
Abstract
Episyrphus balteatus can provide dual ecosystem services including pest control and pollination, which the larvae are excellent predators of aphid pest whereas adults are efficient pollinator. In this study, we assembled a high-quality genome of E. balteatus from northern China geographical population at the chromosome level by using Illumina, PacBio long reads, and Hi-C technologies. The 467.42 Mb genome was obtained from 723 contigs, with a contig N50 of 9.16 Mb and Scaffold N50 of 118.85 Mb, and 90.25% (431.75 Mb) of the assembly was anchored to 4 pseudo-autosomes and one pseudo-heterosome. In total, 14,848 protein-coding genes were annotated, and 95.14% of genes were fully represented in NR, GO, KEGG databases. Besides, we also obtained the mitochondrial genome of E. balteatus of 16, 837 bp in length with 37 typical mitochondrial genes. Overall, this high-quality genome is valuable for evolutionary and genetic studies of E. balteatus and other Syrphidae hoverfly species.
Similar content being viewed by others

Background & Summary
Episyrphus balteatus, also known as the marmalade hoverfly, acts as pollinator and predator, which the larvae can control aphid pests and adults feeding on pollen and nectar, can be used as pollinators in plants (Fig. 1a). As a long-distance migratory insect, it travels high above between high- and low-latitude regions seasonally each year, transports billions of pollen grains, consumes trillions of aphids, and make billions of flower visits on the annual fluxes1. Considering that the populations of many pollinator insects, especially bees (Fig. 1b), are seriously declining2. Hoverflies including E. balteatus are becoming more important in the ecosystem3. To date, plentiful studies about genome, reproduction, behavior, and phylogenetic relationship have been performed on various hoverfly species. However, the lack of high-quality reference genomes has hindered deeper gene function exploration of this species.

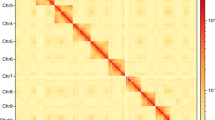
Morphological characteristics of Episyrphus balteatus (a), honeybee (b), and genome scope profiles of 19-mer analysis (c) and Hi-C interactive heatmap of four linkage pseudo-chromosomes in Episyrphus balteatus genome (d). Color indicates the intensity of the interaction signal. The darker the color, the higher the intensity.
In this study, we propose a high-quality genome-assembly at chromosome level and conduct a whole life cycle transcriptome of E. balteatus using a combination of Illumina short-read sequencing, PacBio continuous long read (CLR), and chromosome conformation capture (Hi-C) techniques (Table 1). Through CLR sequencing, 8,740, 850 continuous long reads with N50 of 39.791 kb were obtained, and then a total of 517.92 Mb genome was assembled from 729 contigs with a contig N50 of 9.16 Mb (Tables 2, 3). In Hi-C strategy, 90.25% (467.42 Mb) of the assembly was anchored to 4 chromosomes (Table 4) with a scaffold N50 of 118.85 Mb (Tables 2, 3), while Contig 1996 was proved as the pseudo-X chromosome via whole-genome synteny with other hoverfly species (Fig. 3). We also predicted that transposable elements and tandem repeats accounted for 35.23% (13.90% retroelement and 21.33% DNA transposon) and 8.31% of the total genome (Table 5), respectively. 1611 noncoding RNA, 29 pseudogene, and 14,848 protein-coding genes were obtained (Tables 6, 7), of which 95.14% gene sequences could be annotated to NR, GO, KEGG and other databases (Table 8). We compared the genomic characteristics of E. balteatus with those other insects and identified 1060 expanded gene families and 1535 contracted gene families in the genome of E. balteatus (Fig. 7). In addition to, the mitochondrial genome of E. balteatus was also assembled and annotated with the length of 16837 bp encoding 37 typical mitochondrial genes (Fig. 10; Table 11). Overall, the high-quality E. balteatus nuclear and mitochondrial genome produced here provide a genetic basis for further studies of the biology and ecology of this hoverfly species.
Methods
Sample preparation and genomic DNA sequencing
A pair of E. balteatus female and male adults were originally captured from flowers of Hibiscus syriacus in Anyang City, Henan Province, China, and reared on Megoura crassicauda inbreeding for approximately 10 generations in the laboratory. The insect colony was maintained in the climate chamber at 21 ± 1 °C with 70 ± 2% relative humidity and photoperiod of 14 h Light: 10 h Dark. Individuals were immediately frozen in liquid nitrogen, followed by preservation at −80 °C in the laboratory prior to DNA extraction. Genomic DNA for both Illumina and PacBio CLR sequencing was obtained from 10 newly emerged female adults with surface-sterilized using the Genomic-tip Kit (QIAGEN) according to the manufacturer’s instructions, and for Hi-C sequencing it was obtained from a single newly emerged female adult individual. The determination of genomic DNA’s purity and integrity was conducted using two methods: the NanoDrop 2000 (Thermo Fisher Scientific, USA) and agarose gel electrophoresis (1.2%) respectively.
For Illumina sequencing, the paired-end libraries with a 350 bp length were constructed and sequenced on the Illumina NovaSeq 6000 platform (Illumina, CA, USA), and 57.50 Gb (Table 1) of clean reads were obtained after removing adapter sequences and low-quality reads with HTQC (v1.92.310) software. The short-reads from Illumina platform were quality filtered by Fastp using the parameters is -q 10 -u 50 -y -g -Y 10 -e 20 -l 100 -b 150 -B 150.
To perform PacBio CLR sequencing, we used the Megaruptor®°2 to shear the genomic DNA into fragments of approximately 20 kb. Subsequently, we prepared the SMRTbell library using the SMRTbell Express Template Prep kit 2.0 (Pacific Biosciences) as per the guidelines provided by the manufacturer. Following ligation, the SMRTbell library was digested by exonuclease and purified with 0.45X AMPure PB beads. After library characterization, 15-bells 18 kb were collected using the Sage ELF system (Sage Science, Beverly, MA). Sequencing primers and Sequel II DNA polymerase were annealed and bound to the final SMRTbell library, respectively. Finally, SMRT sequencing was performed using a single 8 M SMRT Cell on the Sequel II System. The sequencing yielded 247.38 Gb (484.58 × coverage) of the continuous long reads (CLR) with an N50 length of 39,791 bp and an average length of 28,301 bp (Table 3).
