minutes after administration of gadolinium by (MOLLI protocol: 4(1)3(1)2). Hematocrit was tested within 24 hours of CMR scanning for extracellular volume (ECV) calculation.
LGE images were acquired at 10–15 minutes after intravenous administration of 0.15 mmol/kg of gadopentetate dimeglumine (Magnevist, Bayer Schering Pharma, Berlin, Germany) using the inversion recovery method with phase-sensitive reconstruction (PSIR) on identical short and long axis views: TR 700 ms; TE 1.56 ms; FA 20°; matrix 256 × 144; inversion time (TI) was individually optimized to null normal myocardial signal using a TI scout sequence.
LV volumes, EF, and mass were analyzed using commercially available software (Qmass 7.6; Medis Medical Imaging Systems, Leiden, the Netherlands) by two experienced observers. LV endocardial and epicardial borders on cine images were manually drawn to define the myocardium, taking care to exclude papillary muscles and the trabeculations from the blood pool. Maximal LV wall thickness was defined as the greatest dimension anywhere within the LV myocardium. LGE is defined quantitatively by a myocardial signal intensity of 6 standard deviations from the normal myocardium and the amount of LGE is semi-automatically quantified using the QMASS 7.6 software. Total LGE was expressed as a proportion of LV myocardium (%LGE).
To quantify T1 values, the endocardial and epicardial contours were traced manually with care not to include trabeculations, blood pool or epicardial fat on the pre-contrast and post-contrast T1-mapping images at the mid LV slice, including regions that were positive for LGE20,21. After fitting the curve using the QMass7.6 software, mean myocardial T1 values was obtained. Blood T1 was obtained by locating a region of interest in the blood pool within the LV cavity in the corresponding pre-contrast and post-contrast T1-mapping images. The ECV was calculated as follow: ECV = (1−HCT) * ([1/T1myo post-1/T1myo pre]/[1/T1blood post-1/T1blood pre]).
Results
Clinical characteristics and phenotypes
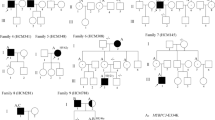
The pedigree of the proband and 35 family members is shown in Fig. 1. The proband (III1) was a 51 years old man who experienced palpitation for 9 years, and had chest pain and mild dyspnea on exertion for two years before admission to our hospital. On admission, the patient presented with a slightly elevated cardiac troponin T level of 21.8 ng/L (normal range 0 to 14 ng/L) and a significantly elevated N-terminal pro-brain natriuretic peptide level of 537 pg./mL (normal range 0 to 125 pg/mL). ECG showed supraventricular tachycardia and non-specific ST-T changes. Coronary angiography showed no significant coronary artery disease. Echocardiography showed significant left atrial enlargement, preserved biventricular systolic function, and severe LV outflow tract obstruction (peak aortic outflow velocity = 4.8 m/s, peak gradient = 92 mmHg). The abnormal morphology and function were confirmed by CMR, which showed a sigmoid septum with LV outflow tract obstruction (Fig. 2III1-B,C) and LV mass index to body surface area (BSA) of 87.2 g/m2 (papillary muscles excluded). The maximal thickness was 23.7 mm on CMR. In addition, CMR also showed LGE in the interventricular septum (Fig. 2III1-D). The proband was successfully managed medically with ß-adrenergic receptor blocker and diuretics.

Pedigree of the family. Squares represent male relatives; circles represent female relatives; filled symbols indicate HCM patients; slants indicate dead members; arrow represent proband; symbol with dots represent mutation carrier with negative phenotype; ? represent that the subject died before our investigation so that we cannot get clinical data and confirm their phenotype; SCD: Sudden cardiac death: G+: positive genotype; P+: positive phenotype; P−: negative phenotype. IV5 was considered as borderline affected with a maximal LV thickness of 13.2 mm.

