
Abstract
Breeding strawberry (Fragaria x ananassa) with enhanced fruit flavour is one of the top breeding goals of many strawberry-producing countries. Although several genes involved in the biosynthetic pathways of key aroma compounds have been identified, the development and application of molecular markers associated with fruit flavour remain limited. This study aims to identify molecular markers closely linked to genes controlling strawberry aroma. A purpose-built Subtracted Diversity Array (SDA) known as Fragaria Discovery Panel (FDP) was used for marker screening. Polymorphic sequences associated with key aroma compounds were identified from two DNA bulks with extreme phenotypes, established using 50 F1 progeny plants derived from Juliette X 07-102-41 cross, two strawberry genotypes differing in aroma profile. A total of 49 polymorphic markers for eight key aroma compounds were detected using genotypic data of the extreme DNA bulks and phenotypic data obtained from gas chromatography-mass spectrometry (GC-MS). A similarity search against the physical maps of Fragaria vesca revealed that FaP1D7 is linked to genes potentially involved in the synthesis of methyl butanoate. A C/T SNP was detected within the feature, which could possibly be converted to a molecular tool for rapid screening of the strawberry accessions for their methyl butanoate production capacity.
Similar content being viewed by others

Introduction
Some studies suggest that the flavour quality of many fruits including strawberry has deteriorated due to conventional breeding practices1,2. Consumers nowadays are also increasingly discriminating and demand highly flavourful fruits. Hence, the development of cultivars with improved flavour is becoming one of the main breeding priorities for many strawberry-producing countries. Strawberry aroma has been studied extensively in the past. Over 300 compounds corresponding to a complex mixture of esters, furanones, aldehydes, alcohols, terpenes, lactones and sulphur compounds have been identified3,4,5,6,7,8. Of these, only about 20 main volatile compounds have been determined to dominate the typical strawberry aroma based on sensory descriptive analysis and their odour activity values9. For instance, the key esters including methyl butanoate, methyl hexanoate, ethyl butanoate and ethyl hexanoate have been attributed to the fruity notes whereas the aldehydes such as (E)-hex-2-enal and hexanal account for the green and grassy notes in strawberry aroma. Furaneol and mesifuranne have been described as the two most important compounds contributing to the sweet and caramel-like flavour in ripe strawberry fruits9,10,11. Apart from chemical compositions, sweetness intensity and fruit firmness also have effects on sensory perception. A previous study demonstrated correlation between sucrose concentration and total volatiles, indicating the dependence of secondary metabolism to primary metabolism during fruit ripening developmental stage. The authors also presented some specific sugar-independent volatiles capable of enhancing the sweetness intensity of strawberry fruits12.
In recent years, strawberry flavour research has progressed from chemical and sensory analyses to the investigation of biosynthesis and genetic control of important aroma compounds13. Genes involved in the biosynthesis of esters and terpenes, the SAAT (strawberry-specific alcohol acyl transferase) and FaNES1 (Fragaria x ananassa nerolidol synthase 1) respectively, have been identified using cDNA microarray technology14,15. Chambers, et al.16 reported the presence/absence of a FaNES1 allele that could mostly predict the linalool-producing and non-producing cultivated and wild strawberry materials, with a few exceptions. Moreover, FaOMT (Fragaria x ananassa O-methyltransferase) and FaQR (Fragaria x ananassa quinone oxidoreductase) genes were also reported to be involved in the formation of mesifuranne and furaneol, respectively17,18,19. Zorrilla-Fontanesi, et al.20 showed that a 30-bp indel in the promoter region of the FaOMT allele is responsible for the high expression of mesifuranne content and may be turned into an important tool for strawberry breeding. Additionally, two recent studies have shown that a fatty acid desaturase FaFAD1 gene was correlated with the production of γ-decalactone in strawberry fruit21 and a PCR-based marker co-segregating with the phenotype was developed22.
Despite the development of PCR-based markers for linalool and γ-decalactone, breeding for improved strawberry flavour has been relatively slow due to the complex genetic control of fruit flavour biogenesis. Therefore, it is crucial to identify genomic regions corresponding to other key aroma compounds for the selection of elite plants towards marker-assisted breeding. We recently developed a Subtracted Diversity Array (SDA)23 called Fragaria Discovery Panel (FDP) as a platform for screening molecular markers associated with agronomically important traits24. We combined the application of FDP with Bulked Segregant Analysis (BSA)25, a rapid method for detecting DNA markers linked to any specific gene in the genome. Taking advantage of the discriminatory power of SDA for genotyping closely related species within the same genus26,27,28, we have successfully identified a putative marker, FaP2E11 possibly involved in controlling day-neutrality in octoploid strawberry24. Here, we extended the utility of the subtracted gDNA microarray-assisted BSA for the identification of polymorphic markers linked to the loci determining strawberry aroma.
Results and Discussion
Variability and distribution of aroma compounds in the parental genotypes and F1 segregating population
Aroma profiling of the fruits from the F1 population derived from a cross between Juliette and 07-102-41 allowed identification of 41 aroma compounds from the headspace of strawberry puree. Of these, 23 compounds (Table 1) were described as aroma-active compounds in strawberry based on gas chromatography quantification, their odour activity values (OAVs) and sensory analysis9,10. The remaining 18 aroma compounds, namely, ethyl propanoate, 4-methyl-2-pentanone, ethyl isobutanoate, methyl 2-methylbutanoate, ethyl tiglate, isobutyl butanoate, isopentyl 3-methylbutanoate, methyl octanoate, ethyl benzoate, (E)-hex-3-enyl butanoate, (Z)-hex-3-enyl butanoate, trans-2-hexenyl butanoate, ethyl octanoate, 3-methylbutyl hexanoate, hexyl hexanoate, (E)-cinnamyl acetate, and (Z)-ethyl cinnamate and benzaldehyde) exhibited a population mean value lower than 0.1% and were detected infrequently in the F1 population. Some of these compounds have been reported to have low broad-sense heritability values in strawberry29. Hence, they were excluded for DNA marker development as environmental factors may have a greater effect in controlling the inheritance of these compounds.