For Hi-C (high-throughput chromatin conformation capture) associated scaffolding, 150 bp paired-end reads with mate mapped to a different contig were constructed firstly. After applying the same filter criteria for short reads, the resulting Hi-C library was sequenced on the Illumina NovaSeq 6000 platform and generated a total of 63.97 Gb (140.14 × coverage) of clean data (Table 1).
Hi-C library preparation and sequencing
The Hi-C technique was used to construct the chromosome-level genome assembly of Episyrphus balteatus, and fresh tissues from one female adult (not including abdomen and wing) were used to construct Hi-C library. Formaldehyde is employed for sample fixation, facilitating the cross-linking of intracellular proteins with DNA and DNA strands with each other. This process ensures the preservation of their interactions and the overall maintenance of the cell’s intricate 3D structure. The DNA undergoes digestion through the restriction enzyme DpnII, leading to the formation of sticky ends on both ends of the crosslink. In the end, the DNA samples were fragmented, ranging from 300 to 700 base pairs (bp). Subsequently, streptavidin magnetic beads were employed to selectively capture DNA fragments that exhibit interactive associations, facilitating the construction of the library. After the library inspection was qualified, the Illumina platform was used for high-throughput sequencing, and the sequencing read length is paired-end 150 bp. The Hi-C library was constructed following the standard library preparation protocol, and 63.97 Gb of clean data was generated (Table 1).
Transcriptome sequencing
Transcriptomic samples were collected from various developmental stages of E. balteatus, which includes the eggs, 1st instar nymphs, 2nd instar nymphs, 3rd instar nymphs, pupae, and female adults of E. balteatus, respectively. Total RNA was extracted from egg, larvae, pupae, and adult samples respectively by using the TRIzol reagent (Thermo Fisher Scientifc, USA). The complementary DNA (cDNA) library was constructed and sequenced on an Illumina Novaseq 6000 platform. Following the construction of the library, the concentration and insert size were determined using Qubit3.0 and Agilent 2100. Moreover, Q-PCR was employed to precisely quantify the effective concentration of the library, ensuring its quality. Ultimately, a total of 123.90 Gb clean RNA-seq data was obtained by removing adapters, low-quality reads, and high-content unknown sequences (Table 1).
Estimation of genomic characteristics
Based on genome survey raw data of 57.50 Gb, the short reads from the Illumina platform were quality filtered by Fastp (version 0.21.0)4 using the parameters was ‘-q 10 -u 50 -y -g -Y 10 -e 20 -l 100 -b 150 -B 150’. The high-quality filtered reads were used for further genome size estimation. To assess potential contamination in the DNA of the collected samples, 10,000 single-ended reads were randomly selected from a 350 bp library sequenced and compared to the NT library by BLAST (ncbi-blast + , version 2.9.0)5 with the parameter set to ‘-num_descriptions 100-num_alignments 100-evalue 1e-05’. The libraries sequenced by Illumina platform were compared with issued plastids of E. balteatus6 by using SOAP (version 2.21)7 with the parameter set of ‘-m 260 -x 440’ to evaluate the extranuclear DNA content in the libraries and ensure the integrity of the genome assembly. Using Jellyfish (version 2.1.4)8, we conducted a count of the 19-kmers. For this analysis, we employed the parameters ‘-h 10000000000’. Subsequently, genome features were calculated using Genomescope (version 2.0)9. The parameters used for this calculation were ‘-k 19 -p 6 -m 100000’. Based on the analysis of diverse ploidy data, it was observed that the Kmer distribution map exhibited optimal fitting accuracy when considered in a diploid context (Fig. 1c), Additionally, the estimated size of an individual genome is approximately 464.94 Mb (Table 2). According to the distribution of kmer, it is estimated that the repetitive sequence content was about 28.05%, the heterozygosity was about 3.7%, and the GC content of the genome was about 30.52% (Fig. 1c).
De novo genome assembly
Genome assembly was completed based on high accuracy CLR data obtained above by using Smartdenovo10 with default parameters, followed by adjust based on Illumina reads for three times via tool Pilon11. In situations where genomes exhibit considerable heterozygosity, the primary assembly might compile all the fragments that bear heterozygous characteristics, leading to a genome size that surpasses the expected value. To resolve the haplotigs and overlaps in the primary assembly, the purge_dups12 was utilized, whereas the assembly was further enhanced through the application of Pilon11 and Racon13 to polish the collected data.
Using Hi-C technology14, the scaffold pipeline of the genome was dismantled into segments of 50 Kb each and subsequently reconstructed. The candidate error regions encompass the locations that could not be restored to their original assembly sequence. Within this region, the identification of error points is based on the Hi-C coverage depth, particularly focusing on areas with low coverage depth. By doing so, the error correction process of the initial assembled genome is successfully accomplished. For anchored contigs, clean read pairs were generated from the Hi-C library and were mapped to the polished Episyrphus balteatus genome using BWA (version 0.7.17)15 with the default parameters.
The paired reads that were mapped to a distinct contig were utilized to perform Hi-C associated scaffolding. Then invalid reads, including self-circle ligation, non-ligation, and various other types such as Dangling Ends, Re-ligation, and Dumped Pairs, were filtered out. HiC-Pro (version2.10.0)16, capable of detecting valid interaction pairs and invalid interaction pairs in Hi-C sequencing outcomes through analysis and comparison, thus was used to facilitate the assessment of Hi-C library quality. Using the agglomerative hierarchical clustering method in LACHESIS (version 2e27abb)16, we were able to cluster 723 contigs into 4 distinct groups (Table 4), which includes 419 sequences with the length of 467, 421,215 bp totally.
Finally, we obtained the high-quality assembled genome of Episyrphus balteatus at the chromosome level. The genome was consisted of 723 contigs with a total length of 467.42 Mb, which was similar to the predicted size of 464.94 Mb, and with a scaffold N50 of 118.85 Mb, maximum length of 120.20 Mb, and GC rate of 31.52% (Table 2). The analysis of Hi-C data aided in the alignment of 419 (57.95%) sequences with the length of 467.42 Mb (90.25%) of genome to 4 pseudo-chromosomes, which demonstrated clear distinctions among them according to the heatmap portraying chromatin interaction. (Table 2 and Table 4; Fig. 1d). In addition, the mitochondrial genome of E. balteatus was assembled through mitoZ17 and NOVOplasty18, and subsequently annotated using MITOS19 and GeSeq20. (Fig. 10; Table 11).