Cardiac morphologies evaluated by CMR; II1 had sigmoid septum (black arrow of II1-C points to basal septal hypertrophy) without LVOT obstruction; II5 had the neutral septum (II5-C: white arrow)with RV involvement (II5-C: black arrow); II7 had neutral septum(II7-B: black arrow) with no RV involvement; III1 had sigmoid septum with LVOT obstruction (III1-B:black arrow); III2 had sigmoid septum without LVOT obstruction; III6 had sigmoid septum without LVOT obstruction; III7 had reversed curvature of the septum; III9 had reversed curvature of the septum; IV5 had symmetric LV hypertrophy; Black arrows of II1-D, II5-D, II7-D, III1-D, III6-D, III7-D, and III9-D point to LGE in the myocardium.
The family included a total of 42 members and five were deceased. One member was not able to be genotyped. Eighteen of the 35 family members (not including the proband) were found to carry the same MYH7 gene mutation and underwent CMR scan. Of those 18 family mutation carriers, 7 were diagnosed with HCM, 1 (IV5) was considered as borderline affected with a maximal LV thickness of 13.2 mm, and 10 were found to not be affected. Details of the demographic, clinical characteristics, and CMR parameters including tissue characteristics of the 8 (including the proband) HCM family members and 1 borderline affected member are shown in Table 1, whereas 10 phenotypically negative mutation carriers are shown in Table 2. In addition, three deceased members (II3, III4, III8) experienced cardiac sudden death by history. Subjects II3 and III8 were asymptomatic without medical history prior to their deaths. Subject II3 died suddenly at age 79 while doing morning exercise, whereas subject III8 died suddenly at age 13 while playing basketball. Subject III4 experienced severe dyspnea on exertion for 1 year and was treated with septal myomectomy at an outside hospital. She succumbed to sudden death 18 months after surgery at age 37. Other family members’ clinical examination, ECG, echocardiography and genetic analyses were normal and were considered unaffected.
Genotype-phenotype investigations
Genotyping confirmed that the family had a mutation in the MYH7 gene. The proband (III1, Fig. 3) and the other 18 family members (Fig. 3) was identified to carry the p.Thr1377Met (c.4130C > T) mutation in MYH7 gene confirmed by the Sanger method. Of the 18 mutation carrier family members, ten proved to be clinically not affected because they experienced no symptoms, and had normal echocardiograms and normal CMR findings (Supplemental Figs 1 and 2). Eight individuals (including the proband) diagnosed with HCM and one borderline affected subject had different HCM phenotypes, including the sigmoid septum with and without LVOT obstruction, neutral septum with right ventricular (RV) involvement, neutral septum without RV involvement, reversed curvature, and symmetrical hypertrophy phenotypes (Fig. 2).

(A) Mutation in MYH7 gene for the proband III1, his other 7 family members with positive phenotype and one borderline affected subject (p.T1377M, c.4130C > T). (B) Mutation in MYH7 gene for other 10 family members of the proband with negative phenotype (p.T1377M, c.4130C > 44T).
T1 mapping and LGE assessment by CMR
Native T1 ranged from 1167 ms to 1287.0 ms and ECV ranged from 24.7% to 30.8% in the phenotypically affected individuals (Table 1). Different degrees of fibrosis in different location of the myocardium were identified by LGE in the phenotypically positive subjects. Native T1 (ranging from 1116.0 to 1262.1 ms) and ECV (ranging from 25.3% to 28.6%) were also found in the 10 phenotypically negative mutation carriers (Table 2).
Discussion
In this study, we report the diverse phenotypes with a single point mutation in MYH7 in a large HCM family. More than half of the mutation carriers were phenotypically negative for HCM. The phenotypically positive HCM subjects exhibited a variety of HCM phenotypes.