Table 1 shows the descriptive statistics of the 23 volatile compounds from the F1 population compared to the parental genotypes. Overall, 14 compounds were detected in either one or both of the parents whereas nine compounds were not identified in any of the parental genotypes but were detected in the F1 plants. Distinctive aroma patterns were observed between parents, with ‘07-102-41’ displaying higher levels of C4 and C6 esters and linalool while the levels of C7 and C8 esters, mesifuranne, (E)-nerolidol and γ-dodecalactone were higher in ‘Juliette’ (Table 1). This result is in agreement with the study which purported that strawberry aroma patterns (i.e., the combination and intensity of aroma compounds) were genotype-dependent30. Furthermore, most of the compounds detected in both parents showed a significant difference in the levels of production except for hexyl acetate and (E)-hex-2-enyl acetate. Nonetheless, diversity in the relative composition could be inferred from a range between minimum and maximum values of the F1 population (Table 1).
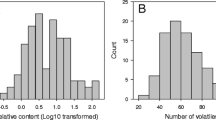
Out of the 23 aroma compounds, we focused our analysis on eight compounds which were considered to have the greatest impact on strawberry aroma due to their low threshold values and high OAVs9,10. The frequency distributions of these eight key aroma compounds, including four esters (methyl butanoate, ethyl butanoate, methyl hexanoate, ethyl hexanoate), one furanone (mesifuranne), two terpenes (linalool and (E)-nerolidol) and one lactone (γ-dodecalactone) are illustrated in Fig. 1. Most of the frequency distributions are highly or moderately skewed towards lower values or zero except for methyl hexanoate which approached a bimodal distribution. This result is in accordance with the frequency distributions for aroma compounds analysed in other fruit crops such as strawberry31; apple32 and peach33 except that none of the compounds found in this study have a frequency distribution skewed towards the higher values. This type of frequency distribution is typical of a trait with polygenic inheritance, indicating that aroma compound production is under the control of more than one gene32. In addition, transgressive segregation, the phenomenon where the individuals in a segregating population exhibit phenotypes that are extreme compared to the parental genotypes34,35,36, was observed for all the compounds assessed except for (E)-nerolidol (Fig. 1). This phenomena could be due to the formation of new combination of alleles at multiple loci underlying quantitative trait differences between the two parents37,38. The high levels of transgressive segregation may be due to the significant differences between parental means (Table 1), that is, the parental genotypes were phenotypically very different from each other39.

Frequency distribution of key aroma compounds measured as relative peak areas in the ‘07-102-41’ x ‘Juliette’ progeny. The mean values of the parents and F1 population are indicated by arrows (D: 07-102-41; J: Juliette; D x J, respectively). x-axis: relative composition (%), y-axis: the number of individual plants.
Overall, the eight key aroma compounds were selected for DNA marker discovery based on: (1) their importance in the characterisation of strawberry aroma9,10,30 and (2) their significant differences in parental means (Table 1). In theory, the probability of detecting the alleles controlling the trait of interest is higher if the parental means are significantly different from each other. Furthermore, the aroma compositions of two Australian-grown cultivars, Albion and Juliette, have been evaluated extensively over ten weeks in one growing season. The inheritance analysis revealed that methyl butanote, ethyl butanoate, mesifuranne and (E)-nerolidol possessed high broad sense heritability values (46.0–74.4%)29. This result coincides with the genotype-by-environment (G × E) analysis, where all selected compounds, except linalool, were found to be predominantly influenced by genotype. These findings also suggest that the yearly phenotypic changes of strawberry plants were not as great as seasonal variations40 and thus, the greater probability to identify molecular markers associated with the selected key aroma compounds. Although no F2 population was available for analysis, it is plausible that the F1 individuals with extreme phenotypes (i.e. much higher or not-detected) can be used as DNA materials in the subsequent Bulked Segregant Analysis (BSA) for the discovery of alleles segregating for the key aroma compounds.
Identification of polymorphic loci associated with key aroma compounds
Signal intensities generated from the hybridisation of the ‘H’ and ‘L’ bulks onto the FDP were used to identify polymorphic loci associated with key aroma compounds. The genotype data was subjected to Discriminant Function Analysis (DFA) using stepwise method to select the most discriminating markers from the original 287 features. It was further used to reduce the number of polymorphic markers and form classification models up to four markers (Table 2). Interestingly, FaP1E7 was detected in more than two key aroma compounds, i.e. methyl butanoate (referred to FaP1E7MB) and methyl hexanoate (FaP1E7MH). The classification of the original cases in the training set based on the discriminant functions showed that only ethyl butanote, mesifuranne and γ-dodecalactone were correctly classified (100%) into the two phenotypic extreme groups. For the other key aroma compounds, the proportion of correctly classified original cases ranged from 83.3% to 95.8% (Table 2). In addition, the accuracy of group membership prediction for the test set by cross-validation ranged from 79.2–100% for the key aroma compounds assessed (Table 2). The high percentage of correct classification implies that all the features selected by DFA are good predictors for the levels of key aroma compound biosynthesis in strawberry.
A similar approach was employed in a previous study which identified AFLP markers associated with stress tolerance index in Sardari wheat ecotypes, where stepwise analysis was firstly used to identify the number of polymorphic markers and then further applied to reduce the number of markers and form classification models of up to 24 markers41. Our results are in accordance with several previous studies, where the rate of misclassification increased as the number of predictors decreased41,42,43. Alwala42 showed that only 61.7–62.2% of correct classification was obtained based on five markers and a minimum of 10 markers were needed to achieve more than 90% of correct classification. The low cross-validation error rate produced in this study implies that the reduced sets of putative DNA markers selected by the DFA possessed high predictive power, hence, a stronger association between the marker and the phenotype43.