Repetitive elements and noncoding RNA annotation
A combination of homology-based and de novo approaches was utilized to identify transposon elements (TE) and tandem repeats. To embark on our analysis, we initiated the generation of a customized repeat library for the genome via RepeatModeler (version 2.0.1)21 (http://www.repeatmasker.org/RepeatModeler/) using the following parameters: “-name & RepeatModeler -pa 12”. This software possesses the capability to automatically employ two distinct de novo repeat discovery programs, namely RECON (version 1.0.8)22 and RepeatScout (version 1.0.6)23. The identification of full-length long terminal repeat retrotransposons (fl-LTR-RTs) was carried out by employing two different approaches. Firstly, the LTR_FINDER (version 1.07)24 tool was employed with parameters “-w 2 -C -D”. Additionally, the LTRharvest (version 1.5.10)25 tool was used with default parameters to complete the identification process. The high-quality intact fl-LTR-RTs and non-redundant LTR library were then produced by LTR_retriever (version 2.9.0)26 with default parameters. Non-redundant species-specific TE library was constructed by combining the de novo TE sequence library above with the well-known Dfam (version 3.5)27 database. We conducted a search for Final TE sequences in the genome of Episyrphus balteatus. These sequences were then classified through the use of a homology search against a library. The software tool used for this analysis was RepeatMasker (version 4.1.2)28 with parameters ‘-nolow -no_is -norna -engine wublast -parallel 8 -qq’. We employed the Tandem Repeats Finder (TRF) (version 409)29 with parameters ‘2 7 7 80 10 50 500 -d -h’ and MIcroSAtellite identification tool (MISA) (version 2.1)30 with default parameters to annotate the tandem repeats. In total, 43.55% of the assembled genome was classified as repetitive sequences in the genome, including transposable elements (TEs) with a sequence length of 182,487,345 bp, accounting for 35.23% of the whole genome, and tandem repeats with a sequence length of 43,054,586 bp, accounting for 8.31% of the whole genome (Table 5).
Non-coding RNAs, such as microRNA, rRNA, tRNA, and other RNAs with unidentified roles, are a class of RNAs that do not possess the ability to synthesize proteins. By utilizing various methods to anticipate non-coding RNAs, several distinct approaches are implemented based on the structural attributes they possess. The identification of tRNA was conducted using tRNAscan-SE (version 1.3.1)31 with default parameters. Prediction of rRNA was primarily performed using barrnap (version 0.9)32 with parameters ‘kingdom euk–threads 1’. For the prediction of miRNA, snoRNA, and snRNA, the Rfam (version 14.5)33 database was utilized through INFERNAL (version 1.1)34 with parameters ‘cpu 3–rfam’. Finally, a total of 1,103 tRNAs, 402 rRNAs, 33 snRNA, 24 snoRNA, and 49 miRNAs were obtained (Table 6).
Gene Prediction and Functional Annotation
This study employed a combination of three methodologies, specifically, de novo prediction, homology search, and transcript-based assembly, to annotate the protein-coding genes present within the genome. Utilizing the genome sequence assembled above, Augustus (version 3.1.0)35 and SNAP (version 2006-07-28)36 were employed to perform ab initio gene prediction with default parameters. For homo-based approaches, GeneModelMapper (GeMoMa) (version 1.7)37 was used with Drosophila melanogaster and Eupeodes corollae as references with parameters ‘run.sh mmseqs’. For the transcript-based methods, we aligned the RNA-seq reads to our previously constructed reference genome using Hisat (version 2.1.0)38 with the parameters ‘dta -p 10’. The assembled reads were transformed into transcripts using Stringtie (version 2.1.4)39 with parameters ‘p 2’. Genes were predicted from the assembled transcripts using GeneMarkS-T (version5.1)40 with default parameters. In this study, we employed the PASA software (version 2.4.1)41 to forecast gene using the unigenes that were assembled through Trinity (version 2.11)36 with default parameters ‘genome_guided_bam’. In the end, we combined predicted genes acquired from homology-based strategies, de novo-derived approaches, and transcripts, to generate the high-confidence gene set by employing the EVidenceModeler (version 1.1.1)42 in combination with PASA (version 2.4.1)41 with default parameters. In total, 14,848 protein-coding genes with an average length of 14629.57 bp were obtained in the assembled E. balteatus genome (Table 7). The average length of coding sequence (CDS) was 1714.47 bp with a total number of CDS of 75,039. The average exon length was 1714.63 bp and the average exon number of each gene was 5.05. A total of 3,095 conserved core genes are included in BUSCO’s diptera database (Table 7). The gene prediction completeness in E. balteatus was evaluated by using Benchmarking Universal Single-Copy Orthologs (BUSCO, version5.2.2)43 with parameters ‘m prot’. Our analysis revealed that 94.22% of the predicted genes (Table 7) contained BUSCO genes, only 0.27% with fragmented BUSCOs and 5.51% with missing BUSCOs, indicating a commendable gene prediction completeness in E. balteatus.
The identification of gene structure and annotations was based on aligning the data with various public databases. These databases included the National Center for Biotechnology Information (NCBI) Non-Redundant (NR), EggNOG, KOG, TrEMBL, InterPro, and Swiss-Prot protein databases. The alignment was carried out using diamond (2.0.4.142) with parameters of ‘masking 0 -e 0.001’. The data were also compared with the Kyoto Encyclopedia of Genes and Genomes (KEGG) database, with an E-value threshold of 1E-5. InterProScan (version 5.34-73.0) utilized the InterPro protein database to annotate the protein domain44. The annotation process was performed with parameters ‘-iprlookup -pa -f xml -dp -t p -cpu 10’. The motifs and domains within gene models were identified by Pfam databases45. In total, around 95.14% (14,126 out of 14,848 total predicted genes) of the predicted genes responsible for encoding proteins could be annotated within the aforementioned databases (Table 8). In conclusion, we separately tallied the count of genes integrated with EVM using three distinct prediction techniques. The outcome revealed that a total of 11,048 genes were predicted consistently across all three methods (Fig. 2b).

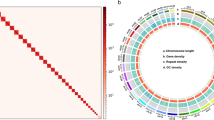
Genome assembly and temporal transcriptome of Episyrphus balteatus. (a) Circle genome landscape of Episyrphus balteatus. Circle a represents chromosomes, while circles b-e indicate TE density, SSR density, SSR, and gene density of each respective chromosome, respectively. (b) Protein-coding gene prediction of Episyrphus balteatus genome through three strategies.