The clinical features of the family is interesting for several reasons. First, a single p.Thr1377Met mutation in the MYH7 gene is shown to induce different phenotypes of HCM or not to induce any abnormal phenotype at all. Although the single point mutation had been previously reported in HCM2, we are the first to report the variable expression in a large family. Second, HCM patients carrying the same point mutation may have different clinical manifestations. The subject II3 had a near normal life span before sudden death but III8 experienced sudden death at a very young age. III4 experienced sudden death18 months after surgical septal myectomy and II5 was recommended to have an ICD implantation. Third, previous works showed that areas of focal fibrosis identified by LGE was associated with prognosis in HCM22, diffuse fibrosis detected by ECV had significant association to fibrosis measured histologically23,24,25, and different morphological subtype of hypertrophic cardiomyopathy could have diverse prognosis26,27,28. In the present study, the 8 HCM phenotypically positive and 1 borderline affected individuals had variable morphological subtypes, focal fibrosis by LGE, and diffuse fibrosis by ECV, which provided strong evidence for variable phenotypes associated with the same genotype. Fourth, the mutation carriers also had a range of native T1 and ECV values in the absence of LGE, which indicated phenotypically negative mutation carriers may have abnormal tissue characteristics as compared to healthy controls29. However, the implication of this for the genotype positive phenotype negative carriers is unknown and future research is needed.
Little data exists describing the genotype and CMR-phenotype relationship in HCM patients. Most of the research focused on the comparison of different phenotypes and prognosis caused by different genetic mutations in HCM3,30,31, or the difference between the HCM patients and genetic carriers with negative phenotype32,33,34. There were reports that described a single site mutation in the MYH7 gene producing not only a classic hypertrophic pattern but also a RCM phenotype35,36,37,38. Despite early hopes that genotyping may predict cardiac morphology and long-term outcomes in HCM, dedicated studies have failed to support this hypothesis39. In addition, there were large families with the same point mutation, which was reported to have 60% penetrance with different degree of LV hypertrophy and clinical presentation from asymptomatic to sudden death40. Genotype may not be an effective way to prognosticate or determine the degree of severity of disease expression of certain genes. Our findings provide imaging evidence that some genotypes may not be useful for the prediction of cardiac phenotypes in HCM.
It is unclear why the same mutation could produce diverse phenotypes. As for the single MYH7 mutation induced phenotypic diversity in different HCM individuals11, additional genetic modifiers could provide an explanation for different phenotypes. It suggested that there might be other factors regulating the expression of pathogenic mutations in the course of disease41. Of HCM modifying genes, such as angiotensin-converting enzyme (ACE)-1, endothelin-1, and tumor necrosis factor-α, their function had not been systematically studied. However, single nucleotide polymorphism (SNPs) and the relationship between the disease progression of HCM and the severity of phenotype has been studied. For example, ACE-1 had at least 13 SNPs. Relative studies had confirmed that the ACE-1 or ACE-2 mutation was associated with the severity of LV hypertrophy in HCM42,43. In addition, other genes such as myosin binding protein H have been investigated as modifiers of the hypertrophic variability in HCM44. Furthermore, complex genetic or a complex hierarchy of genetic, epigenetic, and environmental factors (such as demographic factors including age, sex, and body size, hypertension or obesity) might be involved in determining the phenotype40. In addition to pathogenic genes, background genes and methylation modification might play a very important role on HCM phenotype45,46,47.
Genetic testing in patients with HCM has been used to exclude differential diagnoses including Fabry disease and screen family to identify high risk members. Genetic testing has not been used in the treatment of HCM. Our study adds further evidence that our current understanding of genotype may be insufficient to predict phenotype or outcomes. We need to further explore the mechanisms leading to the diverse phenotypes and identifying high-risk markers for the development of hypertrophy. Because the occurrence of HCM may involve multiple independent signaling pathways, it is important to clarify the specific signaling pathways to effectively treat each subtype of HCM.
The relationship between the genotype, phenotype, and prognosis need further follow-up studies. In addition, the mechanism involved in the different phenotypes still need be explored.