The DFA-selected features were validated using Fisher’s ratio to measure the linear discriminating power of the features. These features were arranged in decreasing order of Fisher’s ratio and only the top 10 features are presented in Table 3. Out of the 20 DFA-selected features, only six (FaP1D7, FaP1D11, FaP1B3, FaP3E12, FaP2D11, and FaP1A7) features showed high Fisher’s ratio values. The other DFA-selected putative markers displayed low Fisher’s ratio values and therefore, were excluded from further statistical analysis. In general, a larger Fisher’s ratio value indicates greater differences between the means of two extreme bulks. Hence, the features with very strong signal intensities in the ‘H’ DNA bulk but very weak signal intensities in the ‘L’ DNA bulk, or vice versa, will potentially generate larger Fisher’s ratio values provided that the background noise inherent in the microarray system is minimum (sum of the variances of the two extreme bulks). Hence, the putative DNA markers with higher Fisher’s ratio could possibly be the best in discriminating between the two phenotypic extreme bulks. The subsequent Independent Samples t-Test showed that the mean differences between the ‘H’ and ‘L’ DNA bulks for eleven putative DNA markers (FaP1D7, FaP1D11, FaP3A2, FaP3B9, FaP1E7MH, FaP1B3, FaP1G2, FaP2E6, FaP3E12, FaP2D11, and FaP1A7) selected by DFA were statistically significant (p < 0.01) (Table 4). This result is also corroborated by six of the features with high Fisher’s ratio (FaP1D7, FaP1D11, FaP1B3, FaP3E12, FaP2D11, and FaP1A7) as putative DNA markers which could be the predictor for their respective key aroma compounds.
The three-way Venn diagram revealed six features at the intersection of the three statistical analyses (Fig. 2). Among the six features, FaP1D7, FaP1D11, and FaP1B3 were found to be putatively associated with ester compounds, i.e. methyl butanoate, ethyl butanoate and ethyl hexanoate, respectively. Additionally, FaP3E12 and FaP2D11 were putatively correlated with linalool and (E)-nerolidol, respectively whereas FaP1A7 was putatively associated with γ-dodecalactone. In general, features exhibiting significant differences of group means are expected to yield a higher Fisher’s ratio value. However, the intersection between DFA and Independent Samples t-Test revealed five other features (FaP3A2, FaP3B9, FaP1E7MH, FaP1G2, and Fa2E6) that differed significantly in their group means were not included in the top 10 features with the highest Fisher’s ratios (Fig. 2). Their low Fisher’s ratio values may be explained by smaller differences between group means or the greater sum of the variances of the two extreme bulks. Some of these features (FaP3A2, FaP3B9, FaP1E7MH, and FaP2E6) displayed low signal intensities as their group means were relatively lower compared to the other features (Table 4). In contrast, the Fisher’s ratio value of FaP1G2 that showed high signal intensities was lowered by a large sum of the variances (data not shown). Moreover, the group means of nine other features (FaP1E7MB, FaP2A11, FaP2G4, FaP3H11, FaP1G8, FaP3F10, FaP2E1, FaP3E8 and FaP3F8) were statistically not significant (p > 0.01). This result further supports the data obtained from the Fisher’s ratio analysis, where all these features have low Fisher’s ratio values (Table 4).

Venn diagram analysis of Fragaria Discovery Panel features selected by three statistical analyses. A three-way Venn diagram showing the putative DNA markers in the intersection of DFA (green), Fisher’s ratio (top 10 features; blue) and Independent Samples t-Test (p < 0.01; red) for all key aroma compounds assessed.
Sequence identity and position of putative molecular markers
The identities of the six putative markers were revealed by DNA sequencing and similarity search against the Fragaria vesca draft genome (v1.1) using the PFR and GDR strawberry servers (GDR, 2009; PFR, 2010). Out of the six features sequenced, FaP1A7 and FaP1D7 appeared to be nuclear-specific whereas FaP1B3, FaP1D11, FaP2D11 and FaP3E12 were chloroplast-specific (Table 5). The nuclear-specific features did not a match any genes in the F. vesca genome database, indicating that they belong to the non-coding DNA region possibly associated to the genes involved in the biosynthesis of aroma compounds. The full sequence of FaP1A7 matched perfectly (100% E-value: 0.0) to a DNA region on linkage group 6 (LG6:21708323..21708764, scf0513196:589686..590127) whereas FaP1D7 was highly similar (E-value: 1e−38) to a genomic region on linkage group 2 (LG2:17544790..17544932, scf0513123:85522. 85664). For chloroplast-specific features, FaP1B3 showed significant similarity (E-value: 5e−45) to gene32946, a chloroplastic-like NAD(P)H-quinone oxidoreductase subunit H located on the scf0510865:52.396. The FaP1D11 was highly similar (E-value: e−133) to gene32967, a chloroplastic-like ATP synthase subunit alpha positioned on the scf0510833:190.1040. In contrast, 77% of FaP2D11 and the full sequence of FaP3E12 matched completely (E-value: 0.0) to scf0510759:1.513 and scf0513205:141.680, respectively. These two features did not correspond to any genes in the chloroplast genome (Table 5).
To identify genes possibly involved in the production of key volatile compounds, the genes situated within 5 cM on either side of the nuclear-specific features (FaP1A7 and FaP1D7) were manually searched on the same linkage group using the PFR Strawberry Server (PFR, 2010). Figure 3a shows a gene corresponding to Arabidopsis thaliana cytosolic acetoacetyl-CoA thiolase II, also known as acetyl-CoA acetyltransferase (ACAT2), was present at approximately 1.6 Mb downstream of FaP1A7. Moreover, an A. thaliana Patatin-related phospholipase A (PLA) gene and an F. x ananassa ethylene receptor (Ers1) gene were found 1.4 kb and 2.0 Mb downstream of FaP1D7, respectively (Fig. 3b).

Landmark of selected putative DNA markers (red arrows) mapped onto the F. vesca draft genome (v1.1) and location of genes associated with key volatile compounds (green arrows). (a) FaP1A7 on LG6:21708323..21708764. ACAT2: acetyl-CoA acetyltransferase (cytosolic). (b) FaP1D7 on LG2:17544790..17544932. PLA: Patatin-related phospholipase A; Ers1: Ethylene receptor.