Genome synteny analysis
Synteny analysis of genes can realize the excavation of genomic structural variation. Diamond (version 0.9.29.130)46 was applied to compare the gene sequences of Episyrphus balteatus and six Syrphidae hoverflies with different pseudo-chromosome number (i.e. 4 pseudo-chromosomes for Eupeodes corollae and Scaeva pyrastri, 5 pseudo-chromosomes for Xylota sylvarum and Syritta pipiens, 6 pseudo-chromosomes for Eristalis tenax and Volucella inanis) and identify similar gene pairs (e < 1e-5, C-score > 0.5). Next, MCScanX47 was employed to assess the chromosomal proximity of comparable gene pairs. Ultimately, it is possible to obtain all the genes within the synteny block with the parameters ‘-m 15’ (Figs. 2a, 3). Each chromosome of E. balteatus have a good relativity with from selected insects above, in which Contig 1996 can correspond to presumptive X-chromosome in these seven hoverflies, indicating that there was a high degree of consistency between them and Contig 1996 is the candidate X chromosome in E. balteatus (Fig. 3).

Whole-genome synteny between Episyrphus balteatus and other hoverfly species. E. balteatus Ay, Episyrphus balteatus population from Anyang City, Henan Province, China; E. balteatus Orf, Episyrphus balteatus population from Wytham Woods, Oxfordshire, UK; E. corollae, Eupeodes corollae; X. sylvarum, Xylota sylvarum; E. tenax, Eristalis tenax; V. inanis, Volucella inanis; S. pyrastri, Scaeva pyrastri; S. pipiens, Syritta pipiens.
Temporal transcriptome of Episyrphus balteatus across egg, nymphal, pupal, adult stage
Samples were collected from all developmental stages of E. balteatus, namely, eggs, 1st instar nymph, 2nd instar nymph, 3rd instar nymph, pupa and female adult. Thirty E. balteatus individuals at each postembryonic stage and 300 egg were placed in each 2.0 mL Eppendorf tube with 3 replicates per stage, respectively. TRIzol reagent (Thermo, USA) was used for RNA extraction. The transcriptome sequences were acquired utilizing the identical methodology outlined within the “Transcriptome sequencing” section aforementioned.
Hisat (version 2.1.0)38 was used to locate the precise position of the clean reads from transcriptome sequencing on the assembled genome of E. balteatus according to the default parameters. Then above reads were assembled by using Stringtie (version 2.1.4)39, and subsequent analysis involved reconstructing the transcripts with default parameters. In conclusion, the expression level of transcripts or genes was quantified using a standardized indicator known as FPKM (Fragments Per Kilobase of transcript per Million mapped fragments)48. Principal components analysis (PCA) and heatmap clustering were utilized together to evaluate the sample relationship between or within groups (Fig. 11).
Here, we define genes as differentially expressed genes (DEGs) if their expression levels vary significantly across distinct development stages. DEGs can be divided into up-regulated and down-regulated genes. Differential expression gene screening and analysis were conducted using DESeq. 2 based on the gene count values observed in each sample49. To minimize the occurrence of false positives resulting from alterations in the expression of numerous genes, the false discovery rate (FDR) was calculated by adjusting the significance p-value of the disparity. Such an adjustment benefits for evaluating the significance of the difference accurately50. Fold change (FC) represents the ratio of genes expression levels between two group of samples. For convenience of comparison, the fold change was expressed as log2FC. In the process of detecting significantly differential expression genes, the criteria employed were |log2FC| ≥ 1 and FDR < 0.01. The more pronounced the disparity in gene expression levels between the two sample groups, the higher absolute value of log2FC and the lower the FDR value. Eventually, the number of differentially expressed genes in the pairwise comparisons of Episyrphus balteatus across egg, 1st instar larvae (L1), 2nd instar larvae (L2), 3rd instar larvae, pupa, and female adult were obtained (Table 9). An enrichment analysis was conducted to detect any over-representation of GO terms and KEGG pathways by utilizing the hypergeometric test with a q-value ≤ 0.05 as the cutoff criterion. The DEGs were then mapped to these GO terms and KEGG pathways to evaluate the reliability and their roles (Figs. 4, 5).

Differential expressed genes in pairwise comparison of Episyrphus balteatus across different developmental stages. (a–e) Volcanic maps of different expressed genes in separate comparison among different developmental stages. The red and green balls represent the significantly up- and down-regulated expressed genes, respectively. (f) Venn diagrams of DEGs among those pairwise comparisons. L1, L2, L3 represent 1st, 2nd, 3rd instar larvae of Episyrphus balteatus, respectively.

Significantly enriched KEGG pathways of DEGs in these pairwise comparison of Episyrphus balteatus at different developmental stages. a, 1st instar larvae vs. egg; b, 2nd instar larvae vs. 1st instar larvae; c, 3rd instar larvae vs. 2nd instar larvae; d, pupa vs. 3rd instar larvae; e, female adult vs. pupae.
Data Records
The dataset of Episyrphus balteatus genomics sequencing is available at NCBI with the accession number of PRJNA104965251 including four subsets (SRR2712810552, SRR2716787653, SRR2712902654, SRR2720421855) as follows: Genomic Illumina sequencing data were deposited in the Sequence Read Archive at NCBI under accession number SRR2712810552. Genomic PacBio sequencing data were deposited in the Sequence Read Archive at NCBI under accession number SRR2716787653. Hi-C sequencing data were deposited in the Sequence Read Archive at NCBI under accession number SRR2712902654. Mitochondrial genome were deposited in the Sequence Read Archive at NCBI under accession number SRR2720421855. RNA-seq data were deposited in the Sequence Read Archive at NCBI under accession number PRJNA105088956. The annotation files of the E. balteatus genome have been deposited at figshare (https://figshare.com/articles/dataset/Genome_annotation_information_of_Episyrphus_balteatus/24797310)57. The final assembled Episyrphus balteatus genome has been submitted to National Center for Biotechnology Information (https://www.ncbi.nlm.nih.gov/) and are publicly accessible under accession number GCA_040182855.158.