In summary, we report a large Chinese families carrying a single MYH7 (p.Thr1377Met, c.4130C > T) genetic mutation which is associated with both the absence of HCM phenotype and diverse phenotypes of HCM. Additional studies are needed to clarify the mechanisms.
References
Maron, B. J. et al. Prevalence of Hypertrophic Cardiomyopathy in a General Population of Young Adults. Circulation 92, 785 (1995).
Richard, Hypertrophic cardiomyopathy. Distribution of disease genes, spectrum of mutations, and implications for a molecular diagnosis strategy (vol 107, pg 2227, 2003). Circulation 109, 3258 (2004).
Lopes, L. R., Rahman, M. S. & Elliott, P. M. A systematic review and meta-analysis of genotype–phenotype associations in patients with hypertrophic cardiomyopathy caused by sarcomeric protein mutations. Heart 99, 1800 (2013).
Ko, Y. L. et al. Malignant familial hypertrophic cardiomyopathy in a family with a 453Arg → Cys mutation in the β-myosin heavy chain gene: Coexistence of sudden death and end-stage heart failure. Hum Genet 97, 585 (1996).
Andersen, P. S. et al. Identification of a novel beta-cardiac myosin heavy chain gene mutation that causes familial hypertrophic cardiomyopathy. AM J Pathol 151, G21 (1997).
Enjuto, M. et al. Malignant hypertrophic cardiomyopathy caused by the Arg723Gly mutation in beta-myosin heavy chain gene. Journal of Molecular & Cellular Cardiology 32, 2307 (2000).
Dohlemann, C., Hebe, J., Meitinger, T. & Vosberg, H. P. Apical hypertrophic cardiomyopathy due to a de novo mutation Arg719Trp of the beta-myosin heavy chain gene and cardiac arrest in childhood. A case report and family study. Z Kardiol 89, 612 (2000).
Miller, G., Colegrave, M. & Peckham, M. N232S, G741R and D778G beta-cardiac myosin mutants, implicated in familial hypertrophic cardiomyopathy, do not disrupt myofibrillar organisation in cultured myotubes. Febs Lett 486, 325 (2000).
Blair, E., Redwood, C. & Watkins, H. Ascertainment strategies and genotype:phenotype correlations in hypertrophic cardiomyopathy. Circulation 108, 24 (2003).
Callis, T. E., Jensen, B. C., Weck, K. E. & Willis, M. S. Evolving molecular diagnostics for familial cardiomyopathies: at the heart of it all. Expert Rev Mol Diagn 10, 329 (2010).
Purushotham, G. et al. The MYH7p.R787H mutation causes hypertrophic cardiomyopathy in two unrelated families. Exp Clin Cardiol 15, 1 (2010).
Wang, H. et al. [The genotype-phenotype correlation of MYH7 gene G15391A mutation and MYBPC3 gene G12101A mutation in familial hypertrophic cardiomyopathy]. Zhonghua Xin Xue Guan Bing Za Zhi 36, 1059 (2008).
Jacoby, D. & McKenna, W. J. Genetics of inherited cardiomyopathy. Eur Heart J 33, 296 (2012).
Meder, B. et al. Targeted next-generation sequencing for the molecular genetic diagnostics of cardiomyopathies. Circ Cardiovasc Genet 4, 110 (2011).
D Argenio, V. et al. DNA Sequence Capture and Next-Generation Sequencing for the Molecular Diagnosis of Genetic Cardiomyopathies. J Mol Diagn 16, 32 (2014).
Millat, G., Chanavat, V. & Rousson, R. Evaluation of a new NGS method based on a custom AmpliSeq library and Ion Torrent PGM sequencing for the fast detection of genetic variations in cardiomyopathies. Clin Chim Acta 433, 266 (2014).
Gersh, B. J. et al. 2011 ACCF/AHA guideline for the diagnosis and treatment of hypertrophic cardiomyopathy: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. J Thorao Cardiovasc Surg 142, e153 (2011).