The FaP1A7 was positively correlated with γ-dodecalactone production in strawberry fruits according to the differences in signal intensities between the ‘H’ (282.66) and ‘L’ (106.71) bulks (Table 4). While the precise lactone biosynthesis pathway in plants remains elusive, it is clear that the formation of γ- and δ-lactones start from β-oxidation of fatty acids44. Based on the fatty acid degradation pathway in the KEGG database, the acetyl-CoA acetyltransferase (ACAT2) gene found in close proximity to FaP1A7 is one of the thiolases that catalyses the reverse reaction in the last step of β-oxidation45,46.
In contrast, FaP1D7 is negatively correlated to methyl butanote based on the differences of hybridisation signal between the ‘H’ (248.23) and ‘L’ (578.61) bulks (Table 4). It has been reported that amino acids and lipids are the most likely precursors of ester formation47. Interestingly, the Patatin-related phospholipase A (PLA) strongly linked to FaP1D7 is one of the members of the phospholipase superfamily that hydrolyses the sn1 and/or sn2 position of the membrane phospholipid to release fatty acids48. The discovery of ethylene receptor (Ers1) at close proximity to FaP1D7 raises an interesting question regarding the role of ethylene in regulating ester formation in non-climacteric fruits, which is yet to be resolved. This result may suggest that a low amount of ethylene accumulation during strawberry fruit ripening is sufficient to regulate the ethylene receptor for the activation of phospholipase A enzyme, resulting in the release of free fatty acids as precursors for methyl ester formation49.
Whilst the role of the ACAT2, PLA and Ers1 genes in controlling the levels of γ-dodecalactone and methyl butanoate in strawberry was not determined in this study, the differences in signal intensities between the ‘H’ and ‘L’ bulks may suggest the presence of allelic variants in the FaP1A7 and FaP1D7 loci. To test this hypothesis, direct amplicon sequencing was performed on the PCR product amplified from the parental genotypes, 07-102-41 and Juliette, to determine the allelic variation of these putative DNA markers.
Detection of Single Nucleotide Polymorphism (SNP)
DNA sequence analysis obtained from FaP1D7 amplification revealed a putative C/T SNP between 07-102-41 and Juliette. By comparing to the original DNA sequence cloned into pGEM®-T Easy vector, the DNA fragment printed on the SDA was possibly derived from Juliette (See Supplementary Fig. S1). This result is in accordance with the SDA data, where the SNR of Juliette (i.e., 513.10) is nearly 1.4 x higher than that of 07-102-41 (i.e., 386.38) and the SNR of the ‘L’ bulk (i.e., 467.90) is almost 1.7 x higher than in the ‘H’ bulk (i.e., 268.67). This is because the DNA sequences with a ‘T’ at the SNP site would bind loosely to the SDA compared to DNA sequences carrying a ‘C’, generating an altered hybridisation pattern50. Based on the aroma profiles of 07-102-41 and Juliette (Table 1), higher levels of methyl butanoate were detected in 07-102-41 (15.4%) compared to Juliette (0.0%). This result suggests that the FaP1D7 marker allele harbouring either a ‘T’ or ‘C’ nucleotide at the SNP site may be associated with high or low levels of methyl butanoate production, respectively. In contrast, difficulty in interpreting the DNA sequence derived from FaP1A7 was encountered due to the overlapping of multiple peaks (chromatogram not shown), indicating the presence of secondary products even after gel purification. Hence, only FaP1D7 was selected for marker validation.
Validation of the FaP1D7 putative marker
In the present study, we validated FaP1D7 putative marker for the levels of methyl butanoate accumulated in four F1 progeny plants exhibiting extreme phenotypes and a wider range of strawberry germplasm. DNA sequencing revealed a ‘T’ nucleotide within FaP1D7 for P38 and P99 exhibiting high levels of methyl butanoate as expected. However, a slight discrepancy was observed for the F1 progeny plants exhibiting low levels of methyl butanoate, where P63 showed a ‘T’ instead of ‘C’ nucleotide as in P1 (Fig. 4). DNA sequence analysis showed that all the commercial cultivars contained a ‘C’ nucleotide within FaP1D7, suggesting that these genotypes should have low levels of methyl butanoate (Fig. 4). These results are in accordance with our GC-MS analysis, where no methyl butanoate was detected in P1 and P63 whereas 24.1% and 25.3% of methyl butanoate was detected in P38 and P99, respectively. The relative composition of methyl butanoate in the commercial cultivars was relatively low at 3.9–5.6% compared to P38 and P99. The parental genotypes, 07-102-41 and Juliette are presented for comparison (Fig. 5). Our observation demonstrated expected marker patterns according to their phenotypic classification into high and low levels of methyl butanoate in all evaluated genotypes except P63. This inconsistency could possibly be due to P63 being misclassified into ‘L’ bulk due to environmental effects. Another possible explanation could be the differences in allele dosage. As the octoploid strawberries are highly heterozygous, therefore more copies of ‘T’ could be present in the genotypes producing high levels of methyl butanoate and vice versa. A similar study performed by Chambers, et al.16 also speculated that there is possibly an allele dosage effect for NES1 allelic variants responsible for the production of linalool in octoploid wild accessions. To address this ambiguity, we propose to detect this genetic variant in different strawberry genotypes using High Resolution Melting (HRM) analysis. This may help to filter out some of the octoploid background effect. Given the data, we also propose to include cultivars or accessions producing high levels of methyl butanoate in further analysis to evaluate and strengthen the prediction power of the C/T SNP.

Detection of the C/T SNP in a wider range of strawberry germplasm. P1 and P63 were selected from the ‘L’ extreme bulk whereas P38 and P99 derived from the ‘H’ extreme bulk.

Relative compositions (%) of methyl butanoate detected in different strawberry genotypes using gas chromatography coupled with mass spectrometry. Parental genotypes: 07-102-41 and Juliette; F1 progeny plants: P1, P38, P63 and P99; commercial cultivars: Albion, Melba, Palomar, San Andreas, Camino Real and Portola. Error bars represent the standard deviation of the mean.