Technical Validation
DNA integrity
The Nanodrop 2000 spectrophotometer (Thermo Fisher Scientific, USA) and QubitTM3Flurometer (USA, Thermo Fisher Scientific) were used to measure the concentration and quality of the extracted DNA. Absorbance of obtained DNA above at 260/280 nm and 260/230 nm were both about 1.8. The quality of genomic DNA was detected by agarose gel electrophoresis. The main band size of DNA fragments was ≥ 23 K, the degradation band was > 5 K. The absence of any contamination in the sample holes substantiated the preservation of DNA molecule integrity throughout this investigation.
Assessment of genome assemblies
Assess the integrity and accuracy of the genome in majorly three ways: To begin, we compared the short sequences acquired via the Illumina sequencing platform with the assembled genome using BWA (version 0.7.17)59. Through statistical comparison rate, proportion of covered genomes, and depth distribution, we assessed both the integrity of the assembled genome and the evenness of sequencing coverage. The results showed that 383,884,382 clean reads were obtained, of which 368,506,902 were located to the reference genome, accounting for 95.99% of all clean reads (Table 10). Secondly, to assess the integrity of the assembled genome, we utilized the Core Eukaryotic Genes Mapping Approach (CEGMA, version 2.5) with default parameters, and a core gene library consisting of 458 genes from eukaryotic model organisms was selected. This approach allows us to evaluate the assembled genome integrality effectively. The results showed that 446 of 458 core genes were identified in the assembled genome, accounting for 97.38% of the total (Table 10). Finally, in the evaluation of the assembled genome, the comparison was carried out between the single-copy gene set generated by BUSCO (version 5.2.2) and our assembled genome through BLAST5. Subsequently, the completeness and ratio of this comparison were assessed. The results showed that the completeness of BUSCO evaluation was 96.71% with only 0.44% fragmented BUSCOs and 2.85% missing BUSCOs (Table 10). All of the above results indicate that our assembled E. balteatus genome has high integrity and accuracy (Table 10). These BUSCO results were also compared with the integrity of other hoverfly species genomes, all of which were comparable (Table 12). Besides, the genome structure statistics of Episyrphus balteatus was compared with those from several representative dipteran insects as well, all of which were comparable (Table 13).
Assessment of genome reliability
In order to evaluate the reliability of genome assembly and annotation of E. balteatus, comparative genomic analysis and phylogenetic reconstruction were performed with genetically close 19 species within the Diptera order (which includes 7 Syrphidae sepecies) and one pollinating insect (Apis mellifera, as the outgroup) (Fig. 6; Table 14). To identify single-copy orthologous genes, the protein sequence of the longest transcript for each gene was retrieved from E. balteatus, as well as those selected 20 different species. For the analysis of gene family clustering, we employed OrthoFinder (version 2.4)60 to compare the protein-coding sequences of 21 species’ genomes (Fig. 6a). The comparison was performed using the diamond method with a threshold e-value of 0.00001. As a result, 10, 037 gene families were constructed for E. balteatus in this work (Table 14), and plentiful of genes families were shared by those species, with especially more number in the comparison of five hoverfly species, than with A. mellifera and other Diptera insects (Fig. 6b,c,d).

Statistics of gene families of Episyrphus balteatus and 19 other insects. Petals of shared and unique gene families of Episyrphus balteatus and 19 other insects (a). Venn diagram of gene families among two hoverfly species (Episyrphus balteatus and E. corollae) and three Diptera insects (b), three other hoverfly species (c), and A. mellifera (d). Gene family of E. balteatus Ref is from ref. 65 (Tables 12 and 14).
To determine the phylogenetic relationships between E. balteatus and other closely related species, we conducted a phylogenetic tree reconstruction. The MAFFT (version 7.205) was utilized61 with the parameters ‘–localpair–maxiterate 1000’to align the protein sequences of the single-copy orthologous genes. Under these criteria, we obtained a total of 545 single-copy genes. We utilized the LG + F + I + G4 model to build the phylogenetic trees using the maximum likelihood method in IQ-TREE (version 1.6.11)62 with bootstrap replicates set to 1000. Then the maximum likelihood method was used to estimate the divergence time by using the MCMCTREE of PAML (version4.9i)63.
The results of phylogenetic analysis indicated that Chironomidae and Culicidae speciated from Diptera ancestral insects firstly before ~248.31 Mya, followed by Psychodidae before ~228.15 Mya, Cecidomyiidae and Sciaridae before ~208.69 Mya, Stratiomyidae before ~179.96 Mya, Diopsidae and Trypetidae before ~ 115.19 Mya, Drosophilidae before ~99.36 Mya, Muscidae and Calliphoridae before ~58.81 Mya (Fig. 7). Interestingly, geographical population from E. balteatus in Anyang (N36°3’, E114°20’, Henan, China) and Oxfordshire (latitude 51.77, longitude –1.34, Berkshire, UK) with approximate 12000 kilometers (Fig. 12) were separated from each other before 5.11 (2.92~7.55) Mya, and were closest relatives to E. corollae (Fig. 7).

Genome evolution of Episyrphus balteatus and 19 other insects. Time-calibrated phylogenetic tree inferred from 545 single-copy orthologs using IQ-TREE version 1.6.11 was constructed based on maximum likelihood. The branch node values indicate the inferred divergence time between species. At the base of the tree is geological time, and at the top of the tree is absolute age in millions of years, with shadows defining each geological period. D. is Devonian; Ca. is Carboniferous; Pe. is Permian; Tr. is Triassic; Ju. is Jurassic; Cr. is Cretaceous; Pa. is Paleogene; N. is Neogene. The numbers of expanded gene families (red) and contracted gene families (blue) are displayed to the right of each species branch.
Based on the determined gene families and the phylogenetic tree created to estimate the divergence time of these species, we used CAFE (version 4.2)64 to predict the expansion and contraction of the gene families in relation to their ancestors. Finally, KEGG pathways enrichment revealed that the 1,060 expanded gene families in E. balteatus genome were maily related to “nucleotide excision repair”, “Toll and Imd signaling pathway”, “starch and sucrose metabolism”(Fig. 8a), and the 1,535 contracted gene families were mostly related to “glycerolipid metabolism”, “fatty acid degradation”, “insect bormone biosynthesis” (Fig. 9a). As for GO enrichment analysis, it was found that expanded gene families in E. balteatus genome mainly involved “DNA integration”, “obsolete membrane part”, and “Ion transmembrane transporter activity” (Figs. 7, 8b,c,d), while the contracted gene families mainly involved “lipid metabolic process”, “plasma membrane”, and “monooxygenase activity” (Figs. 7, 9b,c,d).