Wallis, Y. et al. Practice Guidelines for the Evaluation of Pathogenicity and the Reporting of Sequence Variants in Clinical Molecular Genetics. Association for Clinical Genetic Science (2013).
Cheitlin, M. D. et al. ACC/AHA/ASE 2003 Guideline Update for the Clinical Application of Echocardiography: Summary Article. J AM Soc Echocardiog 16, 1091 (2003).
Messroghli, D. R. et al. Clinical recommendations for cardiovascular magnetic resonance mapping of T1, T2, T2* and extracellular volume: A consensus statement by the Society for Cardiovascular Magnetic Resonance (SCMR) endorsed by the European Association for Cardiovascular Imaging (EACVI). J Cardiovasc Magn R 19, 75 (2017).
Hinojar, R. et al. T1 Mapping in Discrimination of Hypertrophic Phenotypes: Hypertensive Heart Disease and Hypertrophic Cardiomyopathy: Findings From the International T1 Multicenter Cardiovascular Magnetic Resonance Study. Circulation Cardiovascular Imaging 8, e3285 (2015).
Zhen, W. et al. Prognostic Value of LGE-CMR in HCM: A Meta-Analysis. Jacc Cardiovascular Imaging 9, 1392 (2016).
Flett, A. S. et al. Equilibrium contrast cardiovascular magnetic resonance for the measurement of diffuse myocardial fibrosis: preliminary validation in humans. Circulation 122, 138 (2010).
White, S. K. et al. T1 mapping for myocardial extracellular volume measurement by CMR: bolus only versus primed infusion technique. Jacc Cardiovascular Imaging 6, 955 (2013).
Iles, L. M. et al. Histological validation of cardiac magnetic resonance analysis of regional and diffuse interstitial myocardial fibrosis. European Heart Journal - Cardiovascular Imaging 16, 14 (2015).
Rowin, E. J. et al. Hypertrophic Cardiomyopathy With Left Ventricular Apical Aneurysm: Implications for Risk Stratification and Management. J AM Coll Cardiol 69, 761 (2017).
An, S. et al. Comparison of Long-Term Outcome between Apical and Asymmetric Septal Hypertrophic Cardiomyopathy. Cardiology 136, 108 (2017).
Guo, X. et al. The clinical features, outcomes and genetic characteristics of hypertrophic cardiomyopathy patients with severe right ventricular hypertrophy. Plos One 12, e174118 (2017).
Dabir, D. et al. Reference values for healthy human myocardium using a T1 mapping methodology: results from the International T1 Multicenter cardiovascular magnetic resonance study. J Cardiovasc Magn R 16, 69 (2014).
Weisslersnir, A. et al. Lack of Phenotypic Differences by Cardiovascular Magnetic Resonance Imaging in MYH7 (β-Myosin Heavy Chain)- Versus MYBPC3 (Myosin-Binding Protein C)-Related Hypertrophic Cardiomyopathy. Circulation Cardiovascular Imaging 10 (2017).
Choi, J. et al. Long-Term Outcome of 4 Korean Families with Hypertrophic Cardiomyopathy Caused by 4 Different Mutations. Clin Cazrdiol 33, 430 (2010).
Germans, T. et al. Structural Abnormalities of the Inferoseptal Left Ventricular Wall Detected by Cardiac Magnetic Resonance Imaging in Carriers of Hypertrophic Cardiomyopathy Mutations. J AM Coll Cardiol 48, 2518 (2006).
McTaggart, D. R., Ogden, K. J. & Marathe, J. A. A Long Term Follow-up Study of Carriers of Hypertrophic Cardiomyopathy Mutations. Heart, Lung and Circulation 26, 18.
Fujita, T. et al. Increased extent of myocardial fibrosis in genotyped hypertrophic cardiomyopathy with ventricular tachyarrhythmias. J Cardiol 66, 63 (2015).