Conclusions
We report the identification of a putative marker allele, FaP1D7 using an in-house developed subtracted gDNA microarray. FaP1D7 contains a C/T SNP potentially associated with the levels of methyl butanoate in strawberry fruits. While the sample sizes are quite small for validation, and in spite of the complexity of the flavour trait, there appears to be some association between phenotype and genotype. However, the results presented in this study emphasise the need of a validation in determining the effect of allele dosage of the C/T SNP. We proposed to analyse the target SNP in different strawberry germplasms with HRM analysis using real-time PCR. More strawberry genotypes and/or crosses are required to confirm the functionality of the C/T SNP potentially associated with the levels of methyl butanote in strawberry fruits.
Methods
Parental genotypes and segregating population
The idea behind the project is to incorporate unique strawberry aroma from selected breeding lines into one of the Australian cultivars. Juliette, a short day cultivar bred by the Victorian Department of Primary Industries (DPI), is selected as parental genotype because it produces fruits that are sweeter than other cultivars developed through the same breeding program. It is widely accepted by consumers51. It is a bright red strawberry which fruits in the early season (September) in Victoria, Australia. Three promising breeding lines, including 07-102-41, 07-095-35 and 04-069-91 were considered as parental genotypes. Of these, 07-102-41 which has a primarily European genetic background was selected for crossing because it produces unique and very flavourful fruits. It is a short day strawberry with dark red fruits, and genetically diverse from Juliette. The segregating population consisted of 200 F1 plants derived from a cross between ‘Juliette’ and ‘07-102-41’. A subset of 50 F1 individuals was randomly selected as experimental materials based on the healthiness of the plants and the availability of firm fruits per plant. Of these, 37 progeny plants had 07-102-41 as a maternal parent (07-102-41 × Juliette) and the remaining 13 progeny plants were collected from the reciprocal cross (Juliette × 07-102-41) where Juliette was used as the maternal parent. Fully ripe fruits and young leaves from both parental genotypes and F1 population were harvested over the summer of 2011/2012 for aroma profiling and DNA genotyping, respectively. Similarly, fruits and leaves of six commercial cultivars including Melba, Camino Real, Portola, Palomar, San Andreas and Albion were collected across 16 weeks (13/11/2013 – 9/4/2014) for marker validation.
Profiling of strawberry aroma
Extraction of aroma compounds
Aroma compounds were extracted from the fruit puree using Solid Phase Microextraction (SPME) method52. Five large berries per plant were thawed and homogenised with a portable blender. Approximately 1 g of puree was immediately dispensed into individual SPME vials with screw caps and stored at −80 °C. Prior to gas chromatography-mass spectrometry (GC-MS) analysis, the sample was thawed to room temperature for 20 min and pre-equilibrated at 60 °C in a heating block for 10 min. The aroma compounds were extracted using a 65 µm polydimethylxiloxane/divinylbenzene (PDMS/DVB)-coated fiber held in an SPME Holder 57330-U (Supelco, Bellafonte, PA, USA). This fiber was first conditioned at 250 °C for 30 min, and then exposed to the vial headspace for 30 min at 60 °C. After equilibrium, the fiber was removed from the sample and the analytes were thermally desorbed in a GC injector port at 250 °C for 3 min. Aroma profiling for each sample was performed in triplicate.
GC-MS
Aroma profiling for each sample was performed in triplicate using Agilent 6890 GC coupled with a 5973 MS detector (Agilent, CA, USA) through a heated transfer line at 280 °C. Compounds were separated using DB-5ms column with dimensions of 30 m × 0.25 mm I.D. × 0.25 µm film thickness. Helium was used as a carrier gas at a flow rate of 1.5 mL/min. 1.0 µL was injected using the splitless injection mode with a 2.5 min of solvent delay. The oven temperature was programmed initially at 40 °C for 1 min, then increased at a rate of 6 °C/min to 190 °C and kept constant at the same temperature for 26 min with a final isotherm at 190 °C for 4 min. The MS source temperature was 230 °C and the compounds were monitored over the mass range m/z 45–400.
GC-MS data analysis
A similarity search was carried out by comparing the retention times and quality of known compounds in Wiley and Adams mass spectra libraries. All the chromatographic peaks found in two or more technical replicates of the same sample and with a quality greater than 80 were taken into account. The relative composition of aroma compounds (%) in the headspace of the strawberry puree was quantitated based on area normalisation method with two assumptions: (1) detector response is the same for different compounds, and (2) compounds of the sample injected are completely detected and will produce peaks53. The calculation was done according to the equation below:
where Ci = Content of a compound in the sample.
Ai = Area of compound peak in the chromatogram.
At = Total area of the peaks in the chromatogram.
The mean relative composition and standard deviation of each compound were calculated from three technical replicates. Aroma compounds detected by GC-MS and their relative compositions were categorised according to different chemical groups. Target compounds for DNA marker development were chosen based on the distinctive parental aroma profiles and their relative contribution to strawberry flavour as elucidated in a number of publications5,6,9,13,30,54. Application of these selection criteria resulted in the selection of eight compounds of interest including four esters (methyl butanoate, methyl hexanoate, ethyl butanoate and ethyl hexanoate), one furanone (mesifuranne), two terpenes (linalool and (E)-nerolidol) and one lactone (γ-dodecalactone).
Generation of DNA bulks with extreme phenotypes
The number of plants with extreme phenotypes for the selected compounds was identified by generating frequency distributions from the 50 F1 progeny plants along with their parental means using Microsoft Excel. The F1 plants determined from the segregation patterns were selected for BSA. Total genomic DNA was isolated from the leaves of individual plants using QiagenTM DNeasy® Plant Mini Kit (Qiagen, Valencia, CA) according to manufacturer’s instructions. Equal amounts of DNA from F1 progeny plants showing high (H) or low/undetectable (L) levels of key aroma compounds were pooled into the respective ‘H’ and ‘L’ bulks to a final quantity of 2 µg. The number of individuals in each bulk ranged from 3 to 27 plants depending on the key aroma compounds.