KEGG (a) and GO enrichment (b–d) analyses of expanded gene families in Episyrphus balteatus genome.

KEGG (a) and Go (b-d) enrichment analyses of contracted gene families in Episyrphus balteatus genome.

Circular map of Episyrphus balteatus mitochondrial genome with 37 annotated genes of different functional groups.

PCA (principal components analysis) diagrams of transcriptome samples of Episyrphus balteatus at different development periods (a) and corresponding clustering heat maps of transcriptome samples associated with each other at different developmental periods of Episyrphus balteatus (b). Different periods are represented by different colored circles. In terms of correlation, the darker the red color, the higher the correlation, and the darker the blue color, the lower the correlation. L1, L2, L3 represent 1st, 2nd, 3rd instar larvae of Episyrphus balteatus, respectively.

Episyrphus balteatus sample acquisition locations. In this study, a female E. balteatus was originally collected from Anyang City, Henan Province, China and inbreeding reared under controlled laboratory conditions for approximately 10 generations before the start of all experiments. In contrast, in the study of Hawkes and Sivell65, a female E. balteatus specimen was collected from Wytham Woods, Oxfordshire, UK (latitude 51.77, longitude –1.34), and then used for DNA sequencing directly. In Doyle et al. study79, two female Episyrphus (KatzBiotech strain) individuals captured in mountain pass of Bujaruelo (Puerto de Bujaruelo), a 2273 m pass on the French–Spanish border in the Pyrenees, were used for DNA sequencing directly without species identification. (The map image is derived from ArcGIS Online, copyrighted by Esri, and can be used in academic publications).
Code availability
The utilization of bioinformatics software and tools for this research was performed in accordance with published protocols and manuals obtained from public databases. The method provided a description of the software version and parameters without employing any specific code or scripts.
References
Wotton, K. R. et al. Mass seasonal migrations of hoverflies provide extensive pollination and crop protection services. Current biology: CB 29, 2167–2173.e2165, https://doi.org/10.1016/j.cub.2019.05.036 (2019).
Powney, G. D. et al. Widespread losses of pollinating insects in Britain. Nature communications 10, 1018, https://doi.org/10.1038/s41467-019-08974-9 (2019).
Yuan, H. et al. Genome of the hoverfly Eupeodes corollae provides insights into the evolution of predation and pollination in insects. BMC biology 20, 157, https://doi.org/10.1186/s12915-022-01356-6 (2022).
Chen, S., Zhou, Y., Chen, Y. & Gu, J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics (Oxford, England) 34, i884–i890, https://doi.org/10.1093/bioinformatics/bty560 (2018).
Altschul, S. F., Gish, W., Miller, W., Myers, E. W. & Lipman, D. J. Basic local alignment search tool. Journal of molecular biology 215, 403–410, https://doi.org/10.1016/s0022-2836(05)80360-2 (1990).
Pu, D. Q. et al. Mitochondrial genomes of the hoverflies Episyrphus balteatus and Eupeodes corollae (Diptera: Syrphidae), with a phylogenetic analysis of Muscomorpha. Scientific reports 7, 44300, https://doi.org/10.1038/srep44300 (2017).
Li, R., Li, Y., Kristiansen, K. & Wang, J. SOAP: short oligonucleotide alignment program. Bioinformatics (Oxford, England) 24, 713–714, https://doi.org/10.1093/bioinformatics/btn025 (2008).
Marçais, G. & Kingsford, C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics (Oxford, England) 27, 764–770, https://doi.org/10.1093/bioinformatics/btr011 (2011).
Ranallo-Benavidez, T. R., Jaron, K. S. & Schatz, M. C. GenomeScope 2.0 and Smudgeplot for reference-free profiling of polyploid genomes. Nature communications 11, 1432, https://doi.org/10.1038/s41467-020-14998-3 (2020).
Liu, H., Wu, S., Li, A. & Ruan, J. SMARTdenovo: a de novo assembler using long noisy reads. GigaByte (Hong Kong, China) 2021, gigabyte15, https://doi.org/10.46471/gigabyte.15 (2021).
Walker, B. J. et al. Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PloS one 9, e112963, https://doi.org/10.1371/journal.pone.0112963 (2014).
Guan, D. et al. Identifying and removing haplotypic duplication in primary genome assemblies. Bioinformatics (Oxford, England) 36, 2896–2898, https://doi.org/10.1093/bioinformatics/btaa025 (2020).
Vaser, R., Sović, I., Nagarajan, N. & Šikić, M. Fast and accurate de novo genome assembly from long uncorrected reads. Genome research 27, 737–746, https://doi.org/10.1101/gr.214270.116 (2017).
Rao, S. S. et al. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 159, 1665–1680, https://doi.org/10.1016/j.cell.2014.11.021 (2014).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics (Oxford, England) 25, 1754–1760, https://doi.org/10.1093/bioinformatics/btp324 (2009).
Burton, J. N. et al. Chromosome-scale scaffolding of de novo genome assemblies based on chromatin interactions. Nature biotechnology 31, 1119–1125, https://doi.org/10.1038/nbt.2727 (2013).
Meng, G., Li, Y., Yang, C. & Liu, S. MitoZ: a toolkit for animal mitochondrial genome assembly, annotation and visualization. Nucleic acids research 47, e63, https://doi.org/10.1093/nar/gkz173 (2019).
Dierckxsens, N., Mardulyn, P. & Smits, G. NOVOPlasty: de novo assembly of organelle genomes from whole genome data. Nucleic acids research 45, e18, https://doi.org/10.1093/nar/gkw955 (2017).
Bernt, M. et al. MITOS: improved de novo metazoan mitochondrial genome annotation. Molecular phylogenetics and evolution 69, 313–319, https://doi.org/10.1016/j.ympev.2012.08.023 (2013).
Tillich, M. et al. GeSeq - versatile and accurate annotation of organelle genomes. Nucleic acids research 45, W6–w11, https://doi.org/10.1093/nar/gkx391 (2017).
Flynn, J. M. et al. RepeatModeler2 for automated genomic discovery of transposable element families. Proceedings of the National Academy of Sciences of the United States of America 117, 9451–9457, https://doi.org/10.1073/pnas.1921046117 (2020).
Bao, Z. & Eddy, S. R. Automated de novo identification of repeat sequence families in sequenced genomes. Genome research 12, 1269–1276, https://doi.org/10.1101/gr.88502 (2002).