Wu, W. et al. Novel Phenotype-Genotype Correlations of Restrictive Cardiomyopathy with Myosin-Binding Protein C (MYBPC3) Gene Mutations Tested by Next-Generation Sequencing. J AM Heart Assoc 4 (2015).
Kubo, T. et al. Prevalence, Clinical Significance, and Genetic Basis of Hypertrophic Cardiomyopathy With Restrictive Phenotype. J Am coll cardiol 49, 2419 (2007).
Sen-Chowdhry, S., Syrris, P. & Mckenna, W. J. Genetics of restrictive cardiomyopathy. Heart Fail Clin 6, 179 (2010).
Hwang, J. et al. Diverse Phenotypic Expression of Cardiomyopathies in a Family with TNNI3 p.Arg145Trp Mutation. Korean Circulation Journal 47, 270 (2017).
Charron, P. et al. Genetic counselling and testing in cardiomyopathies: a position statement of the European Society of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur Heart J 31, 2715 (2010).
Page, S. P. et al. Cardiac myosin binding protein-C mutations in families with hypertrophic cardiomyopathy: disease expression in relation to age, gender, and long term outcome. Circ Cardiovasc Genet 5, 156 (2012).
Ashrafian, H., McKenna, W. J. & Watkins, H. Disease Pathways and Novel Therapeutic Targets in Hypertrophic Cardiomyopathy. Circ Res 109, 86 (2011).
Marian, A. J. Modifier genes for hypertrophic cardiomyopathy. Curr Opin Cardiol 17, 242 (2002).
van der Merwe, L. et al. Genetic variation in angiotensin-converting enzyme 2 gene is associated with extent of left ventricular hypertrophy in hypertrophic cardiomyopathy. Hum Genet 124, 57 (2008).
Mouton, J. M. et al. MYBPH acts as modifier of cardiac hypertrophy in hypertrophic cardiomyopathy (HCM) patients. Hum Genet 135, 477 (2016).
Clifford, C. P. & Nunez, D. Human β-myosin heavy chain mRNA prevalence is inversely related to the degree of methylation of regulatory elements. Cardiovasc Res 38, 736 (1998).
Wang, L., Seidman, J. G. & Seidman, C. E. Harnessing Molecular Genetics for the Diagnosis and Management of Hypertrophic Cardiomyopathy. Ann Intern Med 152, 513 (2010).
Daw, E. W. et al. Genome-wide mapping of modifier chromosomal loci for human hypertrophic cardiomyopathy. Hum Mol Genet 16, 2463 (2007).
Acknowledgements
Grant support sponsor: National Natural Science Foundation of China; contract grant numbers: 81571638.
Author information
Authors and Affiliations
Contributions
J.W. and K.W. participated in the study design, data analysis and interpretation, performed the statistical analysis, and drafted the manuscript. Y.C.C. contributed to study design, preparation, editing, and review of the manuscript. Y.H. contributed to editing and review of the manuscript. W.H.L. carried out data acquisition and analysis. H.L. analyzed the imaging data. J.Y.S. carried out subject scanning and performed data analysis and interpretation. All the authors read, revised and approved the final manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wang, J., Wan, K., Sun, J. et al. Phenotypic diversity identified by cardiac magnetic resonance in a large hypertrophic cardiomyopathy family with a single MYH7 mutation. Sci Rep 8, 973 (2018). https://doi.org/10.1038/s41598-018-19372-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-19372-4
- Springer Nature Limited
This article is cited by
-
MYH7 in cardiomyopathy and skeletal muscle myopathy
Molecular and Cellular Biochemistry (2024)
-
Cardiovascular Magnetic Resonance Imaging and Heart Failure
Current Cardiology Reports (2021)
-
Left-ventricular outflow tract acceleration time is associated with symptoms in patients with obstructive hypertrophic cardiomyopathy
Journal of Ultrasound (2021)
-
Congenital myopathies are mainly associated with a mild cardiac phenotype
Journal of Neurology (2019)