Microarray-assisted Bulked Segregation Analysis
A 287-feature Fragaria Discovery Panel (FDP) constructed and validated previously was used as a platform for screening of molecular markers associated with key aroma compounds24. Marker discovery was performed by hybridising 16 DNA bulks corresponding to the ‘H’ and ‘L’ extremes of eight key aroma compounds onto the FDP. Target DNA was labelled with Biotin-11-dUTP molecules as described by Gor, et al.24. Hybridisation of the biotinylated DNA targets onto the FDP and fluorescent detection using a biotin-streptavidin system was performed according to Mantri, et al.55. All hybridisations were performed with six technical replicates and two biological replicates to ensure microarray reproducibility, producing a total of 12 data points per feature for subsequent statistical analysis. FDP slide scanning, microarray image capturing and data normalisation were performed based on Gor, et al.24. All microarray experiments were compliant with MIAME guidelines and all data have been deposited in Gene Expression Omnibus (GSE70145).
Statistical analysis
The FDP data was subjected to Discriminant Function Analysis (DFA) to identify molecular markers associated with fruit flavour. The ‘H’ and ‘L’ phenotypic groups and the normalised mean SNR of the 287 FDP features were used as dependent and independent variables, respectively. DFA was performed using IBM SPSS Statistics v. 21 with a stepwise method for the selection of the most discriminative features between the two phenotypic extreme groups. Wilks’ lambda was used to determine the classification efficiency of each feature based on the default F probability values (Entry = 0.05, Removal = 0.10). In this study, six technical replicates (original cases) from the first biological replicate of a phenotypic group were assigned as a training set to predict the group membership of the other six technical replicates (new cases in the test set) from the second biological replicate. All the twelve technical replicates of the selected features were subjected to stepwise analysis again to form classification models using Fisher’s classification function coefficients. The performance of the discriminant function was evaluated using the cross-validation method.
For validation of the DFA-selected features, Fisher’s ratio was employed and calculated according to Lohninger56:
where M1 = Mean of the normalised SNR for each feature in the ‘H’ bulk, M2 = Mean of the normalised SNR for each feature in the ‘L’ bulk, V1 = Variance of the normalised SNR for each feature in the ‘H’ bulk and V2 = Variance of the normalised SNR for each feature in the ‘L’ bulk.
Independent Samples t-Test (IBM SPSS Statistics v. 21) was performed using all the twelve technical replicates of the ‘H’ and ‘L’ bulks as variables. Only the features showing high Fisher’s ratio (top 10) and significant differences between the group means of ‘H’ and ‘L’ bulks (p < 0.01) were retained for further analysis. Finally, a three-way Venn diagram was generated (http://www.pangloss.com/seidel/Protocols/venn.cgi) to identify putative DNA markers that fulfilled all three selection criteria.
DNA sequence analysis
DNA sequencing of putative molecular markers
Plasmids corresponding to the putative DNA markers were sequenced bi-directionally at Macrogen Inc. (Korea) using T7 and Sp6 primers. Similarity search was performed against the Fragaria vesca draft genome (v1.1) using PFR Strawberry Server (https://strawberry.plantandfood.co.nz/) and confirmed with Genome Database for Rosaceae (http://www.rosaceae.org/tools/ncbi_blast). Sequence identity with an E-value < 1e−5 was considered significant. Subsequently, genes located within 5 centiMorgan (cM) on either side of the putative DNA markers were manually searched using PFR Strawberry Server based on previously mapped genes available in Strawberry Genbank and general RefSeq mRNA database. By assuming the genetic length of a normal chromosome as 100 cM57, the physical distance covering 5 cM was calculated following the equation below:
Determination of DNA sequence polymorphism
DNA sequences showing significant similarity (E-value < 1e−5) to F. vesca nuclear sequences were chosen for primer design using Clone Manager Suite v. 7.1 (Sci-Ed Software, Durham, NC). To determine the DNA microstructural variation of the putative markers between the ‘H’ and ‘L’ DNA bulks, PCR amplification was performed on both the parental genotypes (Juliette and 07-102-41) using AccuPrime™ Pfx DNA Polymerase (Invitrogen, NY, USA). Briefly, 50 ng of genomic DNA was used as template in a 50 µL PCR reaction containing 1 X of AccuPrime™ Pfx mix, 40 nM of each sequence-specific forward and reverse primer, 1 U of AccuPrime™ Pfx DNA Polymerase and water. The thermal cycling conditions were as follows: initial denaturation at 95 °C for 2 min; followed by 30 cycles of denaturation at 95 °C for 15 s, annealing at 55 °C for 30 s, extension at 68 °C for 1 min. The integrity and length of PCR products were examined using 2.0% TBE agarose gel electrophoresis. The PCR products were subsequently purified using Qiaquick Gel Extraction Kit (Qiagen, Valencia, CA) and sequenced by Australian Genome Research Facility Ltd. (AGRF) using the sequence-specific primers. All the forward and reverse DNA sequences were aligned for using the Clustal Omega Multiple Sequence Alignment function at https://www.ebi.ac.uk/Tools/msa/clustalo/ to identify any SNPs or indels within the marker. All sequences were deposited in NCBI GeneBank database (KT162989 – KT163008).
Validation of the putative molecular marker
The putative molecular marker that showed sequence polymorphism between the parental genotypes was selected for validation in the F1 segregating population and a wider germplasm. For the purpose of this study, two high level of methyl butanoate producing F1 progeny (P38 and P99) and two low levels of methyl butanoate producing F1 progeny (P1 and P63) along with six commercial cultivars including Melba, Camino Real, Portola, Palomar, San Andreas and Albion were selected for marker validation. The aroma profile for these six commercial cultivars was evaluated using GC-MS as described above. The putative marker sequence was amplified from these plants, cloned into a pGEM-T vector and transformed into E. coli JM109. 10 of the colonies were randomly picked for each genotype and the clones were sequenced bidirectionally using T7 and SP6 primers. The resulting sequences were aligned with sequences derived from the parental genotypes to deduce the association between the polymorphic markers and key aroma compounds.