Price, A. L., Jones, N. C. & Pevzner, P. A. De novo identification of repeat families in large genomes. Bioinformatics (Oxford, England) 21(Suppl 1), i351–358, https://doi.org/10.1093/bioinformatics/bti1018 (2005).
Xu, Z. & Wang, H. LTR_FINDER: an efficient tool for the prediction of full-length LTR retrotransposons. Nucleic acids research 35, W265–268, https://doi.org/10.1093/nar/gkm286 (2007).
Ellinghaus, D., Kurtz, S. & Willhoeft, U. LTRharvest, an efficient and flexible software for de novo detection of LTR retrotransposons. BMC bioinformatics 9, 18, https://doi.org/10.1186/1471-2105-9-18 (2008).
Ou, S. & Jiang, N. LTR_retriever: A Highly Accurate and Sensitive Program for Identification of Long Terminal Repeat Retrotransposons. Plant physiology 176, 1410–1422, https://doi.org/10.1104/pp.17.01310 (2018).
Wheeler, T. J. et al. Dfam: a database of repetitive DNA based on profile hidden Markov models. Nucleic acids research 41, D70–82, https://doi.org/10.1093/nar/gks1265 (2013).
Tarailo-Graovac, M. & Chen, N. Using RepeatMasker to identify repetitive elements in genomic sequences. Current protocols in bioinformatics Chapter 4, 4.10.11-14.10.14, https://doi.org/10.1002/0471250953.bi0410s25 (2009).
Benson, G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic acids research 27, 573–580, https://doi.org/10.1093/nar/27.2.573 (1999).
Beier, S., Thiel, T., Münch, T., Scholz, U. & Mascher, M. MISA-web: a web server for microsatellite prediction. Bioinformatics (Oxford, England) 33, 2583–2585, https://doi.org/10.1093/bioinformatics/btx198 (2017).
Lowe, T. M. & Eddy, S. R. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic acids research 25, 955–964, https://doi.org/10.1093/nar/25.5.955 (1997).
Loman, T. A Novel Method for Predicting Ribosomal RNA Genes in Prokaryotic Genomes. (2017).
Griffiths-Jones, S. et al. Rfam: annotating non-coding RNAs in complete genomes. Nucleic acids research 33, D121–124, https://doi.org/10.1093/nar/gki081 (2005).
Nawrocki, E. P. & Eddy, S. R. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics (Oxford, England) 29, 2933–2935, https://doi.org/10.1093/bioinformatics/btt509 (2013).
Stanke, M., Diekhans, M., Baertsch, R. & Haussler, D. Using native and syntenically mapped cDNA alignments to improve de novo gene finding. Bioinformatics (Oxford, England) 24, 637–644, https://doi.org/10.1093/bioinformatics/btn013 (2008).
Korf, I. Gene finding in novel genomes. BMC bioinformatics 5, 59, https://doi.org/10.1186/1471-2105-5-59 (2004).
Keilwagen, J. et al. Using intron position conservation for homology-based gene prediction. Nucleic acids research 44, e89, https://doi.org/10.1093/nar/gkw092 (2016).
Kim, D., Langmead, B. & Salzberg, S. L. HISAT: a fast spliced aligner with low memory requirements. Nature methods 12, 357–360, https://doi.org/10.1038/nmeth.3317 (2015).
Pertea, M. et al. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nature biotechnology 33, 290–295, https://doi.org/10.1038/nbt.3122 (2015).
Tang, S., Lomsadze, A. & Borodovsky, M. Identification of protein coding regions in RNA transcripts. Nucleic acids research 43, e78, https://doi.org/10.1093/nar/gkv227 (2015).
Haas, B. J. et al. Improving the Arabidopsis genome annotation using maximal transcript alignment assemblies. Nucleic acids research 31, 5654–5666, https://doi.org/10.1093/nar/gkg770 (2003).
Haas, B. J. et al. Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome biology 9, R7, https://doi.org/10.1186/gb-2008-9-1-r7 (2008).
Simão, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V. & Zdobnov, E. M. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics (Oxford, England) 31, 3210–3212, https://doi.org/10.1093/bioinformatics/btv351 (2015).
Jones, P. et al. InterProScan 5: genome-scale protein function classification. Bioinformatics (Oxford, England) 30, 1236–1240, https://doi.org/10.1093/bioinformatics/btu031 (2014).
Mistry, J. et al. Pfam: The protein families database in 2021. Nucleic acids research 49, D412–D419, https://doi.org/10.1093/nar/gkaa913 (2021).
Buchfink, B., Xie, C. & Huson, D. H. Fast and sensitive protein alignment using DIAMOND. Nature methods 12, 59–60, https://doi.org/10.1038/nmeth.3176 (2015).
Wang, Y. et al. MCScanX: a toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic acids research 40, e49-e49, https://doi.org/10.1093/nar/gkr1293 (2012).
Trapnell, C. et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nature biotechnology 28, 511–515, https://doi.org/10.1038/nbt.1621 (2010).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq. 2. Genome biology 15, 550, https://doi.org/10.1186/s13059-014-0550-8 (2014).
Köster, J., Dijkstra, L. J., Marschall, T. & Schönhuth, A. Varlociraptor: enhancing sensitivity and controlling false discovery rate in somatic indel discovery. Genome biology 21, 98, https://doi.org/10.1186/s13059-020-01993-6 (2020).
NCBI Sequence Read Archive. https://identifiers.org/ncbi/bioproject:PRJNA1049652 (2024).
NCBI Sequence Read Archive. https://identifiers.org/ncbi/insdc.sra:SRR27128105 (2024).
NCBI Sequence Read Archive. https://identifiers.org/ncbi/insdc.sra:SRR27167876 (2024).
NCBI Sequence Read Archive. https://identifiers.org/ncbi/insdc.sra:SRR27129026 (2024).
NCBI Sequence Read Archive. https://identifiers.org/ncbi/insdc.sra:SRR27204218 (2024).
NCBI Sequence Read Archive. https://identifiers.org/ncbi/insdc.sra:SRP477240 (2024).
Ji, J. The annotation files of Episyrphus balteatus genome. https://doi.org/10.6084/m9.figshare.24797310.v2 (2023).
Ji, J. Episyrphus balteatus isolate JJ-2024, whole genome shotgun sequencing project. Genbank https://identifiers.org/ncbi/insdc.gca:GCA_040182855.1 (2024).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics (Oxford, England) 25, 1754–1760, https://doi.org/10.1093/bioinformatics/btp324 (2009).
Emms, D. M. & Kelly, S. OrthoFinder: phylogenetic orthology inference for comparative genomics. Genome biology 20, 238, https://doi.org/10.1186/s13059-019-1832-y (2019).
Katoh, K., Asimenos, G. & Toh, H. Multiple alignment of DNA sequences with MAFFT. Methods in molecular biology (Clifton, N.J.) 537, 39–64, https://doi.org/10.1007/978-1-59745-251-9_3 (2009).
Nguyen, L.-T., Schmidt, H. A., von Haeseler, A. & Minh, B. Q. IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies. Molecular Biology and Evolution 32, 268–274, https://doi.org/10.1093/molbev/msu300 (2014).
Yang, Z. PAML: a program package for phylogenetic analysis by maximum likelihood. Computer applications in the biosciences: CABIOS 13, 555–556, https://doi.org/10.1093/bioinformatics/13.5.555 (1997).
Han, M. V., Thomas, G. W. C., Lugo-Martinez, J. & Hahn, M. W. Estimating Gene Gain and Loss Rates in the Presence of Error in Genome Assembly and Annotation Using CAFE 3. Molecular Biology and Evolution 30, 1987–1997, https://doi.org/10.1093/molbev/mst100 (2013).
Hawkes, W., Sivell, O. & Wotton, K. The genome sequence of the Marmalade Hoverfly, Episyrphus balteatus (De Geer, 1776). Wellcome Open Res 8, 106, https://doi.org/10.12688/wellcomeopenres.19073.1 (2023).
Hawkes, W. & Wotton, K. The genome sequence of the drone fly, Eristalis tenax (Linnaeus, 1758). Wellcome Open Res 6, 307, https://doi.org/10.12688/wellcomeopenres.17357.1 (2021).
Hawkes, W., Sivell, O., Sivell, D., Massy, R. & Wotton, K. The genome sequence of the pied hoverfly, Scaeva pyrastri (Linnaeus, 1758). Wellcome Open Res 8, 83, https://doi.org/10.12688/wellcomeopenres.18892.1 (2023).
Crowley, L., Ashworth, M. & Wawman, D. The genome sequence of the Thick-legged Hoverfly, Syritta pipiens (Linnaeus, 1758). Wellcome Open Res, 349, https://doi.org/10.12688/wellcomeopenres.19848.1 (2023).
Hawkes, W. & Wotton, K. The genome sequence of the tapered dronefly, Eristalis pertinax (Scopoli, 1763). Wellcome Open Res 6, 292, https://doi.org/10.12688/wellcomeopenres.17267.2 (2021).
Crowley, L. M., Mitchell, R., Weston, S. T. & Wotton, K. R. The genome sequence of the Lesser Hornet Hoverfly, Volucella inanis (Linnaeus, 1758). Wellcome Open Res 8, 69, https://doi.org/10.12688/wellcomeopenres.18897.1 (2023).
Crowley, L. & Nash, W. The genome sequence of the Golden-tailed Leafwalker, Xylota sylvarum (Linnaeus, 1758). Wellcome Open Res https://doi.org/10.12688/wellcomeopenres.19241.1 (2023).
Sivell, D., Sivell, O., Hawkes, W. L. & Wotton, K. R. The genome sequence of the Vagrant Hoverfly, Eupeodes corollae (Fabricius, 1794). Wellcome Open Res 8, 112, https://doi.org/10.12688/wellcomeopenres.19099.1 (2023).
Hawkes, W., Wotton, K. & Smith, M. The genome sequence of the two-banded wasp hoverfly, Chrysotoxum bicinctum (Linnaeus, 1758). Wellcome Open Res 6, 321, https://doi.org/10.12688/wellcomeopenres.17382.1 (2021).
Hawkes, W. & Wotton, K. The genome sequence of the plain-faced dronefly, Eristalis arbustorum (Linnaeus, 1758). Wellcome Open Res, 61, https://doi.org/10.12688/wellcomeopenres.17580.1 (2022).
Liu, H., Zhao, L., Li, G., He, Y. & Huo, K. The complete mitochondrial genome of Melanostoma mellinum (Linnaeus, 1758) (Diptera: Syrphidae) and phylogenetic analysis. Mitochondrial DNA B Resour 7, 1664–1665, https://doi.org/10.1080/23802359.2022.2107452 (2022).
Sivell, O. & Sivell, D. The genome sequence of a hoverfly, Xanthogramma pedissequum (Harris, 1776). Wellcome Open Res 7, 38, https://doi.org/10.12688/wellcomeopenres.17559.1 (2022).
Falk, S. & Chua, P. The genome sequence of the meadow field syrph, Eupeodes latifasciatus (Macquart, 1829). Wellcome Open Res 7, 253, https://doi.org/10.12688/wellcomeopenres.18113.1 (2022).
Falk, S. The genome sequence of the large burdock Cheilosia, Cheilosia vulpina (Meigen, 1822). Wellcome Open Res 6, 351, https://doi.org/10.12688/wellcomeopenres.17491.1 (2021).
Doyle, T. et al. Genome-wide transcriptomic changes reveal the genetic pathways involved in insect migration. Molecular ecology 31, 4332–4350, https://doi.org/10.1111/mec.16588 (2022).
Acknowledgements
This work is supported by The Science and Technology Innovation 2030 (2023ZD04062), National Key R&D Program of China (2022YFD1400300), Agricultural Science and Technology Innovation Program of Chinese Academy of Agricultural Sciences, and China Agriculture Research System.
Author information
Authors and Affiliations
Contributions
J.J., J.C., J.L., L.W. and X.Z conceived the project; J.J., L.C. and B.L. performed the experiments; J.J. and Y.G. wrote the manuscript; K.Z., C.X., N.H.F., X.G. and M.G. evaluated the results and edited the manuscript; Y.G. and J.J. performed the bioinformatic analyses; D.L. and P.E. reviewed and edited manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Ji, J., Gao, Y., Xu, C. et al. Chromosome-level genome assembly of marmalade hoverfly Episyrphus balteatus (Diptera: Syrphidae). Sci Data 11, 844 (2024). https://doi.org/10.1038/s41597-024-03666-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-024-03666-6
- Springer Nature Limited