References
Bartoshuk, L. M. & Klee, H. J. Better fruits and vegetables through sensory analysis. Current Biology 23, R374–R378 (2013).
Klee, H. J. Improving the flavor of fresh fruits: genomics, biochemistry, and biotechnology. New Phytologist 187, 44–56 (2010).
Dirinck, P. J., De Pooter, H. L., Willaert, G. A. & Schamp, N. M. Flavor quality of cultivated strawberries: the role of the sulfur compounds. Journal of Agricultural and Food Chemistry 29, 316–321 (1981).
Latrasse, A. Fruits III. (Marcel Dekker, 1991).
Pérez, A., Rios, J., Sanz, C. & Olias, J. Aroma components and free amino acids in strawberry variety Chandler during ripening. Journal of Agricultural and Food Chemistry 40, 2232–2235 (1992).
Ulrich, D., Hoberg, E., Rapp, A. & Kecke, S. Analysis of strawberry flavour–discrimination of aroma types by quantification of volatile compounds. Zeitschrift für Lebensmitteluntersuchung und-Forschung A 205, 218–223 (1997).
Zabetakis, I. & Holden, M. A. Strawberry flavour: analysis and biosynthesis. Journal of the Science of Food and Agriculture 74, 421–434 (1997).
Samykanno, K., Pang, E. & Marriott, P. J. Chemical characterisation of two Australian-grown strawberry varieties by using comprehensive two-dimensional gas chromatography–mass spectrometry. Food chemistry 141, 1997–2005 (2013).
Jetti, R., Yang, E., Kurnianta, A., Finn, C. & Qian, M. Quantification of Selected Aroma‐Active Compounds in Strawberries by Headspace Solid‐Phase Microextraction Gas Chromatography and Correlation with Sensory Descriptive Analysis. Journal of food science 72, S487–S496 (2007).
Du, X., Plotto, A., Baldwin, E. & Rouseff, R. Evaluation of volatiles from two subtropical strawberry cultivars using GC–olfactometry, GC-MS odor activity values, and sensory analysis. Journal of agricultural and food chemistry 59, 12569–12577 (2011).
Fukuhara, K., Li, X.-X., Okamura, M., Nakahara, K. & Hayata, Y. Evaluation of odorants contributing to “Toyonoka” strawberry [Fragaria] aroma in extracts using an adsorptive column and aroma dilution analysis. Journal of the Japanese Society for Horticultural Science ( Japan ) (2005).
Schwieterman, M. L. et al. Strawberry flavor: diverse chemical compositions, a seasonal influence, and effects on sensory perception. PLoS One 9, e88446 (2014).
Bood, K. & Zabetakis, I. The biosynthesis of strawberry flavor (II): Biosynthetic and molecular biology studies. Journal of food science 67, 2–8 (2002).
Aharoni, A. et al. Gain and loss of fruit flavor compounds produced by wild and cultivated strawberry species. The Plant Cell Online 16, 3110–3131 (2004).
Aharoni, A. et al. Identification of the SAAT gene involved in strawberry flavor biogenesis by use of DNA microarrays. The Plant Cell Online 12, 647–661 (2000).
Chambers, A., Whitaker, V. M., Gibbs, B., Plotto, A. & Folta, K. M. Detection of the linalool‐producing NES1 variant across diverse strawberry (Fragaria spp.) accessions. Plant Breeding 131, 437–443 (2012).
Raab, T. et al. FaQR, required for the biosynthesis of the strawberry flavor compound 4-hydroxy-2,5-dimethyl-3(2H)-furanone, encodes an enone oxidoreductase. The Plant Cell Online 18, 1023–1037 (2006).
Schwab, W. et al. Genes and enzymes involved in strawberry flavor formation. Vol. 988 (ACS Symposium Series; American Chemical Society, 2008).
Wein, M. et al. Isolation, cloning and expression of a multifunctional O‐methyltransferase capable of forming 2,5‐dimethyl‐4‐methoxy‐3(2H)‐furanone, one of the key aroma compounds in strawberry fruits. The Plant Journal 31, 755–765 (2002).
Zorrilla-Fontanesi, Y. et al. Genetic analysis of strawberry fruit aroma and identification of O-methyltransferase FaOMT as the locus controlling natural variation in mesifurane content. Plant physiology 159, 851–870 (2012).
Sánchez-Sevilla, J. F., Cruz-Rus, E., Valpuesta, V., Botella, M. A. & Amaya, I. Deciphering gamma-decalactone biosynthesis in strawberry fruit using a combination of genetic mapping, RNA-Seq and eQTL analyses. BMC genomics 15, 218 (2014).
Chambers, A. H. et al. Identification of a strawberry flavor gene candidate using an integrated genetic-genomic-analytical chemistry approach. BMC genomics 15, 217 (2014).
Jayasinghe, R. et al. Construction and validation of a prototype microarray for efficient and high‐throughput genotyping of angiosperms. Plant biotechnology journal 5, 282–289 (2007).
Gor, M. C., Mantri, N. & Pang, E. Application of subtracted gDNA microarray-assisted Bulked Segregant Analysis for rapid discovery of molecular markers associated with day-neutrality in strawberry (Fragaria x ananassa). Scientific Reports 6 (2016).
Michelmore, R. W., Paran, I. & Kesseli, R. Identification of markers linked to disease-resistance genes by bulked segregant analysis: a rapid method to detect markers in specific genomic regions by using segregating populations. Proceedings of the National Academy of Sciences 88, 9828–9832 (1991).
Niu, L. et al. Detection of Panax quinquefolius in Panax ginseng using ‘subtracted diversity array’. Journal of the Science of Food and Agriculture 91, 1310–1315 (2011).
Olarte, A. et al. A gDNA microarray for genotyping Salvia species. Molecular biotechnology 54, 770–783 (2013).
Olarte, A., Mantri, N., Nugent, G. & Pang, E. Subtracted diversity array identifies novel molecular markers including retrotransposons for fingerprinting Echinacea species. PLoS One 8, 1–12 (2013).
Samykanno, K. Environmental effects on flavour development in Australian-grown strawberry varieties, RMIT University, (2012).
Hakala, M. A., Lapveteläinen, A. T. & Kallio, H. P. Volatile compounds of selected strawberry varieties analyzed by purge-and-trap headspace GC-MS. Journal of agricultural and food chemistry 50, 1133-1142 (2002).
Olbricht, K., Grafe, C., Weiss, K. & Ulrich, D. Inheritance of aroma compounds in a model population of Fragaria × ananassa Duch. Plant breeding 127, 87–93 (2008).
Dunemann, F., Ulrich, D., Boudichevskaia, A., Grafe, C. & Weber, W. QTL mapping of aroma compounds analysed by headspace solid-phase microextraction gas chromatography in the apple progeny ‘Discovery’ × ‘Prima’. Molecular breeding 23, 501–521 (2009).
Eduardo, I. et al. Genetic dissection of aroma volatile compounds from the essential oil of peach fruit: QTL analysis and identification of candidate genes using dense SNP maps. Tree Genetics & Genomes 9, 189–204 (2013).
deVicente, M. & Tanksley, S. QTL analysis of transgressive segregation in an interspecific tomato cross. Genetics 134, 585–596 (1993).
Rieseberg, L. H., Archer, M. A. & Wayne, R. K. Transgressive segregation, adaptation and speciation. Heredity 83, 363–372 (1999).
Rieseberg, L. H., Widmer, A., Arntz, A. M. & Burke, B. The genetic architecture necessary for transgressive segregation is common in both natural and domesticated populations. Philosophical Transactions of the Royal Society of London. Series B: Biological Sciences 358, 1141–1147 (2003).
Holzman, R. & Hulsey, C. D. Mechanical Transgressive Segregation and the Rapid Origin of Trophic Novelty. Scientific reports 7, 40306 (2017).
Hake, S. & Rocheford, T. Exploiting quantitative trait loci in gene discovery. Genes & development 18, 597–601 (2004).
Bell, M. A. & Travis, M. P. Hybridization, transgressive segregation, genetic covariation, and adaptive radiation. Trends in ecology & evolution 20, 358–361 (2005).
Samykanno, K., Pang, E. & Marriott, P. J. Genotypic and environmental effects on flavor attributes of ‘Albion’ and ‘Juliette’ strawberry fruits. Scientia Horticulturae 164, 633–642 (2013).
Siosemarde, A., Osmani, Z., Bahramnezhad, B., Vahabi, K. & Rouhi, E. Identification of AFLP marker associated with stress tolerance index in Sardari wheat ecotypes. Journal of Agricultural Science and Technology 14, 629–643 (2012).
Alwala, S. Identification of molecular markers associated with resistance to Aspergillus flavus in maize, Louisiana State University, (2007).
Zhang, N., Xu, Y., Akash, M., McCouch, S. & Oard, J. Identification of candidate markers associated with agronomic traits in rice using discriminant analysis. Theoretical and applied genetics 110, 721–729 (2005).
Schwab, W., Davidovich‐Rikanati, R. & Lewinsohn, E. Biosynthesis of plant‐derived flavor compounds. The Plant Journal 54, 712–732 (2008).
Kanehisa, M. & Goto, S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic acids research 28, 27–30 (2000).
Kanehisa, M. et al. Data, information, knowledge and principle: back to metabolism in KEGG. Nucleic acids research 42, D199–D205 (2014).
Beekwilder, J. et al. Functional characterization of enzymes forming volatile esters from strawberry and banana. Plant Physiology 135, 1865–1878 (2004).
Scherer, G. F., Ryu, S. B., Wang, X., Matos, A. R. & Heitz, T. Patatin-related phospholipase A: nomenclature, subfamilies and functions in plants. Trends in plant science 15, 693–700 (2010).
Paliyath, G., Murr, D. P., Handa, A. K. & Lurie, S. Postharvest biology and technology of fruits, vegetables, and flowers. (John Wiley & Sons, 2009).
Lamartine, J. The benefits of DNA microarrays in fundamental and applied bio-medicine. Materials Science and Engineering: C 26, 354–359 (2006).
Brevis, P. Project BS11013: National Strawberry Varietal Improvement Program (Southern Node). (Australia, 2013).
Pawliszyn, J. & Pawliszyn, J. Solid phase microextraction: theory and practice. Vol. 61 (Wiley-Vch New York, 1997).
Shimadzu. Qualitative and quantitative analysis of GC and GCMS. (Singapore, 2006).
Forney, C. F., Kalt, W. & Jordan, M. A. The composition of strawberry aroma is influenced by cultivar, maturity, and storage. HortScience 35, 1022–1026 (2000).
Mantri, N., Olarte, A., Li, C. G., Xue, C. & Pang, E. C. Fingerprinting the Asterid species using Subtracted Diversity Array reveals novel species-specific sequences. PloS one 7, e34873 (2012).
Lohninger, H. Teach/me: Data Analysis. (Springer, 1999).
Kearsey, M. J. & Pooni, H. S. The Genetical Analysis of Quantitative Traits. (Chapman & Hall, 1996).
Acknowledgements
This work was supported by Horticulture Innovation Australia Limited (HIA) and RMIT University.
Author information
Authors and Affiliations
Contributions
E.P. and N.M. supervised the project. M.C.G., E.P. and N.M. collected the samples. M.C.G. constructed the microarray. M.C.G., C.C. and T.D.S. phenotyped the plants. M.C.G. performed microarray-assisted BSA. M.C.G., E.P. and N.M. analysed and interpreted the data and wrote the manuscript. All authors have read and approved the final version of this manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Gor, M.C., Candappa, C., de Silva, T. et al. Identification and validation of FaP1D7, a putative marker associated with the biosynthesis of methyl butanoate in cultivated strawberry (Fragaria x ananassa). Sci Rep 7, 17454 (2017). https://doi.org/10.1038/s41598-017-17448-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-17448-1
- Springer Nature Limited